

Abstract
Genotypes of Mycobacterium tuberculosis complex (MTBC) vary with the geographic origin of the patients and can affect tuberculosis (TB) transmission. This study was aimed to further differentiate spoligotype-defined clusters of drug-resistant MTBC clinical isolates split in Beijing (n = 190) versus non-Beijing isolates (n = 84) from Sichuan region, the second high-burden province in China, by IS6110-restriction fragment length polymorphism (RFLP) and 24-locus MIRU-VNTRs. Among 274 spoligotyped isolates, the clustering ratio of Beijing family was 5.3% by 24-locus MIRU-VNTRs versus 2.1% by IS6110-RFLP, while none of the non-Beijing isolates were clustered by 24-locus MIRU-VNTRs versus 9.5% by IS6110-RFLP. Hence, neither the 24-locus MIRU-VNTR was sufficient enough to fully discriminate the Beijing family, nor the IS6110-RFLP for the non-Beijing isolates. A region adjusted scheme combining 12 highly discriminatory VNTR loci with IS6110-RFLP was a better alternative for typing Beijing strains in Sichuan than 24-locus MIRU-VNTRs alone. IS6110-RFLP was for the first time introduced to systematically genotype MTBC in Sichuan and we conclude that the region-adjusted scheme of 12 highly discriminative VNTRs might be a suitable alternative to 24-locus MIRU-VNTR scheme for non-Beijing strains, while the clusters of the Beijing isolates should be further subtyped using IS6110-RFLP for optimal discrimination.
1. Introduction
Tuberculosis (TB) remains a serious public health issue and a leading cause of adult mortality arising from a single infectious agent. China is not only the top-two country with respect to the estimated number of cases and deaths but also a hotspot of drug resistance [1]. Sichuan, located in southwestern China, has the second-greatest number of TB cases of any Chinese province. The prevalence of drug-resistant TB, especially multidrug-resistant (MDR) TB, is much higher than average for a developed region in eastern China [2–4]. An official report in 2008 revealed that MDR-TB in Sichuan was found in the sputum of 28.3% of all smear-positive, and the proportion of strains showing resistance to at least one drug was 74% [5], a figure maintained at 70% in 2013 [6]. In such a context, investigations on genetic diversity of drug resistant M. tuberculosis clinical isolates may provide with useful information about the origin and transmission of the circulating isolates [7–10].
Although initially considered as the gold standard for identifying epidemiologically linked isolates from patients, the IS6110-restriction fragment length polymorphism (IS6110-RFLP) was time-consuming and expensive and characterized by disadvantages such as requirement of good quality DNA, the presence of strains harboring low/no copy number of IS6110 element in many parts of Asia and certain parts of the world, and difficulties in inter-laboratory comparison of the IS6110-RFLP patterns [11–14]. Subsequently, it was later replaced by PCR-based rapid methods such as mycobacterial interspersed repetitive units-variable number of DNA tandem repeats (MIRU-VNTRs) typing and spoligotyping [15]. Although spoligotyping can identify Beijing family isolates easily [16], classification of M. tuberculosis strains into robust phylogenetic lineages is not always possible and misclassifications may occur occasionally [17, 18]. Considered one of the most successful M. tuberculosis lineages involved in tuberculosis transmission [19], the spoligotyping-defined Beijing clusters are subtyped using more discriminatory tools such as IS6110-RFLP and MIRU-VNTRs to avoid overestimating the clustering rate; and VNTR typing is usually preferred to RFLP on genotyping M. tuberculosis in regions where Beijing genotype isolates make up a low percentage of TB population [20]. However, as was shown recently in China [21], Russia [22], and Japan [23], the 24-locus MIRU-VNTR scheme is not always sufficient for discriminating Beijing isolates in regions with a high prevalence of Beijing strains, leading to the development of region-adjusted complementary typing schemes [22–24], and more recently proposal of a consensus set of Hypervariable MIRU-VNTRs for subtyping Beijing isolates [25].
There is an increasing amount of evidence regarding the importance of subtle regional differences in Beijing genotypes due to evolutionary aspects, for example, Billamas et al. reported that in Thailand, the variations of one or more MIRU-VNTR loci in Beijing isolates of highly similar IS6110-RFLP patterns could imply the occurrence of evolutionary MIRU-VNTR loci among genetically homogeneous M. tuberculosis clinical isolates [26]. Previous studies demonstrated that M. tuberculosis population structure varied geographically in China, mainly from north to south, with southern China showing a relatively smaller proportion of Beijing isolates with a greater distribution frequency of non-Beijing types [27, 28]. Our concomitant research focused on a detailed analysis of spoligotyping-based patterns (HGI = 0.595), underlying a more diverse population structure of drug-resistant M. tuberculosis isolates in Sichuan than in other parts of China [13]. Observed data suggest that subtle differences in M. tuberculosis transmission and epidemiology may exist in Sichuan, making the overall population structure observed quite different from that observed in northern China.
In such a context, the objective of the present study was to further differentiate the spoligotyped drug-resistant M. tuberculosis clinical isolates circulating in Sichuan by IS6110-RFLP and MIRU-VNTRs, so as to develop appropriate genotyping methodology for various M. tuberculosis lineages, particularly the Beijing versus non-Beijing family isolates.
