

Flow cytometry-based diagnostic technique should facilitate the diagnosis of patients with CMCD, with gain-of-function STAT1 mutations.
Keywords: CMCD, STAT1, GOF, pSTAT1
Abstract
CMCD is a rare congenital disorder characterized by persistent or recurrent skin, nail, and mucosal membrane infections caused by Candida albicans. Heterozygous GOF STAT1 mutations have been shown to confer AD CMCD as a result of impaired dephosphorylation of STAT1. We aimed to identify and characterize STAT1 mutations in CMCD patients and to develop a simple diagnostic assay of CMCD. Genetic analysis of STAT1 was performed in patients and their relatives. The mutations identified were characterized by immunoblot and reporter assay using transient gene expression experiments. Patients' leukocytes are investigated by flow cytometry and immunoblot. Six GOF mutations were identified, three of which are reported for the first time, that affect the CCD and DBD of STAT1 in two sporadic and four multiplex cases in 10 CMCD patients from Japan. Two of the 10 patients presented with clinical symptoms atypical to CMCD, including other fungal and viral infections, and three patients developed bronchiectasis. Immunoblot analyses of patients' leukocytes showed abnormally high levels of pSTAT1 following IFN-γ stimulation. Based on this finding, we performed a flow cytometry-based functional analysis of STAT1 GOF alleles using IFN-γ stimulation and the tyrosine kinase inhibitor, staurosporine. The higher levels of pSTAT1 observed in primary CD14+ cells from patients compared with control cells persisted and were amplified by the presence of staurosporine. We developed a flow cytometry-based STAT1 functional screening method that would greatly facilitate the diagnosis of CMCD patients with GOF STAT1 mutations.
Introduction
Patients with CMCD suffer from persistent or recurrent skin, nail, and mucosal membrane infections caused by C. albicans [1, 2]. CMCD is also observed in patients with primary immunodeficiencies, such as T cell deficiencies, AD HIES, AR IL-12p40 deficiency, AR IL-12Rβ1 deficiency, and AR APS-1 [3–6]. Patients with AD HIES have very low levels of IL-17A- and IL-22-producing T cells as a result of an impairment of STAT3-mediated responses [7–10]. The numbers of IL-17A- and IL-22-producing T cells are also low in patients with IL-12p40 and IL-12Rβ1 deficiencies but to a lesser extent than in patients with AD HIES [8]. Patients with APS-1 develop high titers of neutralizing autoantibodies against IL-17A, IL-17F, and/or IL-22 [11, 12]. These findings suggest that human IL-17A, IL-17F, and/or IL-22 may be essential for mucocutaneous immunity to C. albicans [6]. This hypothesis led to the discovery of AR IL-17RA deficiency and AD IL-17F deficiency as genetic etiologies of CMCD without other severe infectious diseases (isolated CMCD) [13]. Thus, inborn errors of IL-17 immunity can underlie CMCD [14–17]. However, no genetic etiology has yet been identified for the vast majority of patients with CMCD.
Heterozygous mutations affecting the CCD of STAT1 were identified recently by next-generation sequencing in patients with AD CMCD [18, 19]. A heterozygous T385M mutation, affecting the DBD of STAT1, was then discovered [20]. These mutations led to increase in Tyr701-pSTAT1, GAF DNA-binding ability, and IFN GAS transcription activity in response to IFN-γ, IFN-α, and IL-27 [18, 20, 21]. Surprisingly, there, GOF STAT1 alleles account for approximately one-half of patients with CMCD in previous studies (ref. [18] and unpublished results). The underlying molecular mechanism was deciphered using inhibitors of kinases and phosphatases and was shown to result from the impaired nuclear dephosphorylation of STAT1 [18]. We report here six GOF STAT1 mutations, three of which have never been described before, in two sporadic and four familial cases from four Japanese families with AD CMCD. We found that CD14+ monocytes from the patients displayed persistent pSTAT1 in response to IFN-γ and in the presence of the tyrosine kinase inhibitor staurosporine. Based on this observation, we developed a flow cytometry-based simple and rapid STAT1 functional screening method to facilitate the diagnosis of CMCD patients with GOF STAT1 mutations.
MATERIALS AND METHODS
Patients
Clinical information is summarized in Table 1. STAT1 mutations identified in patients and familial segregation of STAT1 mutations are shown in Fig. 1B.
Table 1. Summary of 10 Patients with CMCD from Six Kindred.
| Patients | STAT1 mutation | Fungal infections | Viral infection | Other complication |
|---|---|---|---|---|
| A-I-1 | L354M | recurrent tinea unguium, cryptococcal meningitis | recurrent herpes virus infection | recurrent otitis media, bronchiectasis, irritable bowel syndrome, hypothyroidism, persistent, slightly elevated liver enzyme |
| A-II-2 | L354M | recurrent oral aphthous as a result of C. albicans | hypothyroidism, persistent, slightly elevated liver enzyme | |
| B-II-1 | M202V | persistent tinea unguium, recurrent stomatitis as a result of C. albicans | bronchopneumonia | |
| C-I-2 | A267V | onychomycosis, tinea pedis, oral, esophageal, vaginal candidiasis | bronchiectasis, antiparietal cell antibody-positive megaloblastic anemia, hypothyroidism, persistent, slightly elevated liver enzyme | |
| C-II-1 | A267V | onychomycosis | vascular purpura | |
| D-I-2 | R274Q | recurrent oral and esophageal candidiasis, persistent skin candidiasis | ||
| D-II-2 | R274Q | oral candidiasis | ||
| E-II-1 | P329L | oral thrush | ||
| E-II-2 | P329L | recurrent oral thrush | pure red blood cell aplasia, autoimmune hemolytic anemia | |
| F-II-1 | M390T | recurrent oral and skin candidiasis | severe chicken-pox, chronic EBV infection | low serum IgG2 levels, bronchiectasis, mild hypothyroidism, recurrent severe diarrhea, recurrent otitis media, elevated liver enzyme |
Figure 1. Heterozygous mutations affecting the CCD and DBD of STAT1 were identified in two sporadic and four multiplex cases in 10 CMCD patients from Japan.
