

ABSTRACT
Bacterial cell division requires the formation of a mature divisome complex positioned at the midcell. The localization of the divisome complex is determined by the correct positioning, assembly, and constriction of the FtsZ ring (Z-ring). Z-ring constriction control remains poorly understood and (to some extent) controversial, probably due to the fact that this phenomenon is transient and controlled by numerous factors. Here, we characterize ZapE, a novel ATPase found in Gram-negative bacteria, which is required for growth under conditions of low oxygen, while loss of zapE results in temperature-dependent elongation of cell shape. We found that ZapE is recruited to the Z-ring during late stages of the cell division process and correlates with constriction of the Z-ring. Overexpression or inactivation of zapE leads to elongation of Escherichia coli and affects the dynamics of the Z-ring during division. In vitro, ZapE destabilizes FtsZ polymers in an ATP-dependent manner.
IMPORTANCE
Bacterial cell division has mainly been characterized in vitro. In this report, we could identify ZapE as a novel cell division protein which is not essential in vitro but is required during an infectious process. The bacterial cell division process relies on the assembly, positioning, and constriction of FtsZ ring (the so-called Z-ring). Among nonessential cell division proteins recently identified, ZapE is the first in which detection at the Z-ring correlates with its constriction. We demonstrate that ZapE abundance has to be tightly regulated to allow cell division to occur; absence or overexpression of ZapE leads to bacterial filamentation. As zapE is not essential, we speculate that additional Z-ring destabilizing proteins transiently recruited during late cell division process might be identified in the future.
INTRODUCTION
Cell division is a fundamental process that is central to cellular morphology and replication. Accurate division requires precise control of cell enlargement and of the timing and location of the site of division. The first protein found to localize at the midcell during division of prokaryotic cells was FtsZ, a GTPase that is structurally homologous to eukaryotic tubulin (1). FtsZ polymerizes at the midcell, forming a large ring-like network at the cell membrane known as the Z-ring (2). A mature divisome complex subsequently forms at the Z-ring and is composed of at least 10 proteins which are essential for cell division (3). These proteins include FtsZ, FtsA, FtsK, FtsQ, FtsL, FtsB, FtsW, FtsI, and FtsN. It is being increasingly appreciated that a number of accessory proteins are also recruited to the divisome and contribute to cell division and may be essential for growth, depending upon the specific environmental conditions. Several of these Z-ring-associated proteins (Zaps, i.e., ZapA, ZapB, ZapC, and ZapD [4–6]) modulate the dynamics of Z-ring assembly and stability. At present, little is known about cell division during growth under low-oxygen conditions, even though many bacteria thrive in anaerobic environments, particularly within mammalian hosts (7, 8).
Here we identify a further Z-ring-associated protein named ZapE which is required for cell division under low-oxygen conditions. ZapE is an ATPase present in Gram-negative bacteria that appears at the constricting Z-ring late in cell division. ZapE affects FtsZ multimerization and polymerization in vitro. In vivo ZapE recruitment correlates with Z-ring constriction.
RESULTS
Identification of ZapE, required for growth in the absence of oxygen.
While screening a library of Shigella flexneri mutants for their ability to colonize the gastrointestinal (GI) tract (9, 10), we identified a mutant that was defective for colonization with a transposon 12 bp upstream of a 1,128-bp open reading frame (ORF) (accession number b3232; Fig. 1A). The role of this ORF (here designated ZapE) in colonization was confirmed by construction and analysis of a deletion mutant (M90T::ΔzapE) and by complementation (Fig. 1; see also Fig. S1A and Tables S1A and B in the supplemental material). Of note, examination of tissues from infected animals demonstrated that S. flexneri M90T::ΔzapE displayed an elongated cellular phenotype in the GI tract (Fig. 1B).
FIG 1 .
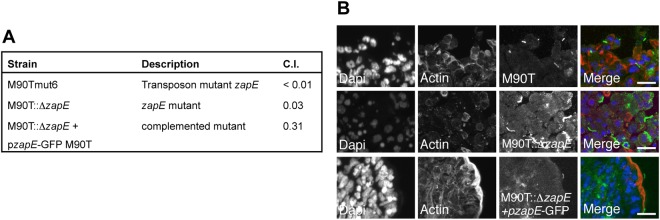
ZapE is required during the Shigella infectious process in vivo. (A) Competitive index (C.I.) of a Shigella flexneri 5A zapE transposon mutant (M90Tmut6), a zapE mutant (M90T::ΔzapE), and a complemented strain (M90T::ΔzapE/pzapE-GFP M90T) in vivo. The C.I. assessed the ability of each mutant to colonize the rabbit ileal loop in comparison with the wild-type strain. A C.I. of 1 indicates no attenuation. Data represent averages of the results of three independent experiments. (B) Immunodetection of the M90T, M90T::ΔzapE, and M90T::ΔzapE/pzapE-GFP strains in the rabbit ileal loop model. DNA was stained with DAPI (blue) and actin with RRX-phalloidin (red). Shigella strains were labeled using α-LPS polyclonal antibody (pAb) (green). Image acquisition was performed using a confocal microscope. Right-hand panels show enlarged areas. Bars are 5 µm.
