

Abstract
Aims
The goal of this study was to determine whether the A1 adenosine receptor (AR) plays a role in atherosclerosis development and to explore its potential mechanisms.
Methods and results
Double knockout (DKO) mice, deficient in the genes encoding A1 AR and apolipoprotein E (apoE), demonstrated reduced atherosclerotic lesions in aortic arch (en face), aortic root, and innominate arteries when compared with apoE-deficient mice (APOE-KO) of the same age. Treating APOE-KO with an A1 AR antagonist (DPCPX) also led to a concentration-dependent reduction in lesions. The total plasma cholesterol and triglyceride levels were not different between DKO and APOE-KO; however, higher triglyceride was observed in DKO fed a high-fat diet. DKO also had higher body weights than APOE-KO. Plasma cytokine concentrations (IL-5, IL-6, and IL-13) were significantly lower in DKO. Proliferating cell nuclear antigen expression was also significantly reduced in the aorta from DKO. Despite smaller lesions in DKO, the composition of the innominate artery lesion and cholesterol loading and efflux from bone marrow-derived macrophages of DKO were not different from APOE-KO.
Conclusion
The A1 AR may play a role in the development of atherosclerosis, possibly due to its pro-inflammatory and mitogenic properties.
Keywords: A1 adenosine receptor; Apolipoprotein E; Cytokines; 1,3-Dipropyl-8-cyclopentylxanthine (DPCPX); IL-13
1. Introduction
Adenosine, which is known to accumulate in many tissues following an onset of ischaemia or inflammation, is well recognized not only as a major local regulator of vascular tone, but also as a significant determinant in the development of inflammation,1,2 smooth muscle proliferation,3,4 oxidative stress,5–7 and apoptosis,8–10 all of which are hallmarks of atherosclerosis. However, the involvement of adenosine in atherosclerosis is controversial. There are four adenosine receptor (AR) subtypes (A1, A2A, A2B, and A3) involved in mediating the effects of extracellular adenosine, and they may interact differentially in multiple pathways relevant to atherosclerosis. All four ARs are found to co-exist in most tissues; however, the dominant receptor subtypes may vary and the other subtypes may play a modulating or a compensatory role. For instance, A2B AR, a low affinity AR, is known to be activated by high concentrations of adenosine and to augment A2A AR-mediated coronary vasodilation, while A1 AR-mediated vasoconstriction has been shown to counter A2A AR-mediated coronary vasodilation. Of the four AR subtypes, A2A, A2B, and A3 AR have been studied directly in relation to a role in atherosclerosis development.8,11 Although exhibiting anti-inflammatory properties, A2A AR was found to be pro-atherogenic through its anti-apoptosis activity in bone marrow-derived macrophage (BMM) and foam cells, possibly through p38 MAPK pathway.8 A2B AR, on the other hand, regulates liver SREBP-1 to reduce plasma and liver cholesterol and triglycerides, hence reducing atherosclerosis development.12 A3 AR did not seem to affect atherosclerosis development at all.11
In this study, using genetic knockout and pharmacological inhibition, we demonstrate, for the first time, that A1 AR also promotes atherosclerosis development, possibly through its pro-inflammatory and mitogenic properties.
2. Methods
2.1. Animals and treatments
A1 AR knockout mice were kindly provided by Dr Jurgen Schnermann from NIDDK/NIH and bred with apolipoprotein E knockout mice (APOE-KO), both on the C57BL/6 background, to generate A1AR and APOE double knockout mice (DKO) and their littermate controls. After genotyping by polymerase chain reaction analysis, littermates from the F2 generation were used to establish DKO breeding pairs, and the resulting progeny were used in our studies. Mice were euthanized using CO2 exposure [for bone marrow (BM) extraction] or over dose sodium pentobarbital i.p. (100 mg/kg, for other experiments). All care and procedures involving mice were done in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ and were approved by the West Virginia University and Cleveland Clinic Animal Care and Use Committees.
2.1.1. High-fat diet and A1 AR antagonist treatment
Initially, to accelerate the atherosclerotic lesion development (the first set of experiments), 8-week-old APOE-KO and DKO mice were fed a high-fat western-type diet (0.2% cholesterol, 21.2% fat, Harlan Teklad TD88137) for a period of 12 weeks.
In the same experiment, two doses of DPCPX (selective A1 AR antagonist), 0.5 and 2.5 mg/kg/day (dissolved in 50% (v/v) DMSO and 50% polyethylene glycol-400), were given to APOE-KO through Alzet osmotic pumps implanted s.c. for 12 weeks, along with the high-fat diet (HFD) treatment. Isoflurane 3% induction in restraining box and 1.5–2% isoflurane maintenance by mask was used for the surgical implantation.
2.2. Experimental protocols
The first experiment utilized five groups of mice (both sexes in equal number in all groups were sacrificed at 20 weeks of age): APOE-KO, APOE-KO fed an HFD (APOE-HFD), DKO fed an HFD (DKO-HFD), low-dose (0.5 mg/kg/day) DPCPX-treated APOE-KO fed an HFD (APOE-HFD-DPCPX-L), and high-dose (2.5 mg/kg/day) DPCPX-treated APOE-KO fed an HFD (APOE-HFD-DPCPX-H).
The second experiment with normal chow-fed mice utilized three groups of male mice: APOE-KO, DKO, and high-dose DPCPX-treated APOE-KO (APOE-DPCPX). All the mice were sacrificed at 16 weeks of age with the DPCPX treatment during weeks 8–16. Only aortic root lesions were assessed in this experiment.
The third experiment was done with APOE-KO and DKO mice (both on normal chow) sacrificed at 50 weeks of age to confirm that the genotype effect was maintained to this age. In addition to aortic arches (en face) and aortic roots, innominate arteries were also analysed in both groups.
On the day of sacrifice, the animals were placed in new cages and fasted 4–5 h. After anaesthesia with sodium pentobarbital (50 mg/kg) i.p., a thoracotomy was performed. Blood was drawn from ventricles and plasma isolated for analysis. The hearts and aortas were excised and placed in PBS.