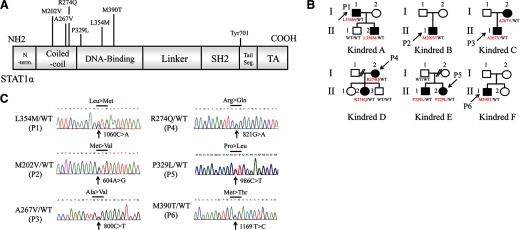
(A) Six mutations were identified in the human STAT1-α isoform. N-term., N-terminus; SH2, Src homology 2; Tail Seg., tail segment; TA, transactivator. (B) Pedigrees of four families with an AD form of CMCD (Kindreds A, C–E) and two sporadic cases with CMCD (Kindreds B and F) are shown. Filled symbols indicate patients with CMCD. The probands are indicated by arrows (P1–P6). (C) Chromatogram of identified mutations of STAT1. All of the mutations were heterozygous and affected the CCD or DBD of STAT1.
Kindred A (L354M/WT).
The proband (A-I-1: P1) is a 45-year-old man who has suffered from recurrent tinea unguium and otitis media since the age of 3 years. He also had several episodes of herpes virus infection, resulting in herpes simplex keratitis, dermatitis herpetiformis, and shingles during his childhood. At the age of 22, he experienced cryptococcal meningitis; he was diagnosed with hypothyroidism and placed on levofloxacin treatment. This patient also suffers from bronchiectasis and irritable bowel syndrome. His son is 7 years old (A-II-2) and suffers from recurrent oral aphthous lesions due to C. albicans. He has experienced no clinical episode suggestive of host susceptibility to viral or invasive fungal infection. At the age of 6 years, he was diagnosed with hypothyroidism and began treatment with levothyroxine. Both P1 and his son (A-II-2) have been found to have persistent, slightly high liver enzyme levels.
Kindred B (M202V/WT).
The patient (B-II-1: P2) is a 34-year-old man with recurrent stomatitis as a result of C. albicans since infancy. He was diagnosed with CMCD at the age of 5 years. Oral itraconazole treatment was initiated at the age of 18 years, but this patient still suffers from persistent tinea unguium and oral candidiasis. He has suffered from bronchopneumonia more than twice yearly since his 20s. Sputum cultures were systematically negative for bacteria, but the bronchopneumonia seemed to respond to treatment with levofloxacin and cefotiam. No hypothyroidism has been detected.
Kindred C (A267V/WT).
The proband (C-II-1: P3) is a 10-year-old boy. He developed onychomycosis at the age of 18 months and was treated with antifungal drugs for 6 months. He has presented no Candida infections since this episode. P3 had vascular purpura at the age of 6 years but does not have hypothyroidism. His mother (C-I-2) is 44 years old. She developed onychomycosis at the age of 1, vaginal candidiasis and tinea pedis at the age of 20, and recurrent oral candidiasis in her 30s, at which time, she was also diagnosed with bronchiectasis. At the age of 42 years, balloon dilatation was performed to relieve esophageal obstruction as a result of esophageal candidiasis. This patient also has hypothyroidism and antiparietal cell antibody-positive megaloblastic anemia. She was diagnosed with persistent slightly high liver enzyme levels.
Kindred D (R274Q/WT).
The proband (D-I-2: P4) is a 32-year-old woman. She developed oral and cutaneous candidiasis at the age of 1 year and pneumonia with pleural effusion at the age of 10 years. The disease-causing pathogen was not identified, but the patient's symptoms improved rapidly after the initiation of antifungal treatments. Oral fluconazole treatment was introduced after this episode. However, the patient suffered from persistent skin candidiasis and recurrent esophageal candidiasis. Her second daughter (D-II-2) also suffers from oral candidiasis. This familial case was reported in a previous manuscript (Kindred H in ref. [18]).
Kindred E (P329L/WT).
The proband (E-II-2: P5) is a 16-year-old girl who has suffered from recurrent oral thrush since infancy. At the age of 14 years, she displayed pure red blood cell aplasia and autoimmune hemolytic anemia, which responded to treatment with steroid and cyclosporine A. Her older brother (E-II-1) is 17 years old and has also suffered from oral thrush. Their father had no STAT1 mutation, and no clinical or genetic information was available for their mother.
Kindred F (M390T/WT).
The patient (F-II-1: P6) is a 30-year-old man with recurrent oral candidiasis since the age of 2 years. He suffered from severe chicken-pox at the age of 1 year, which took 90 days to resolve fully. He developed pneumonia at the age of 3 years, at which point, CMCD was suspected. After this episode, he suffered from recurrent, severe diarrhea, skin and oral candidiasis, and otitis media. The candidiasis was intractable despite treatment with antifungal drugs. At the age of 14 years, this patient was found to have low serum IgG2 levels and was given Ig replacement therapy. At the age of 17 years, he developed an impairment of liver function, and elevated liver enzyme was detected in serum. He also presented recurrent fever of unknown origin. He was found to have a high titer of EBV-transformed B cells in serum (1.9×105 copies/ml; reference range <1×102 copies/ml). He did not produce antibody against EBV nuclear antigen, which neutralizes EBV, during the clinical course of his illness, and chronic EBV infection was therefore suspected. This patient also has bronchiectasis and very mild hypothyroidism that does not require treatment. His parents and his younger sister do not present the clinical phenotype of CMCD.