2. Materials and Methods
2.1. Mycobacterial Isolates
A total of 306 drug-resistant Mycobacterium tuberculosis clinical isolates were randomly selected from isolates showing any sort of resistance. All strains were cultured from samples collected at the Chengdu Antituberculosis Hospital, the only professional antituberculosis hospital in Sichuan, from January 2008 to August 2009. Clinical data were obtained from the subjects' medical records without any invasion of patients' privacy, and the results of this study did not influence patient's treatment in any way.
2.2. Drug Susceptibility
Specimens were collected and disposed in accordance with WHO guidelines. Briefly, strains were cultured on Löwenstein Jensen (LJ) slants at 37°C and MTBC isolates were identified using standard biochemical methods such as susceptibility to p-nitrobenzoic acid (PNB) and to 2-thiopnene carboxylic acid hydrazide (TCH), pyrazinamidase activity (PZA), nitrate reduction, and niacin production. Drug susceptibility testing was done using proportion method with streptomycin (STR), 10 mg/mL; isoniazid (INH), 0.2 mg/mL, rifampin (RIF), 40 mg/mL and ethambutol (EMB), 2 mg/mL.
2.3. DNA Extract and Genotyping
Genomic DNA from clinical isolates was extracted using the CTAB method [29] followed by IS6110-RFLP according to an international protocol [30]. Briefly, genomic DNA was extracted, digested with PvuII, and subjected to agarose gel electrophoresis. After DNA was blotted to a Hybond membrane, DNA fingerprinting was performed using hybridization with the IS6110 insertion sequence and enhanced chemiluminescence assay (Roche). The spoligotyping and MIRU-VNTR information of all drug-resistant isolates were from our previous study [13]. Spoligotyping was performed as described by Kamerbeek et al. [10]. For MIRU-VNTRs, all 24 loci were amplified with corresponding primers as described by Supply et al. [31].
2.4. Database Comparison
Both the spoligotypes and MIRU-VNTR patterns were compared using the SITVIT2 proprietary database of Institut Pasteur de la Guadeloupe, which is an updated version of the previously released SpolDB4 [32] and SITVITWEB databases [33] (available online at http://www.pasteur-guadeloupe.fr:8081/SITVIT_ONLINE/). In this database, spoligotype international type (SIT) and MIRU international type (MIT) designate spoligotype and MIRU patterns shared by 2 or more patient isolates, as opposed to “orphan” which designates patterns reported for a single isolate. The major phylogenetic clades were assigned according to the signatures provided in the database, which defined 62 genetic lineages/sublineages in SpolDB4, with 5 “new rules” for the definition of variants within existing lineages in SITVITWEB and SITVIT2 [32, 33]. These include various M. tuberculosis complex members such as M. bovis, M. caprae, M. microti, M. canetti, M. pinipedi, and M. africanum, as well as rules defining major lineages/sublineages for M. tuberculosis sensu stricto. These include the Beijing clade, the Central Asian (CAS) clade and 2 sublineages, the East African-Indian (EAI) clade and 9 sublineages, the Haarlem (H) clade and 3 sublineages, the Latin American-Mediterranean (LAM) clade and 12 sublineages, the ancestral “Manu” family and 3 sublineages, the S clade, the IS6110-low-banding X clade and 3 sublineages, and an ill-defined T clade with 5 sublineages.
2.5. Statistical Analysis
The analysis of the size of PCR fragments and assignment of the various VNTR alleles were achieved using Quantity One (version 4.6.2) software (Bio-Rad Laboratories). The Hunter-Gaston index (HGI) was calculated as described previously [34]. It was used to evaluate the level of discriminatory power of the typing methods and the allelic diversity of individual VNTR loci. Fingerprints of IS6110 were analyzed using BioNumerics (version 6.1) software (Applied Maths, Kortrijk, Belgium). Similarities between RFLP patterns were calculated using the Dice coefficient, and the dendrogram was produced using an unweighted pair group method with an arithmetic averages algorithm.
3. Results
3.1. Subclustering Beijing and Non-Beijing Isolates by IS6110-RFLP
A subset of 274 isolates with sufficient DNA was available for IS6110-RFLP analysis. The 274 isolates were divided into two groups (Beijing and non-Beijing family) based on spoligotyping results after checking them against the SITVIT2 database. Each group was subjected to IS6110-RFLP analysis. Of the 274 isolates, six clusters and 260 unique patterns were identified using IS6110-RFLP (HGI = 0.999) (Table 1). One of these clusters was split by spoligotyping. The Beijing family strains (n = 190; 69.34%) were subdivided into two clusters and 185 unique patterns (Figure 1), while the non-Beijing family strains (n = 84; 30.66%) were subdivided into three clusters and 75 unique patterns (Figure 2). The clusters varied in size from two to three isolates.
Table 1.
Discriminatory power of the three different typing methods for a subset of 274 drug-resistant M. tuberculosis isolates from Sichuan, China.
| Method | Number of types | Number of unique isolates | Number of clustered isolates | Number of clusters | Range of cluster size (number of isolates) | Number of clustered Beijing isolates | Number of clustered non-Beijing isolates | HGI |
|---|---|---|---|---|---|---|---|---|
| 24-locus MIRU-VNTR | 268 | 262 | 12 | 6 | 2 | 11 | 1 | 0.999 |
| 12 highly discriminatory loci | 267 | 260 | 14 | 7 | 2 | 13 | 1 | 0.999 |
| IS6110-RFLP | 266 | 260 | 14 | 6 | 2-3 | 5 | 9 | 0.999 |
Figure 1.