2.3. Atherosclerotic lesion measurement
Aortic Arch: en face analysis was done as previously described.13
Aortic Root: the analysis was done as previously described.14
Innominate arteries: the innominate arteries were dissected, processed, and paraffin embedded. Five-micrometre-thick sections from each of the paraffin blocks were generated and every fifth section was stained with a modified Movat's pentachrome stain.15,16
2.4. Plaque size and composition analysis of innominate arteries
The cross-sectional lesion area was determined in each Movat's stained section using computer-assisted morphometric analysis (Image Pro, Media Cybernetics, Silver Spring, MD, USA). We also tabulated the frequency of features of plaque composition in each Movat's stained section. These included the following: thin fibrous cap (defined as <3 cell layers), large necrotic core (defined as occupying >50% of the volume of the plaque), intra-plaque haemorrhage (defined as the presence of red blood cells), medial enlargement/erosion (defined as the replacement of the normal media by plaque components), calcification, presence of foam cells, presence of chondrocyte-like cells, and lateral xanthomas (defined as the presence of aggregates of macrophage-derived foam cells situated on the lateral margins of the plaques). These parameters were recorded as binary outcomes and the frequency per lesion for each animal was determined. The total number of cells in the lesions was determined by counting nuclei in each section and the number was then normalized to the area of the lesion. All data collection was done with the operator blinded to the treatment groups. Lesion sizes of the two mouse groups (APOE-KO and DKO) were compared using two-tailed Student's t-test. The lesion components were compared using the Mann–Whitney test.
2.5. Total cholesterol and triglyceride analysis
Total cholesterol and triglycerides in 20 µL mouse plasma samples were assayed using Vitros DT60 Chemistry System (Johnson & Johnson, New Brunswick, NJ, USA) based on the enzymatic method as described by Spayd et al.17
2.6. Real-time RT–PCR
Total RNA was isolated from the aortas of 20-week-old C57, APOE-KO, and DKO using RNAEasy total RNA isolation kit from Qiagen. This was followed by conversion of 0.5 µg of total RNA into cDNA using high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA, USA) according to the instructions of the manufacturer in a total volume of 100 μL. The PCR volume (25 μL) included 12.5 μL of 2× Taqman Universal Mastermix, 1 μL of cDNA, and 1.25 μL of 20× FAM-labelled Taqman gene expression assay master mix solution. For the real-time PCR for ARs and 18S RNA, the Taqman inventoried gene expression product was purchased from Applied Biosystems. 18S ribosomal RNA (rRNA) was used as an endogenous control. The fold difference in expression of target cDNA was determined using the comparative CT method. The ΔCT value was determined in each experiment by subtracting the average 18S RNA CT value from the corresponding average CT for A1, A2A, A2B, and A3 AR in coronary arteries. The fold difference in gene expression of the target was calculated as the average value from 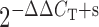 and
and 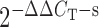 as previously described.18,19
as previously described.18,19
2.7. Western blot
Mouse aorta proteins were isolated in ice-cold tissue lysis buffer consisting of 0.05 M Tris-buffered saline, pH 7.4, 1% Triton X-100, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA, 1 mM phenylmethylsulfonyl, and Halt Protease Inhibitor Cocktail (Thermo Scientific, Waltham, MA, USA) by way of glass mortar and pestle. Samples were then centrifuged for 15 min at 13 000 rpm and the supernatant was stored at −80°C. Protein extracts (30 µg protein per well) were separated on NuPAGE 4–12% Bis–Tris gels (Invitrogen, Carlsbad, CA) along with Novex Sharp Protein Standard, 3.5-260-kDa (Invitrogen) run in parallel. Proteins were then transferred to a PVDF membrane (Millipore, Billerica, MA, USA), blocked by 5% milk for 1 h and then probed with anti-A1 AR rabbit polyclonal IgG antibody (Sigma-Aldrich, St Louis, MO, USA) or anti-PCNA (PC10 clone) mouse monoclonal antibody (Vector Laboratories, Burlingame, CA, USA) with a dilution of 1:1000 in TBST + 0.5% milk overnight at 4°C, or with anti-β-actin antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at a dilution of 1:5000 at room temperature for 1 h. This was followed by incubation with a secondary horseradish peroxidase-conjugated antibody (anti-mouse and anti-rabbit immunoglobulins, respectively; Santa Cruz Biotechnology Inc.) for 1 h at room temperature. For detection of bands, the membranes were treated with an enhanced chemiluminescence reagent (GE Healthcare, Waukesha, WI, USA) for 1 min and subsequently exposed to ECL Hyperfilm (GE Healthcare). Relative band intensities were quantified by densitometry, and each sample was normalized to the β-actin values.
2.8. Cytokine analysis
2.8.1. Multiplex cytokine assay
The plasma levels of IL-5, IL-6, IL-13, TNF-α, and monocyte chemoattractant protein-1 (MCP-1) were measured by a commercial kit from Linco Research (St Charles, MO) using the multiplex Luminex 200 system (Luminex, Austin, TX, USA).
2.9. Bone marrow-derived macrophage cholesterol mass and efflux experiments
2.9.1. Cells
BM cells were flushed from the femur bones of APOE-KO or DKO mice and re-suspended in BM macrophage growth medium (DMEM, 10%FBS, 20% L-cells conditioned media as a source of MSCF), as previously described,20,21 and plated in 24-well (efflux) or 12-well (cholesterol quantification) plates. The media was renewed twice a week until use. After 12 days, the BM cells were confluent and fully differentiated into macrophages and the cells were used for experiments.
2.9.2. Efflux
For basal efflux, BM cells were labelled with 0.5 µCi/mL [3H]-cholesterol in DMEM containing 1% FBS and 10 ng/mL MCSF for 16 h. For efflux from foam cells, FBS was replaced with 50 μg/mL acetylated-low density lipoprotein (AcLDL). After labelling, BM cells were chased for 4 h in DMEM with or without acceptors [10 μg/mL apolipoprotein AI (apoAI) or 100 μg/mL HDL]. At the end of this chase period, the radioactivity in the medium and cells was determined by liquid scintillation counting, and the per cent efflux was calculated as 100 × (medium dpm)/(medium dpm + cell dpm). Finally, percent efflux to acceptors was calculated as (per cent efflux to acceptors) − (percent efflux to DMEM). All treatments were performed in triplicate.
2.9.2.1. Total cholesterol quantification
BM cells from APOE-KO or DKO mice were loaded with 100 μg/mL AcLDL for 48 h (unloaded cells are used as a control) before cholesterol was extracted from cells using hexane:isopropanol (3:2; v:v) and protein extracted using 0.2 N NaOH. Total cholesterol and protein amounts were evaluated as described previously.22 All treatments were performed in triplicate.