Molecular genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes. Complete coding exons of STAT1 and their flanking introns were amplified by PCR and sequenced. We inserted the various alleles of STAT1 into the pcDNA3-V5 vector [18, 22, 23]. We also generated the P329L, L354M, and M390T mutations of STAT1 by PCR-based mutagenesis with mismatched primers. Primer sequences and PCR conditions are available on request.
Flow cytometric analysis
We assessed pSTAT1 in PBMCs from the patients. The MFI of pSTAT1 depended on the timing of the analysis. We therefore systematically assayed PBMCs, 24 h after blood collection. Mononuclear cells were suspended at a density of 104 cells/μl in serum-free RPMI. The cells were incubated with IFN-γ (1000 U/ml) for 15 min. They were then washed and incubated with 0.5 μM staurosporine (for 15 or 30 min for flow cytometry experiments and 15 min for immunoblot assays) in RPMI and subjected to analysis. For flow cytometry, the cells were stained simultaneously with anti-human CD3, CD19, or CD14 FITC antibody (BD PharMingen, San Diego, CA, USA) and treated with staurosporine. They were fixed and permeabilized, according to the BD Phosflow protocol (Protocol III); stained with FITC-conjugated anti-CD3, CD19, or CD14 and PE-conjugated anti-pSTAT1 (BD PharMingen) antibodies; and subjected to flow cytometric analysis. It took 6 h to perform the complete flow cytometric analysis.
Immunoblot analysis and EMSA
STAT1-null U3C fibrosarcoma cells were maintained in DMEM, supplemented with 10% FBS. The cells were harvested and replated at a density of 2.5 × 105 cells/ml in six-well culture plates. After incubation for a further 24 h, plasmid DNA (5 μg/well), carrying the WT or a mutant STAT1 allele, was introduced into the cells by calcium phosphate-mediated transfection. The transfected cells were incubated for 24 h, and 104 IU/ml IFN-γ was then added. The cells were incubated for a further 15 min and then subjected for EMSA and immunoblot analysis. Immunoblot analysis was performed as described previously [24]. The primary antibodies used were an anti-pSTAT1 (pY701) antibody (BD Biosciences, San Jose, CA, USA; Cell Signaling Technology, Danvers, MA, USA); an anti-STAT1 antibody (C-24; Santa Cruz Biotechnology, Santa Cruz, CA, USA); and an anti-β-actin antibody (Sigma-Aldrich, St. Louis, MO, USA). EMSA was carried out as described previously [24]. After IFN-γ stimulation, U3C-transfected cells were subjected to nuclear extraction. We incubated 20 μg nuclear extract with 32P-labeled (α-dATP) GAS (produced under control for the FCGR1 promoter) probe for 30 min.
Luciferase reporter assay
Luciferase assays were performed as described previously [18, 22, 23]. Briefly, transfected U3C cells were stimulated with IFN-α (5000 IU/ml), IL-27 (20 mg/ml), or various concentrations of IFN-γ (1, 5, 10, 50, 100, 500, and 1000 IU/ml) for 8 h and then analyzed. The data are expressed as fold inductions with respect to unstimulated cells. Experiments were performed in triplicate.
qPCR analysis
CD14+ monocytes were purified from PBMCs by magnetic sorting (BD Biosciences) and stimulated with 1000 IU/ml IFN-γ for 2 or 8 h. Then, total RNA was extracted and used for reverse transcription with random primers to generate cDNA. IRF1, CXCL9, and ISG15 mRNA levels were determined by qPCR with Taqman probes. The results were normalized with respect to the values obtained for the endogenous GAPDH cDNA.
Statistical analysis
Statistical significance was analyzed by nonparametric Mann-Whitney U-tests and variance followed by Tukey's post hoc analysis using SPSS software. For all analyses, P < 0.05 was considered statistically significant.
RESULTS
Identification of STAT1 mutations
We investigated five sporadic and five familial cases of CMCD from a total of 15 patients from 10 kindreds. Heterozygous STAT1 mutations were identified in two sporadic and four familial cases (and four additional cases were identified among affected relatives who were subsequently recruited, giving a total of 10 patients). Thus, STAT1 mutations were commonly identified in Japanese patients with CMCD. We identified three previously unknown heterozygous mutations of STAT1: c.1060C > A (L354M) in P1 and his younger son (A-II-2), c.986C > T (P329L) in P5 and her elder brother (E-II-1), and c.1169T > C (M390T) in P6 (Fig. 1C). We also identified previously reported heterozygous mutations: c.604A > G (M202V) in P2 and c.800C > T (A267V) in P3 and his mother (C-I-2) [18, 19]. The c.821G > A (R274Q) mutation was identified in P4 and her second daughter (D-II-2). This familial case is reported in a previous manuscript (Kindred K in ref. [18]). None of these mutations were identified in the healthy relatives tested, suggesting that clinical penetrance was complete. The newly identified mutations, P329L, L354M, and M390T, were not found in the National Center for Biotechnology Information, Ensembl, or dbSNP databases. They were also absent from 1052 controls from 52 ethnic groups in the Centre d'Etude du Polymorphisme Humain and Human Genome Diversity panels. These three mutations are thus rare variants rather than common, irrelevant polymorphisms segregating with AD CMCD.
Impairment of STAT1 Tyr701 dephosphorylation in PBMCs
Previous in vitro studies suggested that CMCD-related STAT1 mutations impair nuclear dephosphorylation [18]. However, only few reports investigate dephosphorylation of STAT1 in primary cells from the patients ex vivo [25, 26]. We analyzed pSTAT1 protein in PBMCs from P4, P5, and two healthy controls. As shown in Fig. 2A, STAT1 protein levels were normal in the CMCD patients' PBMCs. The patients' cells contained some pSTAT1 in the absence of stimulation, and the levels of pSTAT1 increased strongly in response to IFN-γ stimulation, remaining high after treatment with the tyrosine kinase inhibitor staurosporine, which inhibits JAK-STAT signaling upstream of STAT1, probably as a result of impaired nuclear STAT1 dephosphorylation. The upper and lower bands correspond to STAT1α and STAT1β in the pSTAT1 blot, whereas the bands correspond to STAT1α in the STAT1 blot.