To further characterize the contribution of ZapE to cell division, we examined the phenotype of an Escherichia coli MG1655 mutant (K12::ΔzapE) grown under a variety of conditions relevant to survival of bacteria in vivo. Strikingly, while E. coli lacking ZapE had no demonstrable growth defect in aerobic conditions, the mutant was defective for growth in anaerobiosis (Fig. 2A; see also Fig. S1B and C in the supplemental material); complementation analysis confirmed that loss of ZapE was responsible for the failure to grow in a low-oxygen environment. Additionally, growth at elevated temperatures (i.e., 42°C) led to an exaggerated elongation of E. coli lacking ZapE (Fig. 2B). The temperature-dependent elongated phenotype was also observed in Shigella—with or without a virulence plasmid (INV−)—as well as in E. coli, comparing growth at 30°C and 42°C (Student’s t test; P < 0.001) (see Fig. S2A and B). The K12::ΔzapE phenotype was confirmed using a K12::ΔzapE::Km-pzapE strain in the presence of glucose but not of arabinose, allowing functional complementation of the phenotype (see Fig. S3A).
FIG 2 .

zapE inactivation leads to a stress-dependent (anaerobiosis, temperature) elongated phenotype of E. coli. zapE encodes an ATPase. (A) Anaerobiosis-dependent phenotype of E. coli wild-type, ΔzapE mutant, and complemented strains. Growth of K-12, K12::ΔzapE, and K12::ΔzapE/pzapE-GFP strains in minimum media in the presence (+O2) or absence (−O2) of oxygen at 37°C is shown. Scale bars are 10 µm. Arrows indicate elongated bacteria. (B) Temperature-dependent phenotype of K-12, K12::ΔzapE, and K12::ΔzapE/pzapE-GFP strains. Bacteria were grown in rich media at the indicated temperature until an OD600 = 0.5 was reached. Scale bars are 10 µm. Arrows indicate elongated bacteria. **, P < 0.01; *, P < 0.05. (C) ZapE or ZapEK84A ATPase activity assessed by silica layer chromatography. The reaction was performed in a Tris-HCl buffer (50 mM; pH 7.4) containing 10 mCi of radiolabeled ATPγ32 (or GTPγ32), 10 mM ATP, and 2.5 mM MgCl2 in the presence of various ZapE-H6 or ZapEK84A-H6 quantities, as indicated. (D) Most probable atom model of 15 DAMMIN reconstructions fitting the data at up to smax = 0.24 Å − 1 with an x = 1.683 and an NSD value of 0.7 ± 0.024, indicating the stability of the solution. The Walker A motif in the AAA+ ATPase is highlighted in yellow. N-terminal and C-terminal residues of ZapE are highlighted in green and blue.
ZapE is an ATPase that interacts with components of the divisome.
ZapE is found in Gram-negative bacteria (see Table S1C in the supplemental material) and is predicted to contain putative Walker A (GGVGRGK84T) and Walker B nucleotide-binding sites (see Fig. S3B in the supplemental material). Therefore, we examined the capacity of ZapE to hydrolyze ATP and GTP. We found that purified recombinant ZapE hydrolyzed ATP but not GTP (Fig. 2C). Introduction of a single amino acid substitution in the ATP-binding site (in ZapEK84A) abolished ATPase activity (Fig. 2C). In addition, we modeled the organization of the active site of ZapE by small-angle X-ray scattering (SAXS) analysis (Fig. 2D; seen also Fig. S3C to E), using fold recognition methods based on the extended AAA+ ATPase domain of the ATP-dependent metalloprotease FtsH (PDB identification number [ID] 2CE7). As FtsZ is a known FtsH substrate (11, 12), the structural similarity between ZapE and FtsH suggests that ZapE might interact directly with FtsZ.