2.9.3. Gene chip microarray
Aortas from 20-week-old APOE-KO and DKO (n = 3 each) were used in this experiment. RNA was isolated from tissues using RNAse Micro Kit (Qiagen), according to the manufacturer's instructions. RNA quality was ascertained using an Agilent Bioanalyzer (Genomics Core Facility, West Virginia University). One hundred nanogram of each RNA sample with an RIN value >7 was processed using the Ambion WT Expression Kit according to the manufacturer's instructions. cDNA (5.5 µg) was processed for fragmentation and biotin labelling using the Gene Chip WT Terminal Labelling Kit (Affymetrix). The fragmented and biotin-labelled cDNA (50 µL) with added hybridization controls was hybridized to the mouse GeneChip 1.0 ST Gene Arrays (Affymetrix) and detected with the Affymetrix GeneChip Scanner 3000 7G plus. Expression Consol software (Affymetrix) was used to check quality controls of hybridized chips.
2.10. Microarray data analysis
CEL files were uploaded into Partek (St. Louis, MO, USA) for analysis. Raw data were log2 transformed, and then RMA background correction, quantile normalization, and median polish probeset summarization applied. Two-way ANOVA (genotype and chip processing batch effects), assuming non-equal variance, and least significant difference test were performed and, for genes with multiple probesets, the median of individual values calculated. Significance Analysis of Microarrays (SAM) was used on genes (21 759) to determine those with significant changes between APOE-KO and DKO (while also controlling for batch effects), using a false discovery rate (FDR) of 10%. All genes (21 759) were filtered, using a P-value of 0.05 and fold-change ±1.3 (log2 scale), to 235 genes for pathways and gene–gene interaction network analysis using ingenuity pathways analysis (IPA, Redwood City, CA). The network score is based on a hypergeometric distribution and is the −log of a right-tailed Fisher's exact test result. IPA downstream effect analysis includes predictions of regulatory effect for function annotations. For a particular effect, changes in observed gene expression are compared with literature reports. If more genes were observed to change in a direction that would consistently alter the function, that function was predicted to be altered in that direction (that is, either increased or decreased). A z-score statistic is applied to predict whether the observed numbers of genes changed in the direction to produce functional effect would occur by chance. A z-score >2 or <−2 was considered significant.
2.11. Statistics
Data were expressed as mean ± SEM. All experiments (except Micro Array Analysis) were performed using Student's t- test or one-way ANOVA with Bonferroni's multiple comparison post-test. P-value <0.05 was considered significant.
3. Results
Baseline fasting total cholesterol and triglycerides were not different between APOE-KO and DKO; however, triglycerides were higher in DKO after they were fed an HFD (Table 1). In addition, DKO had increased body weight compared with APOE-KO (32.2 ± 8.0 vs. 26.0 ± 4.3 g, respectively, P < 0.05, Table 1). There is no difference in innominate artery lesion composition between 50-week-old APOE-KO and DKO either (Table 1).
Table 1.
Baseline body weight of the mice fed normal diet, total plasma cholesterol and triglyceride, and total plasma cholesterol and triglyceride of the mice fed HFD
| APOE-KO | DKO | |
|---|---|---|
| 20-week-old (mixed sexes) | ||
| Body Weight (g), n = 24 | 26.05 ± 4.35 | 32.18 ± 8.04* |
| Total cholesterol (mg/dL), n = 8 | 464.00 ± 39.30 | 493.20 ± 54.32 |
| Triglyceride (mg/dL), n = 8 | 126.63 ± 33.55 | 164.40 ± 40.07 |
| After HFD, total cholesterol (mg/dL), n = 5 | 992.80 ± 124.66 | 1068.67 ± 134.64 |
| After HFD, total triglyceride (mg/dL), n = 6 | 37.6 ± 4.34 | 64.33 ± 10.73* |
| 50 week old (all males) | APOE (n = 6) | DKO (n = 11) |
| Innominate artery lesion size (µm2) | 174,864.00 ± 69,327.16 | 83,731.12 ± 58,889.82* |
| Composition (frequency, in %) | ||
| Foam cells | 47.50 ± 26.46 | 52.93 ± 32.85 |
| Xanthomas | 10.42 ± 16.61 | 18.57 ± 23.47 |
| Multiple necrotic regions | 52.28 ± 28.88 | 15.75 ± 26.71 |
| Large necrotic core | 3.33 ± 8.16 | 5.30 ± 11.94 |
| Chondrocyte presence | 81.25 ± 15.31 | 50.58 ± 37.73 |
| Minor to moderate calcification | 26.94 ± 29.82 | 18.00 ± 18.08 |
| Large necrotic calcification | 28.06 ± 35.91 | 26.14 ± 39.04 |
| Calcification in media | 33.33 ± 51.64 | 27.89 ± 32.49 |
| Intraplaque-haemorrhage | None | 2.95 ± 6.78 |
Innominate artery lesion size and lesion composition comparisons between APOE-KO and DKO. Data expressed as mean ± SEM.
*Statistical significance from APOE-KO. P < 0.05.
3.1. Aortic arch en face lesions
In 20-week-old APOE-KO mice, feeding the HFD for 12 weeks increased the size of the aortic arch lesions in APOE-KO from 4.6 ± 3.9 to 29.9 ± 2.8% (Figure 1, P < 0.05). DKO fed the HFD had about half the lesion area of APOE-KO on this diet (14.7 ± 4.9%, P < 0.05 vs. APOE-KO HFD). In addition, we observed dose-dependent decrease in lesion area in DPCPX-treated APOE-KO HFD mice, with lesion areas of 20.0 ± 4.1% for the low dose (0.5 mg/kg/h, APOE-HFD-DPCPX-L) and 11.4 ± 3.5% for the high dose (2.5 mg/kg/h, APOE-HFD-DPCPX-H) (both P < 0.05 vs. APOE-KO HFD).
Figure 1.

(A) Sudan IV staining in aortic arch from APOE-KO normal diet (APOE, n = 6), APOE-KO on HFD (APOE-HFD, n = 11), A1AR/APOE-DKO on HFD (DKO-HFD, n = 9), low dose (0.5 mg/kg/day) of DPCPX-treated APOE-KO on HFD (APOE-HFD-DPCPX-L, n = 4), and high dose (2.5 mg/kg/day) of DPCPX-treated APOE-KO on HFD (APOE-HFD-DPCPX-H). (B) Percentage of atherosclerosis lesions calculated from (A). *Significantly different from APOE. #Significantly different from APOE-HFD, $Significantly different from APOE-HFD-DPCPX-L. Data are expressed as mean ± SEM, P < 0.05.