Figure 2. Impairment of dephosphorylation in PBMCs.
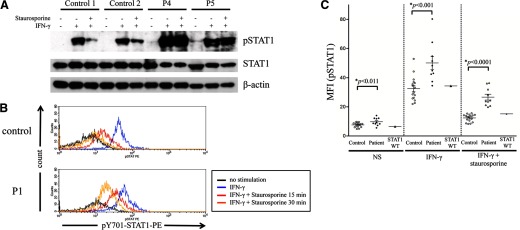
(A) STAT1 dephosphorylation was investigated by immunoblot using PBMCs from controls and patients. PBMCs from P4 and P5 showed an increase in pSTAT1 levels after IFN-γ stimulation. Their pSTAT1 levels were persistently high, even in the presence of staurosporine. Immunoblot analysis was carried out twice to confirm the results. (B and C) PBMCs from the 10 patients, 14 healthy individuals, and one patient with CMCD, carrying a WT STAT1 allele, were stimulated with IFN-γ and incubated in the presence of staurosporine. The cells were stained with anti-CD14 and anti-pY701 STAT1 antibodies and analyzed by flow cytometry with gating on CD14. The figures show a representative histogram of pSTAT1 intensity (B) and a summary of the MFI of pSTAT1 (C). (B) Black line, No stimulation; blue line, IFN-γ for 15 min; red and orange lines, incubation with staurosporine for 15 or 30 min, respectively, after IFN-γ stimulation.
We first investigated IFN-γ-induced pSTAT1 by flow cytometry, using various subsets isolated from PBMCs from healthy controls and patients with CMCD (Supplemental Fig. 1). A high level of pSTAT1 was observed in CD14+ monocytes, whereas it was not obvious in CD3+ T cells or CD19+ B cells. These observations were highly consistent with the data of previous studies [27–30]. Patients' cells showed a higher level of IFN-γ-induced pSTAT1 compared with healthy control. We therefore investigated STAT1 dephosphorylation focusing on CD14+ monocytes. As the STAT1 GOF mutations are thought to be associated with impairment in dephosphorylation of STAT1 [16], we used staurosporine to clarify the difference between STAT1 WT alleles and STAT1 GOF alleles. If the STAT1 dephosphorylation normally occurs in the nucleus, then pSTAT1 should decrease promptly following staurosporine treatment. Most of the patient blood samples were available only 24 h of transport. To control for the effect of time of blood processing on pSTAT1 and STAT1 dephosphorylation, we studied PBMCs from the same donor under two conditions: immediately after collection of blood samples or after a delay of 24 h (Supplemental Fig. 2). The level of pSTAT1 decreased in all of the conditions tested in the blood samples that were delayed. Thus, we decided to perform all assays in PBMCs, isolated and tested 24 h after blood collection. PBMCs from one patient with CMCD, carrying a WT STAT1 allele; 14 healthy individuals; and 10 GOF STAT1 patients—five from Kindreds A–C and five CMCD patients from another cohort carrying GOF STAT1 mutations (nonreported cases)—were incubated with IFN-γ for 15 min. The cells were then washed and incubated with staurosporine-containing media for 15 min prior to analysis. For P1 only, we also measured pSTAT1 after 30 min of staurosporine treatment. Figure 2B shows a representative histogram comparing P1 and control. CD14+ monocytes from P1 had higher levels of pSTAT1 than control cells following IFN-γ stimulation. The CD14+ monocytes from the control displayed rapid STAT1 dephosphorylation, whereas pSTAT1 persisted in the patients' monocytes in the presence of staurosporine. Residual pSTAT1 was found in the cells of P1, even after 30 min of treatment with staurosporine. The summary of MFI values for pSTAT1 obtained by flow cytometry is shown in Fig. 2C. Some overlap was observed, but levels of pSTAT1, in response to IFN-γ, were significantly higher in CD14+ cells from the patients than in those from the controls (*P<0.001). In the absence of stimulation, pSTAT1 levels were higher in the patients' cells than in control cells (*P<0.011). This excess phosphorylation persisted after 15 min of treatment with staurosporine, a tyrosine kinase inhibitor (*P<0.0001). Moreover, in these conditions, there was no overlap in MFI of pSTAT1 between the patients and healthy controls or with a CMCD patient carrying a WT STAT1 allele. The atypical clinical signs observed in the patients did not affect the results of flow cytometric analysis. The difference in the percentage decrease in pSTAT1 levels in the presence of staurosporine between healthy controls and patients was significant too (*P<0.02; Supplemental Fig. 3). These results clearly indicate that the dephosphorylation process is impaired in CMCD patients. The flow cytometric analysis of pSTAT1 levels in monocytes has been established previously as a screening tool to identify IFN-γ signaling defects in patients with MSMDs [31]. Taken together, this flow cytometry-based technique is likely to be useful for the rapid assessment of STAT1 function in CMCD patients.
Induction of ISGs in CD14+ monocytes
We investigated the induction of ISGs in patients' cells by purifying CD14+ monocytes, stimulating them with IFN-γ, and assessing the expression of the downstream ISGs CXCL9, IRF1, and ISG15 by RT-qPCR analysis (Fig. 3). The induction of these three ISGs has been shown to be STAT1-dependent in previous studies [22–24, 32, 33]. CD14+ monocytes from patients with CMCD (P2, P4 and a patient carrying a R274W mutation from another cohort) showed a significantly higher induction of CXCL9 and IRF1 expression than did those of healthy controls. By contrast, we did not see a significant increase in the induction of ISG15 in the patients. These results suggest that the mutated STAT1 alleles identified in CMCD patients are GOF in terms of the induction of transcription for many, but probably not all, downstream ISGs.