To further define the role of ZapE during cell division, we examined whether the protein interacts with components of the divisome, including FtsZ. Using a bacterial two-hybrid (BACTH) system (13), we identified potential interactions between ZapE and FtsQ, FtsL, FtsI, and FtsN (Fig. 3A) (P < 0.01 and P < 0.001, Student’s t test). In contrast, no interaction was observed with FtsA, which forms actin-like protofilaments (14), and FtsK, which coordinates DNA segregation with division (15). A similar result was obtained using ZapE from S. flexneri (M90T) as bait paired with these E. coli division proteins (Fig. 3A). Interactions of ZapE with FtsZ could not be examined by the BACTH system, as expression of both T18-zapE or T25-zapE and T18-ftsZ together was toxic (not shown and reference 16).
FIG 3 .

ZapE is a cytoplasmic protein which interacts with FtsZ in vitro and in vivo. ZapE recruitment correlates with Z-ring constriction. (A) BACTH analysis was performed using the T25-zapE K-12 (E. coli K-12) or T25-zapE M90T (Shigella M90T) versus T18-plasmid constructs with indicated genes. Results are expressed in Miller units and were averaged from three independent experiments. Error bars show the standard deviations (SD). Comparing average activity to that of the T18 negative control, ** indicates P < 0.01 and *** indicates P < 0.001 (Student’s t test). (B) Pulldown assay with K-12 ZapE-H6 and GFP or GFP-tagged proteins in E. coli lysates. The interaction between E. coli FtsZ-GFP (pDSW230) and E. coli ZapE-H6 and ZapEK84A-H6, respectively, was analyzed using a His pulldown assay. (C) Expression and localization of FtsZ-GFP and ZapE-mCherry during a cell division. Time-lapse observation was performed on an LB-agar pad at 30°C, using a 200 M Axiovert epifluorescence microscope (Zeiss). Image acquisition was performed every 3 min (see also Movie S1 and Fig. S4C in the supplemental material for raw fluorescence quantification). This result is representative of five individual observations from three independent experiments. Bars are 2 µm. Max, maximum. (D) Relationship between, respectively, ZapE-mCherry and the FtsZ-GFP mean signal (AU) (K12::ΔzapE pzapE-mCherry) and the Z-ring diameter (pDSW230). Mean FtsZ-GFP and ZapE-mCherry fluorescent signals are represented in Fig. S4C in the supplemental material. n = 5 independent observations; error bars show the SD. *** indicates P < 0.001 (Student’s t test). Max. Const., maximum constriction.
To confirm interactions identified by BACTH analysis, pulldown assays were performed with E. coli His-tagged ZapE bound to beads and green fluorescent protein-FtsQ (GFP-FtsQ), GFP-FtsL, GFP-FtsI, and GFP-FtsN expressed in E. coli (Fig. 3B); no interaction was observed when GFP was expressed on its own. In addition, pulldown assays with bound ZapE or ZapEK84A isolated FtsZ-GFP from cell lysates (Fig. 3B), indicating that the ZapE ATP binding site was not required for FtsZ interaction in vitro.
ZapE was found in the cytoplasm, and further localization by electron microscopy (EM) analysis revealed that most of the signal was detected in the vicinity of the bacterial inner membrane (see Fig. S4A in the supplemental material) (P < 0.001, Student’s t test); this observation is consistent with an interaction with FtsZ.
ZapE localizes to the constricting Z-ring toward the end of cell division.
Next, we performed time-lapse microscopy to determine the temporal and spatial association of ZapE in relation to FtsZ at the single-cell level. To achieve this, ZapE-mCherry was expressed under the control of its native promoter (see Fig. S4B in the supplemental material) in a zapE mutant containing FtsZ-GFP (carried on pDSW230) (17). Of note, ZapE was detected only at the Z-ring and exclusively during the later stages of cell division (Fig. 3C; see also Movie S1 in the supplemental material). Indeed, the appearance of ZapE at the midcell coincided with constriction of the Z-ring and with its subsequent disappearance (see Fig. S4C). In events leading up to the maximalization of Z-ring constriction (defined as t0), FtsZ levels increased in a stepwise fashion as the diameter of the Z-ring diminished; the amount of ZapE present at the Z-ring increased progressively until maximal constriction of the Z-ring occurred (Fig. 3D) (Student’s t test; P < 0.001), demonstrating a clear correlation between the expression and localization of ZapE and Z-ring constriction in vivo.
ZapE abundance modulates Z-ring stability.