IS6110-RFLP based dendrogram of 190 clinical isolates of Beijing family. Gray highlighting indicates isolates with identical IS6110 patterns differentiated by 24-locus MIRU-VNTR. Isolates in blank boxes are fully identical genotypes.
Figure 2.

IS6110-RFLP based dendrogram of 84 clinical isolates of non-Beijing family. Gray highlighting indicates isolates with identical IS6110 patterns. These were differentiated using 24-locus MIRU-VNTR.
The numbers of IS6110 bands ranged from 7 to 17 among the Beijing family and from 1 to 16 among the non-Beijing family (Table 2). One hundred and seventy of the 190 (89.5%) were Beijing family strains with more than 10 copy numbers of IS6110, and the isolates with less than 6 copy numbers constituted about 39.3% of all non-Beijing family, including ill-defined T family (60.6%), Manu2 (9%), LAM (3%), and unknown or orphan clade (15.2%, 12.1%). Three of the 51 ill-defined T family (5.9%), two of the 9 unknown clade, and one of 7 orphan clade strains harbored only one copy of IS6110 element.
Table 2.
IS6110-RFLP analysis of Beijing and non-Beijing isolates from Sichuan.
| Number of IS6110 copies | Number of isolates | |
|---|---|---|
| Beijing isolates (n = 190) |
Non-Beijing isolates (n = 84) |
|
| 1 | 0 | 6 |
| 2–6 | 0 | 27 |
| 7–9 | 20 | 39 |
| 10–14 | 150 | 11 |
| 15–17 | 20 | 1 |
3.2. IS6110 Clusters and VNTR Clusters Subdivided by Each Other or Spoligotyping
Among 274 spoligotype-defined isolates, two shared a fully identical genotype, indicating a concordance of the genotyping methods used, that is, spoligotyping, 24-locus MIRU-VNTR and IS6110-RFLP. Both isolates belonged to the Beijing family (SIT1) harboring 12 copies of IS6110. One RFLP cluster of 2 isolates (SC041 and SC044; both of them had 8 copies of IS6110) was split by either spoligotyping or VNTR approach by two loci (QUB-11b, MIRU39). Other two RFLP clusters were further resolved by VNTR method only. One cluster contained 2 isolates of ill-defined T family harboring 8 copies of IS6110, and the other cluster contained 2 isolates of Beijing family harboring 12 copies of IS6110. Among the non-Beijing family strains, the six isolates harboring only one copy of IS6110 were grouped into two clusters of 3 isolates each, and they were differentiated by multiple MIRU loci and spoligotyping.
The allelic diversity of individual MIRU-VNTR loci was given in Table 3. The HGI value varied significantly from null to 0.852. The 24-locus MIRU-VNTR typing differentiated 274 strains into 6 clusters containing 12 isolates, and there was only one isolate belonged to non-Beijing family (Manu2 clade). Except the fully identical genotypes, one VNTR cluster was differentiated by both spoligotyping and IS6110-RFLP, which differed by 6 RFLP bands. Three out of six VNTR clusters split by RFLP differed by only one RFLP band and another one differed by three RFLP bands. There were 12 highly discriminatory loci (HGI > 0.6) including locus 424, 802, 960, 1644, 1955, 2163b, 2996, 3007, 3192, 3690, 4052, and 4348. Compared with the 24-locus MIRU-VNTR, one more cluster was identified by this 12-locus scheme, which contained 2 isolates (SC169 and SC172) belonging to Beijing family (SIT1). The 12-locus scheme showed a discrimination power equal to that of 24-locus MIRU-VNTR and IS6110-RFLP for the studied sample (the HGI values evaluated for different sets of MIRU-VNTR and IS6110-RFLP were illustrated in Table 1).
Table 3.
Allelic diversity of the 24 MIRU-VNTRs loci in 274 drug-resistant M. tuberculosis isolates from Sichuan, China.