Figure 3. The induction of ISGs in CD14+ monocytes.

CD14+ monocytes from three patients with CMCD and two healthy controls were stimulated with 1000 U/ml IFN-γ (2 or 8 h), and the expression of the downstream ISGs CXCL9, IRF1, and ISG15 was assessed by RT-qPCR. Much stronger induction of CXCL9 and IRF1 was observed in the patients' cells than in control cells, whereas there was no significant increase in ISG15 induction. The expression of ISGs was normalized with respect to that of endogenous GAPDH. The results are representative of three independent experiments. Differences were statistically significant in the cells expressing the mutant STAT1s compared with Control 1 cells (*P<0.05) and Control 2 cells (†P<0.05).
Increased pSTAT1 and GAF DNA-binding ability
For confirmation of the STAT1 gain-of-phosphorylation observed in primary cells from the patients, we investigated STAT1-dependent signaling pathways by transiently transfecting STAT1-null U3C fibrosarcoma cells. STAT1 protein production was normal for the CMCD-related STAT1 mutants (Fig. 4A). Upon IFN-γ stimulation, all of the mutant proteins displayed higher levels of pSTAT1, at least a 1.5-fold increase over WT, as determined by densitometry (Supplemental Fig. 4). As expected, phosphorylation of the MSMD-related mutant, L706S STAT1, was impaired [34]. Thus, all of the CMCD-related mutations identified resulted in a gain-of-phosphorylation in response to IFN-γ. Next, we assessed the DNA-binding ability of the mutant STAT1 proteins to the GAS sequence using the same transfected cells, which were subjected to immunoblot analysis (Fig. 4B). All mutants, including the three mutations identified in the DBD, displayed GOF properties, resulting in an increase of GAF binding to DNA. However, the DNA binding to GAS was abolished in the MSMD-related mutant, L706S STAT1.
Figure 4. Functional analysis of STAT1-mutated alleles using transient gene expression systems.

(A and B) U3C cells expressing WT or mutant STAT1 were stimulated with IFN-γ and subjected to immunoblot (A) and EMSA (B). CMCD-related alleles gave higher levels of pSTAT1 than the WT in response to IFN-γ. The results are representative of two independent experiments. (C–E) Luciferase GAS and ISRE-induced activity in U3C cells. (C) All of the CMCD-related STAT1 mutations, except for M390T, were associated with levels of GAS transcriptional activity in response to IFN-γ, at least twice as high as those in cells transfected with the WT allele. (E) The M390T STAT1 behaved as a GOF protein in response to stimulation with low concentrations of IFN-γ. (D) An increase in ISRE induction in response to IFN-α was also observed in CMCD-related mutants, but this increase was much smaller than that observed for GAS induction. Differences were statistically significant in the cells expressing the mutants compared with the WT-expressing cells (*P<0.05).
Transcriptional activity of the CMCD-related STAT1 mutant protein
Transcriptional activity was studied by transfecting U3C cells with reporter plasmids and plasmids carrying the WT and/or mutant alleles of STAT1. The cells transfected with the CMCD-related alleles, except for M390T, had levels of GAS transcriptional activity in response to IFN-γ, more than twice those of cells transfected with the WT allele (Fig. 4C). The L706S MSMD-related STAT1 allele abolished the transcriptional activity of GAS. The increase in GAS transcriptional activity was only slight for the M390T protein in this condition. However, the GOF became obvious when the cells were stimulated with low concentrations of IFN-γ (Fig. 4E). We also investigated IL-27-induced GAS activation (Supplemental Fig. 5) and confirmed excess GAS induction in all of the CMCD-related mutations. We then assessed ISRE transcription activity in response to IFN-α (Fig. 4D). Some positive effects on ISRE transcription activity were suspected in CMCD-related mutations, but they were not statistically significant. Thus, all of the CMCD-related mutations caused a GOF to GAS-mediated transcription. Moreover, the GOF was more marked at lower concentrations of IFN-γ.
DISCUSSION
In this study, STAT1 mutations were identified with a high frequency (almost 70%) in Japanese CMCD patients; we identified heterozygous STAT1 mutations in two of five sporadic cases and in eight of 10 cases from five multiplex kindreds. In total, we identified 10 CMCD patients with GOF STAT1 mutations from a total of 15 diseased individuals. STAT1 mutations were also found in ∼50% of the patients with CMCD, identified in a previous study investigating large numbers of patients from numerous countries (ref. [18] and unpublished results). It remains possible that the frequency of STAT1 mutations is overestimated here as a result of the limited number of cases in our cohort, but these results nevertheless strongly suggest that STAT1 GOF is the most frequent genetic etiology of CMCD among Japanese patients, accounting for at least one-half of these patients. The clinical penetrance of STAT1 mutations appears to be extremely high, if not complete, as no individual with the morbid genotype but not CMCD has ever been identified [18–21, 35, 36]. We observed differences in clinical signs and severity, even between siblings with the same STAT1 mutation. All of the mutated alleles, including three previously unknown mutations, were gain-of-phosphorylation and GOF in vitro in terms of GAS transcriptional activity. Moreover, all of these mutations affected residues in the CCD or DBD of STAT1. These results are highly consistent with previous findings [18–21, 35, 36].