In 50-week-old mice fed the chow diet, we also observed a 61.3% reduction of aortic arch lesion size in DKO when compared with APOE-KO (Figure 2A, P < 0.05).
Figure 2.

(A) Atherosclerotic lesions in aortas (en face Sudan IV staining) from 50-week-old APOE-KO (APOE) and DKO. Data were expressed as % of aortic arch area. Aortic root lesion comparison between 50-week-old APOE-KO (APOE) and DKO (B); aortic root lesion comparison between 16-week-old APOE-KO (APOE), DKO, and APOE-KO treated with 2.5 mg/kg/day DPCPX (APOE-DPCPX) for 8 weeks (C) Data were measured in µm2. *Significant difference from APOE. Data are expressed as mean ± SEM, P < 0.05.
3.2. Aortic root lesions
In 50-week-old mice fed the chow diet, we also observed a reduction of aortic root lesion size in DKO when compared with APOE-KO (Figure 2B). However, in 20-week-old mice fed the HFD, there was no significant difference in lesion sizes between APOE-KO and DKO (304 826.6 ± 78 174.4 vs. 329 037.7 ± 34 309.6 µm2). We suspected that HFD may overwhelm the genotype effect in this region, so we repeated the experiment in chow-fed mice sacrificed at 16 weeks of age as previously described,23,24 in order to evaluate earlier lesions. Compared with APOE-KO controls, both DKO and high-dose DPCPX-treated APOE-KO had significantly smaller lesions (Figure 2C, P < 0.05 for DKO and DPCPX-treated APOE-KO vs. APOE-KO).
3.3. Innominate artery lesions
In agreement with other lesion sites, the lesion area in DKO was smaller than in APOE-KO at 50 weeks old (Table 1, 83 731 ± 58 889 µm2 in DKO vs. 174 864 ± 69 327 µm2 in APOE-KO, P < 0.05). However, there was no difference between these two groups in the frequency of occurrence of foam cells, necrotic regions, necrotic cores, presence of chondrocytes, and calcification (Table 1).
3.4. RNA and protein analyses
Using real-time RT–PCR, the mRNA levels of A1 AR and A2B AR in aorta were up-regulated in 20-week-old APOE-KO fed an HFD vs. wild-type mice (Figure 3A). Western blot analysis showed up-regulation of both A1 AR and PCNA expression in APOE-KO fed an HFD vs. both wild-type mice and normal chow-fed APOE-KO mice (Figure 3B and C). At 20 weeks of age, there were significant increases of plasma IL-5, IL-6, and IL-13 levels in APOE-KO when compared with the wild-type control that returned to basal levels in the DKO mice. MCP-1 levels also showed a similar trend, but not statistically significant (Figure 4).
Figure 3.

(A) ARs mRNA expression in the aorta from C57BL/6 (C57) and APOE-KO fed an HFD (APOE-HFD) by real-time RT–PCR; (B) A1 AR western blot and densitometry analysis of aortas from C57BL/6 (C57), APOE-KO (APOE), and APOE-KO fed an HFD (APOE-HFD); (C) PCNA western blot and densitometry analysis of aortas from C57, APOE-HFD, and DKO fed an HFD (DKO-HFD). Data are expressed as mean ± SEM. *Significant difference from C57. #Significant difference from APOE in B, from APOE-HFD in C. P < 0.05.
Figure 4.

Plasma levels of IL-5. IL-6, IL-13, and MCP-1 from C57BL/6 (C57), APOE, and DKO on normal chow. Data expressed as mean ± SEM. *Significant difference from C57. #Significant difference from APOE. P < 0.05.
3.5. Microarray analysis
Significant differences in gene expression between DKO and APOE-KO were observed for 24 genes (12 genes up-regulated and 12 down-regulated; see Supplementary material online, Table S1). The top three associated IPA networks are: (i) Endocrine System Disorders, Metabolic Disease, Lipid Metabolism (28 dataset molecules, score 53); (ii) Hematological System Development and Function, Cellular Movement, Immune Cell Trafficking (24 dataset molecules, score 43); (iii) Cell Cycle, DNA Replication, Recombination and Repair, Cellular Compromise (19 dataset molecules, score 31).
In the downstream effect analysis, body mass in DKO was predicted to be significantly increased (z-score = 2.375). This is based on changes in the expression of nine genes in a direction that would increase body mass (BBS4, CISH, IGFBP3, IGFBP6, MAOB, NGF, NR3C2, RETN, VLDLR) and only one gene that would decrease body mass (FASN). Effects predicted to be decreased in DKO (relative to APOE-KO) include functions related to cell migration and movement (z-scores = −2.853 and −2.619), concentration of lipid and fatty acid (z-scores = −2.435 and 2.401), synthesis of fatty acid (z-score = −2.259), and accumulation of triacylglycerol (z-score = −2.000).
Transcription factor analysis predicts activation or inhibition based on changes in gene expression of regulated genes that are consistent with either activation or inhibition of the regulator. Two transcription factors were predicted to be significantly activated in DKO compared with APOE-KO: MED13 (z-score = 2.000) and NFATC2 (z-score = 2.177). Three transcription factors were predicted to be inhibited in DKO compared with APOE-KO: MYC (z-score = −3.133), CEPBA (z-score = −2.559), and NR3C2 (z-score = −2.000).
3.6. Macrophage cholesterol metabolism
APOE-KO macrophages were labelled with [3H]-cholesterol in the presence or absence of cholesterol loading with AcLDL, and efflux of cholesterol to apoAI or HDL was determined. Efflux to HDL in the unloaded state was 9.79 ± 0.46% and was not significantly modified by cholesterol loading of the cells (10.11 ± 1.17%, P = 0.89, Figure 5A). However, efflux to apoAI was 3.7 times higher when cells were loaded, presumably due to ABCA1 induction (from 1.66 ± 0.17 to 6.15 ± 0.47%, P = 0.038, Figure 5A vs. B). Cholesterol efflux from DKO macrophages to apoAI or HDL was not significantly different than that observed from the APOE-KO cells (Figure 5B). Cholesterol mass assays were also performed in macrophages from APOE-KO and DKO mice. APOE-KO unloaded cells have most of their total cholesterol in free cholesterol (TC = 47.0 ± 1.8 µg/mg cell protein and FC = 46.0 ± 0.5 µg/mg cell protein) with barely detectable levels of cholesterol esters (CE) (Figure 5C). After AcLDL loading, TC levels increased 6.6 times to 309.4 ± 26.8 µg/mg cell protein with the majority stored in lipid droplets as CE (235.1 ± 24.2 µg/mg cell protein), while FC levels increased to 74.2 ± 4.2 µg/mg cell protein. DKO macrophages had cholesterol levels similar to APOE-KO cells under both basal and AcLDL-loaded conditions (Figure 5D).