| Schemes of VNTR locus | VNTR locus | VNTR alias | No. of alleles | Range of repeats | Allelic diversity (HGI) for | ||
|---|---|---|---|---|---|---|---|
| Beijing isolates (n = 190) |
Non-Beijing isolates (n = 84) |
All isolates (n = 274) |
|||||
| Discriminatory loci | 424 | Mtub04 | 6 | 0–4, 6 | 0.722 | 0.705 | 0.745 |
| 577 | ETR-C | 5 | 2–6 | 0.111 | 0.159 | 0.126 | |
| 580 | MIRU4 | 7 | 1–6, 8 | 0.185 | 0.65 | 0.367 | |
| 802 | MIRU40 | 5 | 1–5 | 0.558 | 0.727 | 0.643 | |
| 960 | MIRU10 | 6 | 0–5 | 0.589 | 0.596 | 0.627 | |
| 1644 | MIRU16 | 6 | 0–5 | 0.664 | 0.689 | 0.673 | |
| 1955 | Mtub21 | 9 | 1–9 | 0.706 | 0.73 | 0.803 | |
| 2163b | QUB-11b | 15 | 0–10, 19–22 | 0.783 | 0.852 | 0.829 | |
| 2165 | ETR-A | 5 | 1–5 | 0.262 | 0.678 | 0.502 | |
| 2401 | Mtub30 | 5 | 2–6 | 0.140 | 0.265 | 0.438 | |
| 2996 | MIRU26 | 10 | 1–10 | 0.727 | 0.724 | 0.79 | |
| 3192 | MIRU31 | 6 | 1–6 | 0.586 | 0.697 | 0.708 | |
| 3690 | Mtub39 | 6 | 1–6 | 0.626 | 0.792 | 0.708 | |
| 4052 | QUB-26 | 11 | 1–11 | 0.759 | 0.828 | 0.782 | |
| 4156 | QUB-4156 | 7 | 0–7 | 0.605 | 0.479 | 0.571 | |
| Additional loci | 154 | MIRU2 | 3 | 1–3 | 0 | 0.07 | 0.022 |
| 2059 | MIRU20 | 2 | 1-2 | 0.031 | 0.069 | 0.043 | |
| 2347 | Mtub29 | 4 | 2–5 | 0.052 | 0.093 | 0.064 | |
| 2461 | ETR-B | 3 | 1–3 | 0.031 | 0.488 | 0.236 | |
| 2531 | MIRU23 | 7 | 1, 3–8 | 0.176 | 0.199 | 0.183 | |
| 2687 | MIRU24 | 1 | 1 | 0 | 0 | 0 | |
| 3007 | MIRU27 | 5 | 0–4 | 0.599 | 0.662 | 0.624 | |
| 3171 | Mtub34 | 4 | 1–4 | 0.120 | 0.137 | 0.125 | |
| 4348 | MIRU39 | 6 | 1–6 | 0.734 | 0.696 | 0.751 | |
In bold: the HGI > 0.6.
The 12-loci-VNTR scheme was developed based on the highly discriminatory loci (HGI > 0.6) for all isolates. Both discriminatory and additional loci were from Supply et al. [31].
4. Discussion
The ongoing transmission of M. tuberculosis in certain settings is heavily influenced by the prevailing population structure of tubercle bacilli, and certain genetic families of this species have attracted more attention due to their global dissemination and/or remarkable pathogenic properties [35], such as the predominance of a highly homogeneous group like the Beijing family in northern China, Japan, and South Korea, where its proportions can be as high as 70–92.59% [22, 28, 36]. This differs notably from the predominance of LAM strains in South America and all subregions of Africa (except AFRI being the most predominant in Western Africa), M. africanum in West-Africa, Haarlem, X, and T in Europe, and CAS and EAI in the Indian subcontinent [32, 33]. These regional variations in M. tuberculosis population structure require that a baseline value of circulating genotypes is established to facilitate typing schemes suitable for each geographical setting. We considered it desirable to evaluate the relative efficiency of the 24-locus MIRU-VNTRs and the IS6110-RFLP for differentiating spoligotype-defined clusters of drug-resistant M. tuberculosis clinical isolates from Sichuan.
A recent study reported that the Beijing family strains acquired drug resistance in vitro more rapidly and were preferentially associated with resistance to multiple drugs than Euro-American lineage [37]. In Sichuan, spoligotyping-based analysis showed that Beijing family represented 69.28% of all isolates and so constituted the largest group (66.24%) of MDR-TB [13]; a finding that required further differentiation of the clinical isolates using genotyping methods of higher discriminatory power. The high variability in copy number and location of IS6110, as well as its stability over time, renders IS6110-RFLP typing as a useful diagnostic and epidemiological tool [19], but it fails to adequately differentiate M. tuberculosis strains with identical patterns of low copy numbers of IS6110 isolates [11, 14, 38, 39]. As compared to other M. tuberculosis strains, the Beijing isolates are characterized by specific IS6110 insertion points [19]; in our study 89.5% of Beijing family isolates contained >10 copies of IS6110, and the proportion of low-copy-number (≤6) isolates was null, showing an excellent discriminatory power, an observation that was consistent with other studies [40–42].
The discriminatory power of 24-locus MIRU-VNTR was reported to be comparable to that of IS6110-RFLP when combined with spoligotyping [43, 44]. Nonetheless, the rate of clustering is not a unique index to compare two typing methods due to the different characteristics involved in case of over-clustering observed by 2 different methods. Thus, even though VNTR typing is certainly useful as a secondary means of typing M. tuberculosis with low copy numbers of IS6110 [11], it cannot alone define all unique isolates. This is particularly true for discriminating the Beijing genotype strains, and some loci beyond the 24-locus MIRU format have permitted to further subdivide the Beijing clusters in several countries, including Russia, Japan, and Kyrgyzstan [23, 25, 45, 46]. In a comparative 5-year nationwide survey of IS6110-RFLP versus 24-locus VNTR typing in the Netherlands [20], the authors showed that although the level of discrimination did not differ substantially among the two methods, the global concordance—defined as isolates labeled unique or identically distributed in clusters by both methods—amounted to only 78.5%. Of the remaining cases, 12% were only clustered by VNTRs, 7.7% only by RFLP, and 1.8% revealed altogether different cluster compositions by the two approaches. A similar observation may exist in our study by comparing these two methods, and apparently there was a slight tendency of over-clustering by 24-locus VNTRs as compared to IS6110-RFLP for Beijing strains. All clusters were related to the Beijing family by 24-locus VNTRs (Table 1), while three of the potential VNTR over-clusters were split by RFLP, though the subclusters were apparently closely related since they differed by only one RFLP band.