Levels of pSTAT1 were found to be persistently high in the patients' CD14+ monocytes by flow cytometric analysis. STAT1 does not appear to be hyperphosphorylated in lymphocytes but rather, in myeloid cells from patients with CMCD. A high level of pSTAT1 can be explained by the strong IFN-γR2 surface expression, specifically on monocytes, whereas IFN-γR1 is expressed ubiquitously [30]. The patients' cells had high basal levels of pSTAT1, a finding confirmed by immunoblot analysis of the patients' PBMCs. The amount of pSTAT1 in the cell is determined by the balance between pSTAT1 and STAT1 dephosphorylation [37]. CMCD-related mutations impair STAT1 nuclear dephosphorylation, potentially leading to some accumulation of pSTAT1 even in the absence of stimulation. Following stimulation with IFN-γ, CD14+ monocytes contained significantly larger amounts of pSTAT1 than control cells. Moreover, this difference in pSTAT1 levels was even greater when staurosporine was added after IFN-γ stimulation. Higher levels of IFN-γ-induced pSTAT1 in monocytes of CMCD patients overlap with levels detected in cells from normal subjects, and the addition of staurosporine is required to discriminate the CMCD patients from control subjects. This technique is likely to be useful for the rapid assessment of STAT1 function in CMCD patients with unknown genetic etiology or those carrying functionally uncharacterized STAT1 mutations. Furthermore, the results obtained are not affected by clinical diversity in the patients. Given the high frequency of GOF mutations of STAT1, the establishment of a rapid screening system based on STAT1 function should greatly facilitate the diagnosis of patients with CMCD.
Two patients presented atypical clinical signs in addition to CMCD. P1 suffered from cryptococcal meningitis, shingles, herpes simplex keratitis, and dermatitis. P6 presented severe, prolonged chicken-pox and chronic EBV infection. Cryptococcus is an opportunistic fungal pathogen, and IFN-γ signaling may play an important role in protective immunity against this microbe [38]. Indeed, IFN-γ administration increases the rate of clearance of HIV-associated cryptococcal infection from the cerebrospinal fluid when combined with antifungal drugs [39]. The CMCD-related STAT1 mutations observed here were GOF in terms of IFN-γ-induced GAS transcriptional activity in vitro, so the cryptococcal meningitis observed in this patient is paradoxical. In addition to our study, recurrent cutaneous fusariosis and disseminated coccidioidomycosis and histoplasmosis have been reported in patients with GOF STAT1 mutation [40, 41]. Viral infections have also been reported in CMCD patients: one case of symptomatic cytomegalovirus infection and one familial case with recurrent herpes simplex virus and varicella infections have been reported [19, 25, 36]. STAT1 mediates the IFN-α/β-induced transcription of ISRE, which plays an important role in antiviral immunity. Therefore, severe viral infection is one of the typical symptoms in patients with AR STAT1 deficiency, which is caused by loss-of-function or hypomorphic mutations of STAT1 [23, 32, 42–44]. As the CMCD-related STAT1 mutations presented normal activity in terms of IFN-α-induced ISRE transcriptional activity in vitro, viral infections observed in two patients are also paradoxical. We observed higher levels of induction for CXCL9 and IRF1, but not ISG15, in response to IFN-γ in CD14+ monocytes from CMCD patients than in those from controls. These results suggest that STAT1 GOF mutations may not induce an increase in the transcription of all downstream ISGs, potentially even causing paradoxical patterns of regulation for some target genes. There may be similar paradoxical responses to IFN-α/β, at least for some target genes. Moreover, a recent report suggests that the induction of CXCL9 and CXCL10 is impaired in STAT1-deficient U3A cells expressing GOF STAT1 alleles upon specific conditions, such as the restimulation with IFN-γ [41]. Further studies are required to determine whether and how STAT1 GOF mutations confer a predisposition to pathogens, for which clearance generally requires normal IFN-γ and/or IFN-α signaling.
Surprisingly, a very recent report identified GOF mutations in STAT1 in patients with forkhead box P3 (FOXP3) WT immune dysregulation, polyendocrinopathy, autoimmune enteropathy, and X-linked-like syndrome [25]. These patients present a broad, infectious phenotype that may be explained partially by progressive lymphopenia, finally resulting in hypogammaglobulinemia [26]. Although progressive lymphopenia was not observed, the patients in our cohort also presented a broad, infectious phenotype. Taken together with the clinical cases presented in our current study, these findings suggest that GOF mutations of STAT1 may be associated with a much broader infectious phenotype than thought initially.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported, in part, by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (22591161 to M.K.) and by Research on Measures for Intractable Diseases funding from the Japanese Ministry of Health, Labor and Welfare (H22-Nanchi-ippan-078 to M.K.). This study was also supported, in part, by The Rockefeller University and grants from the National Center for Research Resources; National Center for Advancing Sciences (NCATS), U.S. National Institutes of Health (Grant Number 8UL1TR000043); and St. Giles Foundation. S.C. was supported by the AXA Research Fund and V.L.B and X-F.K., by the Stony Wold-Herbert Fund. Sequence analysis was supported by the Analysis Center of Life Science, Natural Science Center for Basic Research and Development, Hiroshima University.