We next examined whether modulating levels of ZapE within cells affected their shape and the appearance of Z-rings. Initially, the location of FtsZ-GFP during cell division was examined in either the presence or the absence of ZapE. FtsZ-GFP does not fully complement an ftsZ mutant, but localizes to the midcell, and does not impair normal cell division when expressed at basal levels in wild-type bacteria (18). Loss of ZapE in a strain expressing FtsZ-GFP led to marked filamentation, with the loss of ordered Z-rings in cells (Fig. 4A [rich media] and B [minimum media]; see also Fig. S5A and Movie S2 in the supplemental material [live microscopy]). This filamentation was more pronounced during growth at higher temperatures (Fig. 4A) and during the stationary phase (Fig. 4C). We confirmed that filamentation in cells lacking ZapE was more marked following overexpression of native FtsZ in a dose-dependent manner (Fig. 4D).
FIG 4 .

Effect of ZapE inactivation on K-12 shape upon FtsZ-GFP and FtsZ expression modulation. (A) Localization of FtsZ-GFP (pDSW230) in K-12 and K12::ΔzapE strains grown in rich media (LB) at 37°C or 42°C in the absence of IPTG until an OD600 = 0.5 was reached. Bars are 5 µm. (B) Localization of FtsZ-GFP (pDSW230) in K-12 and K12::ΔzapE strains grown in minimum media (M9) at 37°C in the absence of IPTG until an OD600 = 0.5 was reached. Bars are 2 µm. (C) FtsZ-GFP (pDSW230) localization in K-12 and K12::ΔzapE strains during the stationary phase performed in LB rich media at 37°C or 42°C. These observations are representative of the results of at least three independent experiments. Bars are 1 µm. (D) Effect of FtsZ-H6 (WM971) overexpression on K-12 and K12::ΔzapE shape. Bacteria were grown in LB at 37°C in the presence of the indicated concentrations of IPTG until an OD600 = 0.5 was reached. These observations are representative of the results of three independent experiments. Bars are 10 µm.
Loss of ZapE in a strain expressing FtsZ-mCherry led to the formation of multiple disorganized FtsZ rings found sporadically along the length of the extended bacteria compared to the wild-type strain (Fig. 5A). Furthermore, we examined the effect of overexpressing ZapE under the control of an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible promoter. In wild-type cells, ZapE overexpression also resulted in dose-dependent filamentation (Fig. 5B and C; see also Fig. S5B in the supplemental material). However, no Z-rings (detected with FtsZ-mCherry) were detected along the length of filaments when ZapE overexpression was induced with high levels of IPTG (Fig. 5A; see also Fig. S5C [as a control, without IPTG]), although additional adverse effects of ZapE overexpression could not be ruled out. This was not observed in cells overexpressing ZapEK84A, which exhibited normal bacterial shape and single ordered Z-rings (Fig. 5A). Taken together, these data are consistent with ZapE and FtsZ having antagonizing roles at the Z-ring inside cells.
FIG 5 .

ZapE level of expression perturbates Z-ring stability and bacterial shape. (A) Effect of zapE inactivation and ZapE or ZapEK84A overexpression on Z-ring stability (FtsZ-mCherry, pAKF133). Bacteria were grown in minimum media at 37°C in the presence of arabinose (0.01%) in addition to IPTG (1 mM) when indicated. Data are representative of the results of four independent observations (see also Fig. S5C in the supplemental material). (B) ZapE-H6 level of expression in strain K12-pzapE-H6 grown in LB at 37°C in the presence of the indicated concentrations of IPTG until an OD600 = 0.5 was reached. The level of expression of ZapE-H6 was assessed by Western blot analysis on bacterial whole extract using a polyclonal α-ZapE antibody. α-RecA was used as a control. (C) Phenotypic characterization of strain K12-pzapE-H6 grown under the conditions described for panel B. These observations are representative of the results of three independent experiments. Bars are 2 µm. (D) Polymerization of FtsZ (10 µM) with FtsZ-GFP (5 µM) in vitro was performed over 3 min, in the presence of 10 mM CaCl2, as described in references 16 and 19. ZapE-H6 or ZapEK84A-H6 (10 µM) was subsequently added to the reaction mixture (t = 0), with ATP added when indicated. Confocal imaging was performed at 0 and 3 min. Data are representative of the results of three independent experiments. Bars are 5 µm.
ZapE impairs the stability of polymerized FtsZ in vitro.
Next we examined the effect of ZapE on polymerized FtsZ in vitro using purified recombinant proteins. As described previously (19), in the presence of GTP and Ca2+ purified FtsZ and FtsZ-GFP form small polymers in vitro (Fig. 5D; see also Fig. S5D in the supplemental material). The addition of ZapE promoted the polymerization of FtsZ/FtsZ-GFP through large helical structures (Fig. 5D; see also Movie S3). These structures were no longer stable when ATP was added with ZapE after a 3-min incubation (Fig. 5D), while ZapEK84A had no effect (Fig. 5D), indicating that active ZapE reduces the stability of FtsZ polymers in the presence of ATP through a molecular mechanism which remains to be defined.