Among non-Beijing family strains, two isolates sharing identical IS6110-RFLP and spoligotypes (SIT3238) profiles and 6 isolates in two clusters with a single IS6110 copy were completely differentiated by VNTRs. The general experience is that M. tuberculosis isolates contain multiple IS6110 elements and that strains with the same IS6110 fingerprints are epidemiologically related [39], but many M. tuberculosis isolates in South and Southeast Asia, including Thailand and India, have only a few copies or even single copy of IS6110 [11, 47, 48] making the discriminating power of IS6110-RFLP too low to be useful for inferring epidemiological linkages [11]. We too encountered a similar situation; nonetheless almost all the isolates with less than 6 copy numbers of IS6110 belonged to non-Beijing family in our study. The low-copy-number isolates in our study ranked in the following order: orphan strains, 4/7 or 57.1%; unknown clade, 5/9 or 55.6%; LAM9 1/2 or 50%; ill-defined T family, 20/51 or 39.2%; Manu2, 3/10 or 30%. Three out of 6 isolates harboring only one copy of IS6110 element belonged to ill-defined T family, which was the second most frequent family (18.6%).
However, despite a high clustering rate by IS6110-RFLP in many parts of Asia and certain parts of the world [11, 12, 49, 50], the switch to IS6110-RFLP typing may present downsides such as overestimating the proportion of clustered M. tuberculosis isolates of the non-Beijing family (e.g., the ill-defined T family in Sichuan). Thus 24-locus VNTR typing should be maintained for differentiating M. tuberculosis isolates with low copy numbers of IS6110. Although the discriminatory power of VNTR typing can be increased by implementing more MIRU-VNTR loci scattered through genome of M. tuberculosis, the phylogeographically diverse population structure of M. tuberculosis may make first-line typing sets country- and region-specific, facilitating the inclusion of specific MIRU-VNTR loci in different typing schemes [40, 45]. Since both 24-locus MIRU-VNTR and spoligotyping had a tendency to overestimate the proportion of clustered isolates of Beijing family which could be easily overcome by IS6110-RFLP; combining 12 highly discriminatory MIRU-VNTR loci with IS6110-RFLP could be a suitable alternative to 24-locus scheme for typing Beijing strains in Sichuan (Figure 3). On the other hand, IS6110-RFLP does not seem appropriate for differentiating non-Beijing strains due to over-clustering, which are equally and sufficiently discriminated both by 24-locus MIRU-VNTR and 12 highly discriminatory MIRU-VNTR loci (Table 1). In such as a case, the 12 highly discriminatory loci scheme consisting of the most polymorphic loci and requiring less time and labor deems suitable for non-Beijing family strains (Figure 3).
Figure 3.
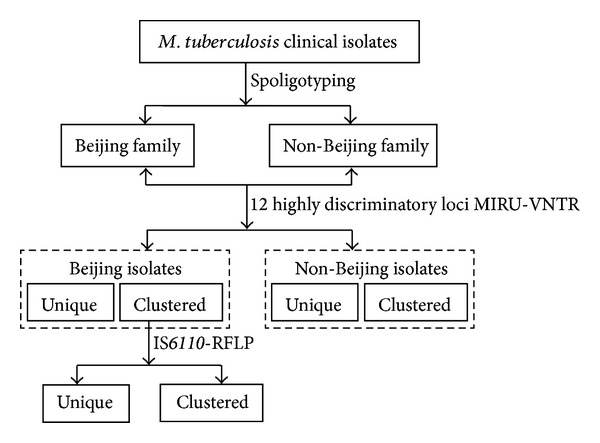
Schematic overview of the typing scheme developed to differentiate M. tuberculosis clinical isolates.
Systematic genotyping may help predict the spreading of MDR strains and improve the management of TB [50]. It has been suggested that isolates with higher IS6110 copies may evolve increased selective advantages (such as drug resistance, virulence, and efficient transmission) over those with fewer copies [41, 51–53]. Whether studies aiming to infer the potential implication of the IS6110 elements as underlying mechanisms for successful emergence of Beijing genotype strains would fully answer the genotype and phenotype relationships and explain the current transmission patterns and spread of drug-resistant TB remains debatable [19, 54–56]. In our study, 150 (78.94%) Beijing isolates were found to harbor 10 to 14 IS6110 copies, 20 harbor 7 to 9 copies, and only 20 harbor 15 to 17 IS6110 copies (Table 2). This may indicate that Beijing family strains in Sichuan were prone to moderate copies of IS6110 elements, disclosing a different IS6110-RFLP profile distinct from those observed in Beijing and Myanmar [23, 43]. On average, the W-Beijing family possesses a higher number of IS6110 copies (around 21) than any other lineage [41]. In our study, multidrug-resistance was observed among all non-Beijing isolates [29]. Five out of 6 non-Beijing isolates with only one IS6110 copy were MDR-TB (Figure 2). These results may support the fact that low-copy-number strains that belonging primarily to the non-Beijing family also evolve selective advantages and cause the transmission of drug-resistance and even outbreaks [51, 52].