Figure 5.

Cholesterol efflux from unloaded (A), or acetylated-low density lipoprotein (AcLDL)-loaded (B) BMMs from APOE-KO or DKO mice to apolipoprotein AI (ApoAI) and HDL. Cholesterol levels in unloaded (C), or AcLDL-loaded (D) BMMs from APOE-KO and DKO. TC, total cholesterol; FC, free cholesterol; CE, cholesterol esters.
4. Discussion
Adenosine is well known for its anti-inflammatory properties, mainly through the activation of A2A AR; however, other receptor subtypes also play a role in various inflammatory processes. However, the role of inflammation in AR-mediated atherosclerosis development is far from clear. Despite the higher proinflammatory cytokine levels, atherosclerotic lesion size in the aortas of A2A AR/APOE DKO mice was decreased compared with APOE-KO mice.8 The role of inflammation in APOE-KO mice with deleted A2B or A3 ARs were not studied in previous reports,11,25 while in our study, this is the first report to show clear correlation of reduced atherosclerotic lesions along with reduced inflammation in response to A1 AR deletion or pharmacological inhibition. Our results strongly suggest the pro-inflammatory property of A1 AR that may pay a significant role in atherosclerosis development. What is intriguing in our study is the profound higher level of IL-13, a Th2 cytokine, found in APOE-KO (405.12 ± 107.60 pg/mL) and the significant reduction in DKO mice (144.61 ± 192.50 pg/mL) (Figure 4). In a study of plasma markers in patients with coronary artery disease, IL-13 is increased.26 A correlation was present between the severity of the coronary lesions and serum IL-13 concentrations in patients with unstable angina treated with coronary angioplasty.27 IL-13 treatment has been shown to damage endothelium, induces leucocyte attachment and infiltration through compliment-activated antibody-dependent cytotoxicity, and enhances atherosclerosis development in APOE-KO.28 Previous study also demonstrates that IL-13 is strongly induced in adenosine deaminase-deficient mice.29 Significant increases in A1, A2B, and A3 AR in IL-13 overexpressing lung also underscore the importance of the AR subtypes involved.29 Thus, there is a clear link between A1 AR and IL-13 production. Further study of the source (inflammatory cell type) of IL-13 and other cytokines in the A1 AR-mediated pro-atherosclerotic effect is needed.
Smooth muscle cell proliferation is one of the major mechanisms in atherosclerosis development. Adenosine is also known to mediate mitogenic response in smooth muscle cells. A1 AR stimulation increases cellular DNA content, protein synthesis, cell number, and protein proliferating cell nuclear antigen (PCNA) staining in experiments using porcine coronary smooth muscle cells, suggesting mitogenic action of A1 AR (possibly through PI3/AKT/MAPKS pathway).3,4 Using western blot analysis, our study showed PCNA expression was clearly reduced in DKO mice (Figure 3C), which correlates with the decreased size of the atherosclerotic lesions. These results imply that the A1 AR-mediated smooth muscle proliferation may play a role in A1 AR-mediated atherosclerosis development.
IL-13 also plays a significant role in vascular smooth muscle proliferation.30–32 Studies in rat aortic vascular smooth muscle cells indicate that IL-13 and IL-4 stimulate up-regulation of ornithine decarboxylase (ODC) and arginase I expression, possibly through cAMP and JAK/STAT6 pathway.32,33 ODC is the first rate-controlling enzyme in the synthesis of polyamines and is essential for normal cell growth, while arginase I has been shown to promote cell proliferation.33,34 Combined with the fact that A1 AR may be involved in Th17 to Th2 conversion, which will release IL-13,35 and the plasma level of IL-13 is reduced in A1 AR knockout mice in our previous study36 and DKO in our current study (Figure 4), we suspect that, besides the previous known A1AR→PKC→PI3→AKT→MAPK pathway, a possible A1AR→IL-13→cAMP→JAK/STAT6→arginase I pathway may also play a significant role under pathophysiologial condition, such as atherosclerosis.
The infiltration of blood-borne monocytes into nascent atherosclerotic lesions is one of the earliest responses to the subendothelial matrix retention of apolipoprotein B, which is the universal initiating event of atherosclerosis. The monocytes differentiate into macrophages in the subendothelium and begin to ingest the retained lipoproteins.37 ATP-binding cassette 1 (ABCA1) has been strongly implicated as one of the most important regulators of cholesterol efflux in macrophage.38 Previous reports suggest an important role of cAMP as modulating murine atherosclerotic plaque progression, by inducing the ABCA1 secretory pathway, thus removing excess free cholesterol from macrophages and reducing circulating levels of cholesterol.39,40 Indeed, CGS-21680, an A2A agonist that increases cAMP, elevates the expression of ABCA1 and this effect is inhibited by A2A AR antagonist.41 Hence, A1 AR, which is known to reduce cAMP levels, may have the potential to reduce ABCA1 expression, and thus reduce cholesterol efflux and promote foam cell formation. However, we did not observe any difference between BMMs from APOE-KO and DKO. Thus, ABCA1 and cholesterol efflux mechanism may not play any role in A1 AR-mediated atherosclerosis development. In addition, the existence of A1 AR on macrophages is controversial42,43 and macrophages are known for their heterogeneity, and change their phenotype based upon the environmental conditions.44 For instance, a recent study in BMM/osteoclast demonstrates that A1 AR expression increases day after day in BMM after stimulation by CSF-1 and RANKL.43 The actual phenotypes of the macrophage in the plaque is not known. Therefore, this issue needs further investigation.