DISCUSSION
Here, we identified and characterized a novel Z-ring-associated protein named ZapE, which is an ATPase found among Gram-negative bacteria (see Table S1C in the supplemental material). ZapE is not essential in E. coli or in Shigella during growth under standard laboratory conditions. However, in the absence of oxygen or at temperatures over 37°C, the contribution of ZapE to cell division becomes evident, with cells lacking this protein displaying a growth defect and elongated phenotype (Fig. 2A and B). The latter was detected in tissue sections of the GI tract infected with a Shigella zapE mutant; as a likely consequence of the cell division defect, zapE is required for efficient colonization of GI track (Fig. 1A and B). Therefore, the identification of ZapE from studies of bacteria in vivo indicates that examining bacteria in the environments that they encounter outside the laboratory could uncover the function of other accessory cell division proteins. In the future, studies of bacterial cell division under other growth conditions such as low pH, oxidative stress, or amino acid starvation will reveal novel aspects of cell division regulation allowing bacterial survival during host invasion.
Several ATPases targeting FtsZ during cell division such as FtsH, MinD, and KaiC are involved in cell division functions. FtsH is an ATP-dependent zinc metalloprotease targeting FtsZ in vitro (11), although this activity could not be confirmed in vivo (12). MinD belongs to the Min system involved in Z-ring positioning (20). KaiC inhibits Z-ring formation controlling the circadian clock in Synechococcus elongatus (21). None of these ATPases have proven functions in the dynamics of Z-ring constriction in vivo. Indeed, if starting events of cell division (Z-ring assembly and positioning) have been well characterized (22), events leading to the disappearance of the ring and completion of division have not been defined in as much detail and remain to be discussed (23). This might be explained by the transiency of the Z-ring constriction phenomenon. Consistently, ZapE fusion (mCherry) recruitment at the Z-ring could be seen in this study only by live microscopy (Fig. 3C; see also Movie S1 in the supplemental material).
We observed that in the presence of ATP and Ca2+, ZapE promoted bundling of FtsZ/FtsZ-GFP polymers into unstable three-dimensional structures (Fig. 5D; see also Movie S3 in the supplemental material). A structural homology between ZapE and FtsH was found by SAXS analysis (Fig. 2D). This observation should support further investigations on how ZapE directly or indirectly affects the effect of activity on Z-ring constriction, accounting for the destabilization observed in vitro (Fig. 5D) and in vivo upon ZapE overexpression (Fig. 4A).
The precise control of the ZapE level appears to be crucial for Z-ring stability and dynamics as we showed that either ZapE loss or overexpression (Fig. 4A) altered Z-ring stability and led to bacterial filamentation. Further studies should aim at defining ZapE abundance regulation (expression and degradation) during the cell division process and its consequences for Z-ring stability to better characterize the role of ZapE in the Z-ring dynamic in vivo.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are described in Table S1A in the supplemental material. Shigella strains were grown in Trypticase soy (TCS) broth or on TCS agar plates supplemented with 0.01% Congo red (Sigma). E. coli strains were grown in LB media. Anaerobic growth experiments were performed in a Mini Macs MG-250 chamber (Don Withley). d-Glucose and l-arabinose were used at a concentration of 0.2% to modulate the expression of genes cloned under the control of the pBAD33 promoter.
Expression plasmid construction.
The pSUC vector construction was achieved by amplifying the mCherry fusion from the pmCherry-N1 vector using the SG150 and SG151 primer pair (see Table S1B in the supplemental material), introducing the BamHI and EcoRI restriction sites. The pSU19 vector was digested with BamHI and EcoRI restriction enzymes prior ligation of the digested mCherry amplified fragment, leading to the generation of the pSUC vector (see Fig. S4B). This expression vector allows the expression of mCherry protein fusions in the C terminus under the control of the promoter of the gene of interest in E. coli and in Shigella.
Expression of the FtsZ-GFP fusion under the control of a lac promoter was performed using either the pDSW230 and pDSW231 construct (kindly provided by David Weiss) (described in Table S1A in the supplemental material). FtsZ-mCherry fusion expression under the control of an arabinose-inducible promoter was performed with pAKF133 (described in Table S1A).
DNA manipulations.