5. Conclusion
This is the first investigation to differentiate M. tuberculosis isolates from Sichuan province by IS6110-RFLP. The results obtained showed that combining 12 highly discriminatory MIRU-VNTR loci with IS6110-RFLP was a better alternative for typing Beijing strains in Sichuan than 24-locus MIRU-VNTRs alone. Furthermore, the 12 highly discriminative loci scheme developed in this study showed a resolution equal to that of IS6110-RFLP and 24-locus MIRN-VNTR for discrimination of non-Beijing strains.
Acknowledgments
The authors are grateful to the financial support by National Nature Science Foundation of China (30972524, J1103518). The authors also thank Philip Supply for kindly providing the VNTR manual and the correspondence tables. Véronique Hill and Nalin Rastogi acknowledge help of their colleagues David Couvin and Thierry Zozio (Institut Pasteur de la Guadeloupe) for helping with the SITVIT2 database which benefited from research Grants from the Regional Council of Guadeloupe (Grant CR/08-1612). Yuding Zhao is cofirst author.
Ethical Approval
This study was approved by the Ethics Committee of National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention. All patients included in this study provided written informed consent for the use of clinical samples.
Conflict of Interests
The authors declare no conflict of interest.
References
- 1.World Health Organization. WHO Report. Geneva, Switzerland: World Health Organization; 2010. Global tuberculosis control. [Google Scholar]
- 2.Liu J, Jiang S, Cheng S. Analysis of current status of tuberculosis control and its strategy in China. The Journal of The Chinese Antituberculosis Association. 2003;25:129–131. [Google Scholar]
- 3.Liu Y, Duan W, Xiong G, et al. The dynamic change of drug resistance of tuberculosis in Sichuan Province. Journal of Medical Informatics. 2002;18:9–10. [Google Scholar]
- 4.National Technical Steering Group of the Epidemiological Sampling Survey for Tuberculosis (NTSG) Report on nationwide random survey for the epidemiology of tuberculosis in 2000. The Journal of The Chinese Antituberculosis Association. 2002;24:65–108. [Google Scholar]
- 5.Yang XY, Zhang NM, Diao X, Mao X, Li YP. Epidemiological analysis of pulmonary tuberculosis in Sichuan Province, China, 2000–2006. International Journal of Infectious Diseases. 2008;12(5):534–554. doi: 10.1016/j.ijid.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Tang K, Sun H, Zhao Y, et al. Characterization of rifampin-resistant isolates of Mycobacterium tuberculosis from Sichuan in China. Tuberculosis. 2013;93(1):89–95. doi: 10.1016/j.tube.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Durmaz R, Zozio T, Gunal S, et al. Genetic diversity and major spoligotype families of drug-resistant Mycobacterium tuberculosis clinical isolates from different regions of Turkey. Infection, Genetics and Evolution. 2007;7(4):513–519. doi: 10.1016/j.meegid.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Easterbrook PJ, Gibson A, Murad S, et al. High rates of clustering of strains causing tuberculosis in Harare, Zimbabwe: a molecular epidemiological study. Journal of Clinical Microbiology. 2004;42(10):4536–4544. doi: 10.1128/JCM.42.10.4536-4544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filliol I, Driscoll JR, Van Soolingen D, et al. Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. Journal of Clinical Microbiology. 2003;41(5):1963–1970. doi: 10.1128/JCM.41.5.1963-1970.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamerbeek J, Schouls L, Kolk A, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. Journal of Clinical Microbiology. 1997;35(4):907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thong-On A, Smittipat N, Juthayothin T, et al. Variable-number tandem repeats typing of Mycobacterium tuberculosis isolates with low copy numbers of IS6110 in Thailand. Tuberculosis. 2010;90(1):9–15. doi: 10.1016/j.tube.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Joseph BV, Soman S, Radhakrishnan I, et al. Molecular epidemiology of Mycobacterium tuberculosis isolates from Kerala, India using IS6110-RFLP, spoligotyping and MIRU-VNTRs. Infection, Genetics and Evolution. 2013;16:157–164. doi: 10.1016/j.meegid.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Feng Q, Tang K, et al. The population structure of drug-resistant Mycobacterium tuberculosis clinical isolates from Sichuan in China. Infection, Genetics and Evolution. 2012;12(4):718–724. doi: 10.1016/j.meegid.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 14.Radhakrishnan I, Manju YK, Kumar RA, Mundayoor S. Implications of low frequency of IS6110 in fingerprinting field isolates of Mycobacterium tuberculosis from Kerala, India. Journal of Clinical Microbiology. 2001;39(4, article 1683) doi: 10.1128/JCM.39.4.1683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathema B, Kurepina NE, Bifani PJ, Kreiswirth BN. Molecular epidemiology of tuberculosis: current insights. Clinical Microbiology Reviews. 2006;19(4):658–685. doi: 10.1128/CMR.00061-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kremer K, Glynn JR, Lillebaek T, et al. Definition of the Beijing/W lineage of Mycobacterium tuberculosis on the basis of genetic markers. Journal of Clinical Microbiology. 