We thank Dr. Yoshiko Hasii (Osaka University) for referring patients. We thank Dusan Bogunovic, Alexandra Kreins, Marcela Moncada Velez, Ruben Martinez-Barricarte, and Michael Ciancanelli for helpful discussions and critical reading. We thank the members of the laboratory, Yelena Nemirovskaya and Eric Anderson, for secretarial assistance and Tiffany Nivare for technical assistance.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- AD
- autosomal-dominant
- APS-1
- autoimmune polyendocrinopathy type 1 syndrome
- AR
- autosomal-recessive
- CCD
- coiled-coil domain
- CMCD
- chronic mucocutaneous candidiasis disease
- DBD
- DNA-binding domain
- GAF
- γ-activating factor
- GAS
- γ-activated sequence
- GOF
- gain-of-function
- HIES
- hyper-IgE syndrome
- IRF
- IFN regulatory factor
- ISG
- IFN-stimulated gene
- ISRE
- IFN-stimulated response element
- MFI
- mean fluorescence intensity
- MSMD
- mendelian susceptibility to mycobacterial disease
- pSTAT1
- phosphorylated STAT1
- qPCR
- quantitative PCR
AUTHORSHIP
Y.M., M.T., S.O., O.H., and S.M. were involved in research design and data analysis. Y.M. and S.O. wrote the manuscript. K.I., N.H., H.M., S.K., Y.O., T. Imai, S.T., T.O., and T. Ito treated the patients. S.Y., Y.T., V.L.B., X-F.K., S.C., S.B-D., A.P., J-L.C., T.M., and M.K. directed experiments and edited the paper. S.O., T.M., and M.K. supervised all work.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Kirkpatrick C. H. (2001) Chronic mucocutaneous candidiasis. Pediatr. Infect. Dis. J. 20, 197–206 [DOI] [PubMed] [Google Scholar]
- 2. Lilic D. (2002) New perspectives on the immunology of chronic mucocutaneous candidiasis. Curr. Opin. Infect. Dis. 15, 143–147 [DOI] [PubMed] [Google Scholar]
- 3. Minegishi Y., Saito M., Tsuchiya S., Tsuge I., Takada H., Hara T., Kawamura N., Ariga T., Pasic S., Stojkovic O., Metin A., Karasuyama H. (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062 [DOI] [PubMed] [Google Scholar]
- 4. Holland S. M., DeLeo F. R., Elloumi H. Z., Hsu A. P., Uzel G., Brodsky N., Freeman A. F., Demidowich A., Davis J., Turner M. L., et al. (2007) STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357, 1608–1619 [DOI] [PubMed] [Google Scholar]
- 5. De Beaucoudrey L., Samarina A., Bustamante J., Cobat A., Boisson-Dupuis S., Feinberg J., Al-Muhsen S., Jannière L., Rose Y., de Suremain M., et al. (2010) Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 89, 381–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Puel A., Picard C., Cypowyj S., Lilic D., Abel L., Casanova J. L. (2010) Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr. Opin. Immunol. 22, 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Minegishi Y., Saito M., Nagasawa M., Takada H., Hara T., Tsuchiya S., Agematsu K., Yamada M., Kawamura N., Ariga T., Tsuge I., Karasuyama H. (2009) Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J. Exp. Med. 206, 1291–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Beaucoudrey L., Puel A., Filipe-Santos O., Cobat A., Ghandil P., Chrabieh M., Feinberg J., von Bernuth H., Samarina A., Janniere L., et al. (2008) Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med. 205, 1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Milner J. D., Brenchley J. M., Laurence A., Freeman A. F., Hill B. J., Elias K. M., Kanno Y., Spalding C., Elloumi H. Z., Paulson M. L., Davis J., Hsu A., Asher A. I., O'Shea J., Holland S. M., Paul W. E., Douek D. C. (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma C. S., Chew G. Y., Simpson N., Priyadarshi A., Wong M., Grimbacher B., Fulcher D. A., Tangye S. G., Cook M. C. (2008) Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 205, 1551–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kisand K., Boe Wolff A. S., Podkrajsek K. T., Tserel L., Link M., Kisand K. V., Ersvaer E., Perheentupa J., Erichsen M. M., Bratanic N., et al. (2010) Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Puel A., Doffinger R., Natividad A., Chrabieh M., Barcenas-Morales G., Picard C., Cobat A., Ouachee-Chardin M., Toulon A., Bustamante J., et al. (2010) Autoantibodies against IL-17A, IL-17F, and. IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 207, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Puel A., Cypowyj S., Bustamante J., Wright J. F., Liu L., Lim H. K., Migaud M., Israel L., Chrabieh M., Audry M., et al. (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cypowyj S., Picard C., Marodi L., Casanova J. L., Puel A. (2012) Immunity to infection in IL-17-deficient mice and humans. Eur. J. Immunol. 42, 2246–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marodi L., Cypowyj S., Toth B., Chernyshova L., Puel A., Casanova J. L. (2012) Molecular mechanisms of mucocutaneous immunity against Candida and Staphylococcus species. J. Allergy Clin. Immunol. 130, 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Puel A., Cypowyj S., Marodi L., Abel L., Picard C., Casanova J. L. (2012) Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 12, 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alcais A., Quintana-Murci L., Thaler D. S., Schurr E., Abel L., Casanova J. L. (2010) Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann. N. Y. Acad. Sci. 1214, 18–33 [DOI] [PubMed] [Google Scholar]
- 18. Liu L., Okada S., Kong X. F., Kreins A. Y., Cypowyj S., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P., et al. (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208, 1635–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van de Veerdonk F. L., Plantinga T. S., Hoischen A., Smeekens S. P., Joosten L. A., Gilissen C., Arts P., Rosentul D. C., Carmichael A. J., Smits-van der Graaf C. A., Kullberg B. J., van der Meer J. W., Lilic D., Veltman J. A., Netea M. G. (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365, 54–61 [DOI] [PubMed] [Google Scholar]
- 20. Takezaki S., Yamada M., Kato M., Park M. J., Maruyama K., Yamazaki Y., Chida N., Ohara O., Kobayashi I., Ariga T. (2012) Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J. Immunol. 189, 1521–1526 [DOI] [PubMed] [Google Scholar]