The initial transposon insertion in S. flexneri M90T in zapE was performed as described previously (9). The construction of inactivated zapE mutants was then performed in E. coli and S. flexneri.
ΔzapE mutant construction.
In MG1655, E. coli K-12 P1vir page lysate was prepared on the JW3201 donor strain from the Keio collection (1, 24). In the JW3201 strain, the zapE ORF is substituted by the kanamycin (Km) resistance marker (ΔzapE::Km) (2, 24). The ΔzapE::Km cassette was introduced into MG1655 by P1 transduction (3, 25), and selection for kanamycin-resistant (Kmr) colonies was made on LB plates containing kanamycin (50 µg/ml). After reisolation, several clones were verified by PCR to confirm the right chromosomal structure of the ΔzapE::Km deletion. One clone was chosen and named K12ΔzapE::Km.
K12::ΔzapE was then obtained from K12ΔzapE::Km by removing the kanamycin resistance marker from the ΔzapE::Km cassette. In this ΔzapE::Km cassette, the antibiotic resistance marker is flanked by two direct frt repeats, which are the recognition targets for the site-specific recombinase FLP (4, 24). Therefore, to get rid of the resistance marker from the K12::ΔzapE::Km chromosome, pCP20, a temperature-sensitive plasmid that encodes the FLP recombinase, was used (5, 26). Briefly, K12Δop1::Km cells were transformed with pCP20, and chloramphenicol-resistant (Cmr) colonies were selected at 30°C on LB plates containing the corresponding antibiotic (30 µg/ml). Several of these clones were grown overnight on antibiotic-free LB plates at 42°C. Ten independent colonies were selected, and after single-colony passage at 30°C, all 10 colonies were no longer Cmr and Kmr, indicating simultaneous loss of pCP20 and the kanamycin resistance marker from the bacterial chromosome. This FLP-catalyzed excision created an in-frame deletion of the zapE ORF, leaving behind a 102-bp scar sequence (ΔzapE::frt) (6, 24). To confirm the correct chromosomal structure of the ΔzapE::frt deletion, several Cms and Kms clones were tested by PCR using the NWpr40 and NWpr41 primer pair (see Table S1B in the supplemental material). After confirmation, one clone was chosen and named K12::ΔzapE.
Alternatively, the ΔzapE::Km cassette was introduced into MG1655 expressing pzapE (pBAD33-zapE) by P1 transduction (7, 25), and selection for kanamycin-resistant (Kmr) colonies was performed on LB plates containing kanamycin (50 µg/ml). The K12::ΔzapE::Km pzapE strain was obtained.
In order to inactivate zapE in Shigella flexneri (M90T), a one-step chromosomal inactivation method was used to target homologous region for integration. Therefore, we generated PCR products with much longer flanking sequence using the K12::ΔzapE null mutant as the template. The M90T was transformed with PCR products amplified from K12::ΔzapE::Km mutant genomic DNA using primers NWpr23 and NWpr24 (see Table S1B in the supplemental material). The NWpr23 and NWpr24 primers were designed to include 50 bp of upstream and downstream sequence flanking zapE. This product was transformed into M90T::pKD46, which resulted in all kanamycin-resistant colonies containing the 1.50-kb kanamycin resistance gene as analyzed by PCR. Thus, an S. flexneri null mutant (M90T::ΔzapE) was successfully generated.
Expressing, respectively, a zapE-GFP and a zapE-mCherry fusion under the control of the zapE promoter resulted in the complementation of the M90T::ΔzapE and K12::ΔzapE mutants. In order to express a ZapE-GFP fusion, the zapE genes of Shigella and E. coli and their promoters (~500 bp) were amplified with the SG127 and SG128 primer pair (see Table S1B in the supplemental material) and cloned into pFpV25 vector digested with the BamHI and NdeI restriction enzymes. The pzapE-GFP M90T and pzapE-GFP K-12 constructs were obtained and sequenced (Table S1A).
In order to express zapE-mCherry, the zapE gene and its promoter (~500 bp) were amplified with the SG219 and SG155 primer pair (see Table S1B in the supplemental material) and cloned in pSUC vector digested with the HindIII and XbaI restriction enzymes (see Fig. S4B). The K84A point mutation of zapE was performed using the SG114 and SG115 primer pair (see Table S1B). pzapE-mCherry and pzapEK84A-mCherry constructs were obtained and sequenced (see Table S1A).