2004;42(9):4040–4049. doi: 10.1128/JCM.42.9.4040-4049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenner L, Malla B, Ninet B, et al. ‘Pseudo-Beijing’: evidence for convergent evolution in the direct repeat region of Mycobacterium tuberculosis . PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0024737.e24737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flores L, Van T, Narayanan S, DeRiemer K, Kato-Maeda M, Gagneux S. Large sequence polymorphisms classify Mycobacterium tuberculosis strains with ancestral spoligotyping patterns. Journal of Clinical Microbiology. 2007;45(10):3393–3395. doi: 10.1128/JCM.00828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alonso H, Samper S, Martin C, et al. Mapping IS6110 in high-copy number Mycobacterium tuberculosis strains shows specific insertion points in the Beijing genotype. BMC Genomics. 2013;14, article 422 doi: 10.1186/1471-2164-14-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Beer JL, van Ingen J, de Vries G, et al. Comparative study of is6110 restriction fragment length polymorphism and variable-number tandem-repeat typing of Mycobacterium tuberculosis isolates in The Netherlands, based on a 5-year nationwide survey. Journal of Clinical Microbiology. 2013;51(4):1193–1198. doi: 10.1128/JCM.03061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei WJ, Mokrousov I, Gui ZS, et al. Evaluation of new variable-number tandem-repeat systems for typing Mycobacterium tuberculosis with Beijing genotype isolates from Beijing, China. Journal of Clinical Microbiology. 2008;46(3):1045–1049. doi: 10.1128/JCM.01869-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mokrousov I, Narvskaya O, Vyazovaya A, et al. Mycobacterium tuberculosis Beijing genotype in Russia: in search of informative variable-number tandem-repeat loci. Journal of Clinical Microbiology. 2008;46(11):3576–3584. doi: 10.1128/JCM.00414-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwamoto T, Yoshida S, Suzuki K, et al. Hypervariable loci that enhance the discriminatory ability of newly proposed 15-loci and 24-loci variable-number tandem repeat typing method on Mycobacterium tuberculosis strains predominated by the Beijing family. FEMS Microbiology Letters. 2007;270(1):67–74. doi: 10.1111/j.1574-6968.2007.00658.x. [DOI] [PubMed] [Google Scholar]
- 24.Murase Y, Mitarai S, Sugawara I, Kato S, Maeda S. Promising loci of variable numbers of tandem repeats for typing Beijing family Mycobacterium tuberculosis . Journal of Medical Microbiology. 2008;57(7):873–880. doi: 10.1099/jmm.0.47564-0. [DOI] [PubMed] [Google Scholar]
- 25.Allix-Béguec C, Wahl C, Hanekom M, et al. Proposal of a consensus set of hypervariable mycobacterial interspersed repetitive-unit-variable-number tandem-repeat loci for subtyping of Mycobacterium tuberculosis Beijing isolates. Journal of Clinical Microbiology. 2014;52(1):164–172. doi: 10.1128/JCM.02519-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Billamas P, Smittipat N, Juthayothin T, et al. Evolution of some variable-number tandem repeat loci among a group of Beijing strains of Mycobacterium tuberculosis . Tuberculosis. 2007;87(6):498–501. doi: 10.1016/j.tube.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Dong HY, Liu ZG, Lv B, et al. Spoligotypes of Mycobacterium tuberculosis from different provinces of China. Journal of Clinical Microbiology. 2010;48(11):4102–4106. doi: 10.1128/JCM.00549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Xu P, Shen X, et al. Non-beijing strains of Mycobacterium tuberculosis in China. Journal of Clinical Microbiology. 2011;49(1):392–395. doi: 10.1128/JCM.00754-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Soolingen D, Hermans PWM, de Haas PEW, Soll DR, van Embden JDA. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. Journal of Clinical Microbiology. 1991;29(11):2578–2586. doi: 10.1128/jcm.29.11.2578-2586.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Embden JDA, Cave MD, Crawford JT, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. Journal of Clinical Microbiology. 1993;31(2):406–409. doi: 10.1128/jcm.31.2.406-409.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis . Journal of Clinical Microbiology. 2006;44(12):4498–4510. doi: 10.1128/JCM.01392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brudey K, Driscoll JR, Rigouts L, et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiology. 2006;6, article 23 doi: 10.1186/1471-2180-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demay C, Liens B, Burguière T, et al. SITVITWEB—a publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infection, Genetics and Evolution. 2012;12(4):755–766. doi: 10.1016/j.meegid.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. Journal of Clinical Microbiology. 1988;26(11):2465–2466. doi: 10.1128/jcm.26.11.2465-2466.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mokrousov I. The quiet and controversial: ural family of Mycobacterium tuberculosis . Infection, Genetics and Evolution. 2012;12(4):619–629. doi: 10.1016/j.meegid.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 36.Park YK, Bai GH, Kim SJ. Restriction fragment length polymorphism analysis of Mycobacterium tuberculosis isolated from countries in the western pacific region. Journal of Clinical Microbiology. 2000;38(1):191–197. doi: 10.1128/jcm.38.1.191-197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford CB, Shah RR, Maeda MK, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nature Genetics. 2013;45(7):784–790. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahadevan R, Narayanan S, Paramasivan CN, Prabhakar R, Narayanan PR. Restriction fragment length polymorphism typing of clinical isolates of Mycobacterium tuberculosis from patients with pulmonary tuberculosis in Madras, India, by use of direct-repeat probe. Journal of Clinical Microbiology. 1995;33(11):3037–3039. doi: 10.1128/jcm.33.11.3037-3039.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Soolingen D, de Haas PEW, Hermans PWM, Groenen PMA, van Embden JDA. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis . Journal of Clinical Microbiology. 1993;31(8):1987–1995. doi: 10.1128/jcm.31.8.1987-1995.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valcheva V, Mokrousov I, Narvskaya O, Rastogi N, Markova N. Utility of new 24-locus variable-number tandem-repeat typing for discriminating Mycobacterium tuberculosis clinical isolates collected in Bulgaria. Journal of Clinical Microbiology. 2008;46(9):3005–3011. doi: 10.1128/JCM.00437-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McEvoy CR, Falmer AA, van Pittius NCG, Victor TC, van Helden PD, Warren RM. The role of IS6110 in the evolution of Mycobacterium tuberculosis . Tuberculosis. 2007;87(5):393–404. doi: 10.1016/j.tube.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 42.Houben RM, Glynn JR. A systematic review and meta-analysis of molecular epidemiological studies of tuberculosis: development of a new tool to aid interpretation. Tropical Medicine and International Health. 2009;14(8):892–909. doi: 10.1111/j.1365-3156.2009.02316.x. [DOI] [PubMed] [Google Scholar]
- 43.Phyu S, Stavrum R, Lwin T, Svendsen SS, Ti T, Grewal HMS. Predominance of Mycobacterium tuberculosis eai and beijing lineages in yangon, Myanmar. Journal of Clinical Microbiology. 2009;47(2):335–344. doi: 10.1128/JCM.01812-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shamputa IC, Lee J, Allix-Béguec C, et al. Genetic diversity of Mycobacterium tuberculosis isolates from a tertiary care tuberculosis hospital in south Korea. Journal of Clinical Microbiology. 2010;48(2):387–394. doi: 10.1128/JCM.02167-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niemann S, Diel R, Khechinashvili G, Gegia M, Mdivani N, Tang Y-W. Mycobacterium tuberculosis Beijing lineage favors the spread of multidrug-resistant tuberculosis in the Republic of Georgia. Journal of Clinical Microbiology. 2010;48(10):3544–3550. doi: 10.1128/JCM.00715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mokrousov I, Valcheva V, Sovhozova N, Aldashev A, Rastogi N, Isakova J. Penitentiary population of Mycobacterium tuberculosis in Kyrgyzstan: exceptionally high prevalence of the Beijing genotype and its Russia-specific subtype. Infection, Genetics and Evolution. 2009;9(6):1400–1405. doi: 10.1016/j.meegid.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Mathuria JP, Sharma P, Prakash P, Samaria JK, Katoch VM, Anupurba S. Role of spoligotyping and IS6110-RFLP in assessing genetic diversity of Mycobacterium tuberculosis in India. Infection, Genetics and Evolution. 2008;8(3):346–351. doi: 10.1016/j.meegid.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Palittapongarnpim P, Luangsook P, Tartsuphaswadikul S, Chuchottaworn C, Prachaktam R, Sathapatayavongs B. Restriction fragment length polymorphism study of Mycobacterium tuberculosis in Thailand using IS6110 as probe. International Journal of Tuberculosis and Lung Disease. 1997;1(4):370–376. [PubMed] [Google Scholar]
- 49.Chauhan DS, Sharma VD, Parashar D, et al. Molecular typing of Mycobacterium tuberculosis isolates from different parts of India based on IS6110 element polymorphism using RFLP analysis. Indian Journal of Medical Research. 2007;125(4):577–581. [PubMed] [Google Scholar]
- 50.Perizzolo PF, Dalla Costa ER, Ribeiro AW, et al. Characteristics of multidrug-resistant Mycobacterium tuberculosis in southern Brazil. Tuberculosis. 2012;92(1):56–59. doi: 10.1016/j.tube.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Valway SE, Sanchez MPC, Shinnick TF, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis . The New England Journal of Medicine. 1998;338(10):633–639. doi: 10.1056/NEJM199803053381001. [DOI] [PubMed] [Google Scholar]
- 52.Victor TC, Streicher EM, Kewley C, et al. Spread of an emerging Mycobacterium tuberculosis drug-resistant strain in the Western Cape of South Africa. International Journal of Tuberculosis and Lung Disease. 2007;11(2):195–201. [PubMed] [Google Scholar]
- 53.Gutacker MM, Mathema B, Soini H, et al. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. Journal of Infectious Diseases. 2006;193(1):121–128. doi: 10.1086/498574. [DOI] [PubMed] [Google Scholar]
- 54.Parwati I, van Crevel R, van Soolingen D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. The Lancet Infectious Diseases. 2010;10(2):103–111. doi: 10.1016/S1473-3099(09)70330-5. [DOI] [PubMed] [Google Scholar]
- 55.Sun YJ, Lee AS, Wong SY, et al. Genotype and phenotype relationships and transmission analysis of drug-resistant tuberculosis in Singapore. International Journal of Tuberculosis and Lung Disease. 2007;11(4):436–442. [PubMed] [Google Scholar]
- 56.Toungoussova OS, Sandven P, Mariandyshev AO, Nizovtseva NI, Bjune G, Caugant DA. Spread of drug-resistant Mycobacterium tuberculosis strains of the Beijing genotype in the Archangel Oblast, Russia. Journal of Clinical Microbiology. 2002;40(6):1930–1937. doi: 10.1128/JCM.40.6.1930-1937.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]