- 21. Smeekens S. P., Plantinga T. S., van de Veerdonk F. L., Heinhuis B., Hoischen A., Joosten L. A., Arkwright P. D., Gennery A., Kullberg B. J., Veltman J. A., Lilic D., van der Meer J. W., Netea M. G. (2011) STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One 6, e29248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chapgier A., Boisson-Dupuis S., Jouanguy E., Vogt G., Feinberg J., Prochnicka-Chalufour A., Casrouge A., Yang K., Soudais C., Fieschi C., et al. (2006) Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kong X. F., Ciancanelli M., Al-Hajjar S., Alsina L., Zumwalt T., Bustamante J., Feinberg J., Audry M., Prando C., Bryant V., et al. (2010) A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood 116, 5895–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsumura M., Okada S., Sakai H., Yasunaga S., Ohtsubo M., Murata T., Obata H., Yasumi T., Kong X. F., Abhyankar A., et al. (2012) Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum. Mutat. 33, 1377–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Uzel G., Sampaio E. P., Lawrence M. G., Hsu A. P., Hackett M., Dorsey M. J., Noel R. J., Verbsky J. W., Freeman A. F., Janssen E., et al. (2013) Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J. Allergy Clin. Immunol. 131, 1611–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Romberg N., Morbach H., Lawrence M. G., Kim S., Kang I., Holland S. M., Milner J. D., Meffre E. (2013) Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J. Allergy Clin. Immunol. 131, 1691–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernabei P., Coccia E. M., Rigamonti L., Bosticardo M., Forni G., Pestka S., Krause C. D., Battistini A., Novelli F. (2001) Interferon-γ receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J. Leukoc. Biol. 70, 950–960 [PubMed] [Google Scholar]
- 28. Bach E. A., Aguet M., Schreiber R. D. (1997) The IFN γ receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 15, 563–591 [DOI] [PubMed] [Google Scholar]
- 29. Van Boxel-Dezaire A. H., Stark G. R. (2007) Cell type-specific signaling in response to interferon-γ. Curr. Top. Microbiol. Immunol. 316, 119–154 [DOI] [PubMed] [Google Scholar]
- 30. Kong X. F., Vogt G., Itan Y., Macura-Biegun A., Szaflarska A., Kowalczyk D., Chapgier A., Abhyankar A., Furthner D., Khayat C. D., et al. (2013) Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum. Mol. Genet. 22, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fleisher T. A., Dorman S. E., Anderson J. A., Vail M., Brown M. R., Holland S. M. (1999) Detection of intracellular phosphorylated STAT-1 by flow cytometry. Clin. Immunol. 90, 425–430 [DOI] [PubMed] [Google Scholar]
- 32. Chapgier A., Kong X. F., Boisson-Dupuis S., Jouanguy E., Averbuch D., Feinberg J., Zhang S. Y., Bustamante J., Vogt G., Lejeune J., Mayola E., de Beaucoudrey L., Abel L., Engelhard D., Casanova J. L. (2009) A partial form of recessive STAT1 deficiency in humans. J. Clin. Invest. 119, 1502–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bogunovic D., Byun M., Durfee L. A., Abhyankar A., Sanal O., Mansouri D., Salem S., Radovanovic I., Grant A. V., Adimi P., et al. (2012) Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dupuis S., Dargemont C., Fieschi C., Thomassin N., Rosenzweig S., Harris J., Holland S. M., Schreiber R. D., Casanova J. L. (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293, 300–303 [DOI] [PubMed] [Google Scholar]
- 35. Hori T., Ohnishi H., Teramoto T., Tsubouchi K., Naiki T., Hirose Y., Ohara O., Seishima M., Kaneko H., Fukao T., Kondo N. (2012) Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J. Clin. Immunol. 32, 1213–1220 [DOI] [PubMed] [Google Scholar]
- 36. Toth B., Mehes L., Tasko S., Szalai Z., Tulassay Z., Cypowyj S., Casanova J. L., Puel A., Marodi L. (2012) Herpes in STAT1 deficiency. Lancet 379, 2500. [DOI] [PubMed] [Google Scholar]
- 37. Ten Hoeve J., de Jesus Ibarra-Sanchez M., Fu Y., Zhu W., Tremblay M., David M., Shuai K. (2002) Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 22, 5662–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen G. H., McDonald R. A., Wells J. C., Huffnagle G. B., Lukacs N. W., Toews G. B. (2005) The γ interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans. Infect. Immun. 73, 1788–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jarvis J. N., Meintjes G., Rebe K., Williams G. N., Bicanic T., Williams A., Schutz C., Bekker L. G., Wood R., Harrison T. S. (2012) Adjunctive interferon-γ immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS 26, 1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X., Lin Z., Gao L., Wang A., Wan Z., Chen W., Yang Y., Li R. (2013) Exome sequencing reveals a signal transducer and activator of transcription 1 (STAT1) mutation in a child with recalcitrant cutaneous fusariosis. J. Allergy Clin. Immunol. 131, 1242–1243 [DOI] [PubMed] [Google Scholar]
- 41. Sampaio E. P., Hsu A. P., Pechacek J., Bax H. I., Dias D. L., Paulson M. L., Chandrasekaran P., Rosen L. B., Carvalho D. S., Ding L., et al. (2013) Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 131, 1624–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I. Z., Al-Jumaah S., Yang K., Chapgier A., Eidenschenk C., Eid P., Al Ghonaium A., Tufenkeji H., Frayha H., Al-Gazlan S., Al-Rayes H., Schreiber R. D., Gresser I., Casanova J. L. (2003) Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388–391 [DOI] [PubMed] [Google Scholar]
- 43. Chapgier A., Wynn R. F., Jouanguy E., Filipe-Santos O., Zhang S., Feinberg J., Hawkins K., Casanova J. L., Arkwright P. D. (2006) Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J. Immunol. 176, 5078–5083 [DOI] [PubMed] [Google Scholar]
- 44. Boisson-Dupuis S., Kong X. F., Okada S., Cypowyj S., Puel A., Abel L., Casanova J. L. (2012) Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 24, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.