In order to overproduce the ZapE-H6 and ZapEK84A-H6 protein fusions in an IPTG-dependent manner, the corresponding zapE DNA fragments were amplified by PCR prior cloning in the pKJ1 plasmid digested using the NcoI and BamHI restriction enzymes (see Table S1A in the supplemental material). zapE was amplified using the SG90 and SG91 primer pair (see Table S1B), and zapEK84A was obtained using the SG114 and 1G115 primer pair to introduce a K84A single point mutation (see Table S1B). The resulting pzapE-H6 and pzapEK84A-H6 constructs were analyzed by PCR and sequenced.
Rabbit ligated ileal loop model.
New Zealand White rabbits weighting 2.5 to 3 kg (Charles River Breeding Laboratories, Wilmington, MA) were used for experimental infections. For each animal, up to 12 intestinal ligated loops, each 5 cm in length, were prepared as described previously (8, 9, 27). For the evaluation of the competitive index (C.I.), equal quantities of the wild-type strain and of the mutant were injected in each loop (corresponding to a total dose of 105 CFU per loop). After 16 h, animals were sacrificed and the luminal fluid was aspirated and S. flexneri recovered. The C.I. was calculated as the proportion of mutant to wild-type bacteria recovered from animals divided by the proportion of mutant to wild-type bacteria in the inoculums, and results are expressed as the means of the results determined with at least 4 loops from two independent animals. The experimental protocol was approved by the French Ethic Committee Paris 1 (number 20070004, 9 December 2007).
For immunohistochemical staining, infected rabbit ileum samples were washed in phosphate-buffered saline (PBS), incubated at 4°C in PBS containing 12% sucrose for 90 min and then in PBS–18% sucrose overnight, and frozen in optimum cutting temperature (O.C.T.) compound (Sakura) on dry ice. Sections (7 µm in thickness) were obtained using a CM-3050 cryostat (Leica). Fluorescent staining was performed using a rabbit anti-Shigella lipopolysaccharide (LPS) primary antibody (P. Sansonetti, Institut Pasteur) (1:200 dilution) and an anti-rabbit fluorescein isothiocyanate (FITC)-conjugated secondary antibody (1:1,000). Epithelium cell nuclei were stained with DAPI (4[prime],6-diamidino-2-phenylindole) (1:1,000) and actin stained with Rhodamine Red-X (RRX)–phalloidin (1:1,000). Image acquisition was performed using laser scanning confocal microscopy. Image analysis was performed using ImageJ software.
Two-hybrid screen.
We used the BACTH system that is based on the interaction-mediated reconstitution of an adenylate cyclase (AC) enzyme in the otherwise defective DHM1 E. coli strain (9, 13, 28). This system is composed of two replication-compatible plasmids, pKT25 and pUT18, respectively encoding the intrinsically inactive N-terminal T25 domain and C-terminal T18 domain of the AC enzyme. E. coli and Shigella zapE was amplified using the NG1281 and NG1282 primer pair and cloned in pKT25 vector.
pKT25 and pUT18 plasmids were subsequently doubly transformed to DHM1 to search for the AC reconstitution that turns on β-galactosidase production, leading to the production of blue color after 2 days of growth at 30°C on indicator plates containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (Eurobio) (40 mg ml-1), isopropyl-1-thio-β-d-galactopyranoside (Invitrogen) (0.5 mM), Ap, Km, and nalidixic acid. β-Galactosidase activity was measured as described before, averaged from three independent experiments, and expressed as Miller units (10, 13, 29).
Antibody—α-ZapE antibody production.
An α-ZapE rabbit polyclonal antibody was collected from two New Zealand White rabbits challenged with purified ZapE-H6 (2 mg/ml solution) on four occasions separated by 2 weeks rest. The first injection (intradermal; 500 µl) was performed with the purified protein (125 µg) and with complete Freund’s adjuvant. The second injection was performed following a similar procedure in the presence of incomplete Freund’s adjuvant. The third and the last injections were performed with no adjuvant. The final blood collection was performed by cardiac puncture using a heparin-free tube. Sera were separated from blood cells by centrifugation (14,000 rpm, 30 min). α-FtsZ rabbit polyclonal antibody was kindly provided by Kenn Gerdes (4, 13, 17).
TLC analysis.
ATPase and GTPase assays were performed in the presence of bovine serum albumin (BSA) (Sigma) (1.25 mg/ml), ATPγ32 or GTPγ32 (PerkinElmer) (30 μCi), ATP or GTP (Sigma) (50 mM), and 0.1 to 10 µg of purified ZapE-H6 and ZapEK84A-H6 as indicated. The final reaction mixture volume was 20 µl in TMD buffer (25 mM Tris [pH 7.4], 10 mM MgCl2, 1 mM dithiothreitol [DTT]). The reaction was run during 10 min at 37°C and stopped by the addition of 20 µl methanol. When indicated, chromatography was performed on thin-layer chromatography (TLC) plates (Thomas Scientific), with a mobile phase containing a mixture of lithium chloride (LiCl) and formic acid. After migration, a film was exposed on the plate and further developed. Radiolabeled Pi presence through ATPγ32 hydrolysis was then revealed.