Up-regulation of A1 AR is found under pathological conditions, such as oxidative stress, ischaemia, diabetes, obesity, and inflammation.45–52 In this study, we also found the same up-regulation of A1 AR in atherosclerotic mouse aorta at both the RNA and protein levels. We also found increased A2B AR mRNA in atherosclerotic aorta (Figure 3). The simultaneous increase of A1AR and A2B AR in APOE-KO fed HFD animals may indicate an antagonistic balance between the two receptor subtypes. In a prior publication, the transcription factor SREBP-1 and its downstream effectors, which are indirectly required for cholesterol biosynthesis and for uptake and fatty acid biosynthesis, are elevated in livers of A2B AR/APOE DKO mice fed an HFD.12 In our gene chip analysis, decreased expression of four genes (Cfd, Fasn, Retn, and Thrsp) that up-regulate SREBP-1 was observed in DKO (see Supplementary material online, Table S1), suggesting that a counter effect may exist between A1 AR and A2B AR in this pathway. In addition, total cholesterol levels were elevated in A2B AR/APOE DKO mice fed an HFD,12 but were not altered in the DKO fed an HFD in the current study (Table 1), suggesting that the up-regulated A2B AR in DKO may be involved in lowering total cholesterol when A1 AR was deleted.
Another interesting finding is that DKO weighed more than APOE-KO (Table 1). A previous study has shown up-regulation of A1 AR in adipose tissue from obese American women of European and African descent.52 Indeed, A1 AR is known to play a significant role in lipolysis and reduces circulating free fatty acids;53 and some A1 AR partial agonists have been tested in clinical trials as insulin-sensitizing agents.54 It has also been shown that phenyl–isopropyl–adenosine (PIA), an A1 AR agonist, causes reduction in free fatty acid and triglyceride concentrations in normal and hyper-triglyceridemic rats.55 We did not find significant differences in plasma total cholesterol and triglyceride levels between chow-fed APOE-KO and DKO. However, in HFD-fed mice, plasma triglyceride levels were significantly higher in DKO (Table 1), supporting the previous finding that A1 AR plays a significant role in triglyceride metabolism. In addition, stimulation of oxidative stress by agents such as H2O2, A1 AR was up-regulated in hamster ductus deferens (DDT1 MF-2) smooth muscle cells5 and in chinchilla cochlea via activation of NF-κΒ.56,57 We speculate that the A1 AR up-regulation may partly (by hyperlipidemic-induced oxidative stress) contribute to atherosclerosis, as a positive feedback loop.
In summary, the removal or inhibition of A1 AR in APOE-KO clearly reduced atherosclerosis development in all the vascular sites measured (aortic arch, aortic root, and innominate arteries). However, feeding HFD may affect the efficacy of A1 AR inhibition in aortic root lesions. Although total baseline cholesterol and triglyceride levels were not different between APOE-KO and DKO fed normal chow diet, higher triglyceride levels were observed in DKO fed an HFD. Higher body weight was also observed in DKO. An increase in plasma cytokines in APOE-KO and a reduction in DKO suggest the pro-inflammatory properties of A1 AR that may play a role in atherosclerosis development. In addition, A1 AR-mediated mitogenic responses may also be involved.
Although the use of genetic knockout mice serves as a great tool in narrowing possible pathway(s) in our study, the limitation of this study is also the use of mouse model because many times the findings may not be translated to humans. Further studies of these proposed mechanisms are needed in human tissue/subject.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by West Virginia University [Research Funding Development Grant to B.T.]; and National Institute of Health [HL 094447, U54GM104942, and HL 027339 to S.J.M., HL 74001 to R.R.M., HL 098193 to J.D.S., and GM103434 to M.E.D.].
Supplementary Material
Acknowledgements
We thank Mr Kevin Roush, Dr Daniel Fil, Ms Wioletta Szeszel-Fedorowicz, Ms Sherry Xie, and Mr Jerry L. Ricks for their excellent technical expertise.
Conflict of interest: none declared.
References
- 1.Wilson CN, Nadeem A, Spina D, Brown R, Page CP, Mustafa SJ. Adenosine receptors and asthma. Handb Exp Pharmacol. 2009;193:329–362. doi: 10.1007/978-3-540-89615-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowaluk EA. Adenosine modulation: a novel approach to analgesia and inflammation. Expert Opin Investig Drugs. 1998;7:535–543. doi: 10.1517/13543784.7.4.535. [DOI] [PubMed] [Google Scholar]
- 3.Shen J, Halenda SP, Sturek M, Wilden PA. Cell-signaling evidence for adenosine stimulation of coronary smooth muscle proliferation via the A1 adenosine receptor. Circ Res. 2005;97:574–582. doi: 10.1161/01.RES.0000181159.83588.4b. [DOI] [PubMed] [Google Scholar]
- 4.Shen J, Halenda SP, Sturek M, Wilden PA. Novel mitogenic effect of adenosine on coronary artery smooth muscle cells: role for the A1 adenosine receptor. Circ Res. 2005;96:982–990. doi: 10.1161/01.RES.0000165800.81876.52. [DOI] [PubMed] [Google Scholar]
- 5.Ramkumar V, Hallam DM, Nie Z. Adenosine, oxidative stress and cytoprotection. Jpn J Pharmacol. 2001;86:265–274. doi: 10.1254/jjp.86.265. [DOI] [PubMed] [Google Scholar]
- 6.Hayes ES. Adenosine receptors and cardiovascular disease: the adenosine-1 receptor (A1) and A1 selective ligands. Cardiovasc Toxicol. 2003;3:71–88. doi: 10.1385/ct:3:1:71. [DOI] [PubMed] [Google Scholar]
- 7.El-Awady MS, Ansari HR, Fil D, Tilley SL, Mustafa SJ. NADPH oxidase pathway is involved in aortic contraction induced by A3 adenosine receptor in mice. J Pharmacol Exp Ther. 2011;338:711–717. doi: 10.1124/jpet.111.180828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Zhang W, Zhu C, Bucher C, Blazar BR, Zhang C, et al. Inactivation of the Adenosine A2A Receptor Protects Apolipoprotein E-Deficient Mice From Atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1046–1052. doi: 10.1161/ATVBAHA.109.188839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pingle SC, Jajoo S, Mukherjea D, Sniderhan LF, Jhaveri KA, Marcuzzi A, et al. Activation of the adenosine A1 receptor inhibits HIV-1 tat-induced apoptosis by reducing nuclear factor-kappaB activation and inducible nitric-oxide synthase. Mol Pharmacol. 2007;72:856–867. doi: 10.1124/mol.106.031427. [DOI] [PubMed] [Google Scholar]
- 10.Peyot ML, Gadeau AP, Dandre F, Belloc I, Dupuch F, Desgranges C. Extracellular adenosine induces apoptosis of human arterial smooth muscle cells via A(2b)-purinoceptor. Circ Res. 2000;86:76–85. doi: 10.1161/01.res.86.1.76. [DOI] [PubMed] [Google Scholar]
- 11.Jones MR, Zhao Z, Sullivan CP, Schreiber BM, Stone PJ, Toselli PA, et al. A(3) adenosine receptor deficiency does not influence atherogenesis. J Cell Biochem. 2004;92:1034–1043. doi: 10.1002/jcb.20122. [DOI] [PubMed] [Google Scholar]
- 12.Koupenova M, Johnston-Cox H, Vezeridis A, Gavras H, Yang D, Zannis V, et al. The A2b adenosine receptor regulates hyperlipidemia and atherosclerosis. Circulation. 2012;125:354–363. doi: 10.1161/CIRCULATIONAHA.111.057596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baglione J, Smith JD. Quantitative assay for mouse atherosclerosis in the aortic root. In: Wang QK, editor. Methods in Molecular Medicine, Vol 129: Cardiovascular Disease: Methods and Protocols. Totowa, NJ: Humana Press; 2006. pp. 83–95. [DOI] [PubMed] [Google Scholar]
- 15.Elbadawi A. Hexachrome modification of Movat's stain. Stain Technol. 1976;51:249–253. doi: 10.3109/10520297609116713. [DOI] [PubMed] [Google Scholar]
- 16.Russell HK., Jr A modification of Movat's pentachrome stain. Arch Pathol. 1972;94:187–191. [PubMed] [Google Scholar]
- 17.Spayd RW, Bruschi B, Burdick BA, Dappen GM, Eikenberry JN, Esders TW, et al. Multilayer film elements for clinical analysis: applications to representative chemical determinations. Clin Chem. 1978;24:1343–1350. [PubMed] [Google Scholar]
- 18.Morrison RR, Teng B, Oldenburg PJ, Katwa LC, Schnermann JB, Mustafa SJ. Effects of targeted deletion of A1 adenosine receptors on postischemic cardiac function and expression of adenosine receptor subtypes. Am J Physiol Heart Circ Physiol. 2006;291:H1875–H1882. doi: 10.1152/ajpheart.00158.2005. [DOI] [PubMed] [Google Scholar]
- 19.Nadeem A, Fan M, Ansari HR, Ledent C, Jamal Mustafa S. Enhanced airway reactivity and inflammation in A2A adenosine receptor-deficient allergic mice. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1335–L1344. doi: 10.1152/ajplung.00416.2006. [DOI] [PubMed] [Google Scholar]
- 20.Austin PE, McCulloch EA, Till JE. Characterization of the factor in L-cell conditioned medium capable of stimulating colony formation by mouse marrow cells in culture. J Cell Physiol. 1971;77:121–134. doi: 10.1002/jcp.1040770202. [DOI] [PubMed] [Google Scholar]
- 21.Knight KR, Vairo G, Hamilton JA. Regulation of pinocytosis in murine macrophages by colony-stimulating factors and other agents. J Leukoc Biol. 1992;51:350–359. doi: 10.1002/jlb.51.4.350. [DOI] [PubMed] [Google Scholar]
- 22.Robinet P, Wang Z, Hazen SL, Smith JD. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res. 2010;51:3364–3369. doi: 10.1194/jlr.D007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandrasekharan UM, Mavrakis L, Bonfield TL, Smith JD, DiCorleto PE. Decreased atherosclerosis in mice deficient in tumor necrosis factor-alpha receptor-II (p75) Arterioscler Thromb Vasc Biol. 2007;27:e16–e17. doi: 10.1161/01.ATV.0000255551.33365.22. [DOI] [PubMed] [Google Scholar]
- 24.Smith JD, Peng DQ, Dansky HM, Settle M, Baglione J, Le Goff W, et al. Transcriptome profile of macrophages from atherosclerosis-sensitive and atherosclerosis-resistant mice. Mamm Genome. 2006;17:220–229. doi: 10.1007/s00335-005-0099-7. [DOI] [PubMed] [Google Scholar]
- 25.Koupenova M, Johnston-Cox H, Vezeridis A, Gavras H, Yang D, Zannis V, et al. A2b adenosine receptor regulates hyperlipidemia and atherosclerosis. Circulation. 2012;125:354–363. doi: 10.1161/CIRCULATIONAHA.111.057596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jha HC, Divya A, Prasad J, Mittal A. Plasma circulatory markers in male and female patients with coronary artery disease. Heart Lung. 2010;39:296–303. doi: 10.1016/j.hrtlng.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Brunetti ND, Pepe M, Munno I, Tiecco F, Quagliara D, De Gennaro L, et al. Th2-dependent cytokine release in patients treated with coronary angioplasty. Coron Artery Dis. 2008;19:133–137. doi: 10.1097/MCA.0b013e3282f3fbcb. [DOI] [PubMed] [Google Scholar]
- 28.Foteinos G, Afzal AR, Mandal K, Jahangiri M, Xu Q. Anti-heat shock protein 60 autoantibodies induce atherosclerosis in apolipoprotein E-deficient mice via endothelial damage. Circulation. 2005;112:1206–1213. doi: 10.1161/CIRCULATIONAHA.105.547414. [DOI] [PubMed] [Google Scholar]
- 29.Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, et al. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margulis A, Nocka KH, Brennan AM, Deng B, Fleming M, Goldman SJ, et al. Mast cell-dependent contraction of human airway smooth muscle cell-containing collagen gels: influence of cytokines, matrix metalloproteases, and serine proteases. J Immunol. 2009;183:1739–1750. doi: 10.4049/jimmunol.0803951. [DOI] [PubMed] [Google Scholar]
- 31.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–690. doi: 10.1067/mai.2003.1333. quiz 691. [DOI] [PubMed] [Google Scholar]
- 32.Wei LH, Jacobs AT, Morris SM, Jr, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- 33.Wei LH, Yang Y, Wu G, Ignarro LJ. IL-4 and IL-13 upregulate ornithine decarboxylase expression by PI3K and MAP kinase pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2008;294:C1198–C1205. doi: 10.1152/ajpcell.00325.2007. [DOI] [PubMed] [Google Scholar]
- 34.Wei LH, Wu G, Morris SM, Jr, Ignarro LJ. Elevated arginase I expression in rat aortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci USA. 2001;98:9260–9264. doi: 10.1073/pnas.161294898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon HG, Tae YM, Kim YS, Gyu Jeon S, Oh SY, Song Gho Y, et al. Conversion of Th17-type into Th2-type inflammation by acetyl salicylic acid via the adenosine and uric acid pathway in the lung. Allergy. 2010;65:1093–1103. doi: 10.1111/j.1398-9995.2010.02352.x. [DOI] [PubMed] [Google Scholar]
- 36.Ponnoth DS, Nadeem A, Tilley S, Mustafa SJ. A1 Adenosine Receptor Causes Inflammation and Impaired Vasorelaxation in a Murine Model of Asthma. Am J Respir Crit Care Med. 2008;177:A532. [Google Scholar]
- 37.Tabas I, Seimon T, Timmins J, Li G, Lim W. Macrophage apoptosis in advanced atherosclerosis. Ann N Y Acad Sci. 2009;1173((Suppl. 1):E40–E45. doi: 10.1111/j.1749-6632.2009.04957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krause BR, Auerbach BJ. Reverse cholesterol transport and future pharmacological approaches to the treatment of atherosclerosis. Curr Opin Investig Drugs. 2001;2:375–381. [PubMed] [Google Scholar]
- 39.Fantidis P. The role of intracellular 3′5′-cyclic adenosine monophosphate (cAMP) in atherosclerosis. Curr Vasc Pharmacol. 2010;8:464–472. doi: 10.2174/157016110791330843. [DOI] [PubMed] [Google Scholar]
- 40.Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 41.Reiss AB, Rahman MM, Chan ES, Montesinos MC, Awadallah NW, Cronstein BN. Adenosine A2A receptor occupancy stimulates expression of proteins involved in reverse cholesterol transport and inhibits foam cell formation in macrophages. J Leukoc Biol. 2004;76:727–734. doi: 10.1189/jlb.0204107. [DOI] [PubMed] [Google Scholar]
- 42.Streitova D, Hofer M, Hola J, Vacek A, Pospisil M. Adenosine A(1), A(2a), A(2b), and A(3) receptors in hematopoiesis. 2. Expression of receptor mRNA in resting and lipopolysaccharide-activated mouse RAW 264.7 macrophages. Physiol Res. 2010;59:139–144. doi: 10.33549/physiolres.931724. [DOI] [PubMed] [Google Scholar]
- 43.Kara FM, Chitu V, Sloane J, Axelrod M, Fredholm BB, Stanley ER, et al. Adenosine A1 receptors (A1Rs) play a critical role in osteoclast formation and function. FASEB J. 2010;24:2325–2333. doi: 10.1096/fj.09-147447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoger JL, Goossens P, de Winther MP. Macrophage heterogeneity: relevance and functional implications in atherosclerosis. Curr Vasc Pharmacol. 2010;8:233–248. doi: 10.2174/157016110790886983. [DOI] [PubMed] [Google Scholar]
- 45.Funakoshi H, Zacharia LC, Tang Z, Zhang J, Lee LL, Good JC, et al. A1 adenosine receptor upregulation accompanies decreasing myocardial adenosine levels in mice with left ventricular dysfunction. Circulation. 2007;115:2307–2315. doi: 10.1161/CIRCULATIONAHA.107.694596. [DOI] [PubMed] [Google Scholar]
- 46.Grden M, Podgorska M, Szutowicz A, Pawelczyk T. Diabetes-induced alterations of adenosine receptors expression level in rat liver. Exp Mol Pathol. 2007;83:392–398. doi: 10.1016/j.yexmp.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Lai DM, Tu YK, Liu IM, Cheng JT. Increase of adenosine A1 receptor gene expression in cerebral ischemia of Wistar rats. Neurosci Lett. 2005;387:59–61. doi: 10.1016/j.neulet.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 48.Liu IM, Tzeng TF, Tsai CC, Lai TY, Chang CT, Cheng JT. Increase in adenosine A1 receptor gene expression in the liver of streptozotocin-induced diabetic rats. Diabetes Metab Res Rev. 2003;19:209–215. doi: 10.1002/dmrr.369. [DOI] [PubMed] [Google Scholar]
- 49.Nie Z, Mei Y, Ford M, Rybak L, Marcuzzi A, Ren H, et al. Oxidative stress increases A1 adenosine receptor expression by activating nuclear factor kappa B. Mol Pharmacol. 1998;53:663–669. doi: 10.1124/mol.53.4.663. [DOI] [PubMed] [Google Scholar]
- 50.Pawelczyk T, Grden M, Rzepko R, Sakowicz M, Szutowicz A. Region-specific alterations of adenosine receptors expression level in kidney of diabetic rat. Am J Pathol. 2005;167:315–325. doi: 10.1016/S0002-9440(10)62977-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rogachev B, Ziv NY, Mazar J, Nakav S, Chaimovitz C, Zlotnik M, et al. Adenosine is upregulated during peritonitis and is involved in downregulation of inflammation. Kidney Int. 2006;70:675–681. doi: 10.1038/sj.ki.5001609. [DOI] [PubMed] [Google Scholar]
- 52.Barakat H, Davis J, Lang D, Mustafa SJ, McConnaughey MM. Differences in the expression of the adenosine A1 receptor in adipose tissue of obese black and white women. J Clin Endocrinol Metab. 2006;91:1882–1886. doi: 10.1210/jc.2005-2109. [DOI] [PubMed] [Google Scholar]
- 53.Dhalla AK, Wong MY, Voshol PJ, Belardinelli L, Reaven GM. A1 adenosine receptor partial agonist lowers plasma FFA and improves insulin resistance induced by high-fat diet in rodents. Am J Physiol Endocrinol Metab. 2007;292:E1358–E1363. doi: 10.1152/ajpendo.00573.2006. [DOI] [PubMed] [Google Scholar]
- 54.Elzein E, Zablocki J. A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin Investig Drugs. 2008;17:1901–1910. doi: 10.1517/13543780802497284. [DOI] [PubMed] [Google Scholar]
- 55.Hoffman BB, Dall'Aglio E, Hollenbeck C, Chang H, Reaven GM. Suppression of free fatty acids and triglycerides in normal and hypertriglyceridemic rats by the adenosine receptor agonist phenylisopropyladenosine. J Pharmacol Exp Ther. 1986;239:715–718. [PubMed] [Google Scholar]
- 56.Ford MS, Maggirwar SB, Rybak LP, Whitworth C, Ramkumar V. Expression and function of adenosine receptors in the chinchilla cochlea. Hear Res. 1997;105:130–140. doi: 10.1016/s0378-5955(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 57.Ramkumar V, Whitworth CA, Pingle SC, Hughes LF, Rybak LP. Noise induces A1 adenosine receptor expression in the chinchilla cochlea. Hear Res. 2004;188:47–56. doi: 10.1016/S0378-5955(03)00344-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.