Protein expression and purification.
ZapE-H6 and ZapEK84A-H6 protein fusions were expressed using the pzapE-H6 K-12 and pzapEK84A-H6 K-12 constructs (see Table S1A in the supplemental material) expressed in an E. coli BL21(DE3) strain. Overnight cultures were subcultured in fresh LB media (1:100) and grown at 37°C until an optical density at 600 nm (OD600) of 0.5 was reached. Overexpression was induced by the addition of 0.5 mM IPTG and was performed overnight at room temperature (RT). His-tagged proteins were purified on Talon beads (Clontech) and further purified by gel filtration using a Hiload 16/60 Superdex 200 column (GE) and a Tris buffer (50 mM; pH 7.5) containing 5 mM MgCl2, 1 mM EDTA, and 0.1 M NaCl. FtsZ and FtsZ-GFP were purified by ion exchange on a Hiload 16/10 DEAE column using a Tris buffer (50 mM; pH 7.5) containing 5 mM MgCl2, 1 mM EDTA, and 0.1 M NaCl (buffer 1) and a Tris buffer (50 mM; pH 7.5) containing 5 mM MgCl2, 1 mM EDTA, and 1 M KCl (buffer 2) as described previously (14, 30), followed by a gel filtration, as described above.
His pulldown assay.
ZapE-H6 or ZapEK84A-H6 fusions (30 µg) were incubated on Talon beads for 1 h at 4°C in resuspension buffer (20 mM Tris-HCl [pH 8], 100 mM NaCl), supplemented with a protease inhibitor cocktail (Roche). Unbound proteins were removed by washing twice using the resuspension buffer. Extracts containing GFP fusions (FtsZ, FtsQ, FtsL, FtsI, and FtsN) were prepared from bacterial cultures grown in LB at 37°C in the presence of 1 mM IPTG for 3 h. Bacterial pellets were resuspended in a Tris-HCl buffer (50 mM) supplemented with a protease inhibitor cocktail (Roche) and sonicated. The resulting suspensions were centrifuged at 12,000 × g for 30 min, and supernatants were collected. In each pulldown experiment, 10 µg of E. coli soluble extract containing GFP fusions (FtsZ, FtsQ, FtsL, FtsI, and FtsN) was incubated during 1 h at 4°C. After 3 washes with resuspension buffer, 2× sample buffer was added and the beads were boiled and subjected to SDS-PAGE gel analysis. As controls, extracts were incubated with Talon beads only.
FtsZ/FtsZ-GFP polymerization in vitro.
Fluorescent FtsZ/FtsZ-GFP polymers were generated in a buffer containing 50 mM HEPES, 50 mM KCl, 5 mM MgCl2, and 10 mM CaCl2 in addition to 1 mM GTP. Polymerization of FtsZ (10 µM) and FtsZ-GFP (5 µM) occurred during 0 to 3 min at 30°C on glass slides. Then, ZapE-H6 or ZapEK84A-H6 (10 µM) and ATP (1 mM) were added when indicated to reach a final volume of 100 µl. Polymer imaging was performed using a TCS SP5 confocal microscope (Leica).
Western blot analysis.
Bacterial extracts were prepared as follows. For FtsZ, ZapE, and RecA detection in K-12 and K12::ΔzapE strains, overnight bacterial cultures were subcultured in 100 ml LB liquid media at 37°C until the OD at A600 reached 0.3. Bacteria were harvested by centrifugation for 15 min at 3,000 × g, washed, and then resuspended in 10 ml PBS. Cells were spun again for 5 min at 3,000 × g and resuspended in 10 ml of PBS. Membrane-associated (Pellet) and soluble (Sol.) proteins were separated by centrifugation for 20 min at 12,000 × g.
Total protein concentrations were measured by the method of Bradford (Bio-Rad). Proteins were separated by 16% SDS-PAGE, transferred to nitrocellulose membranes, and incubated with the primary antibodies diluted in PBS–5% milk–0.01% Tween 20 (Sigma) overnight. Membranes were washed in PBS three times and then incubated with secondary antibodies for 1 h before washing. Antibody binding was detected with chemiluminescence (ECL kit; GE Healthcare).
FtsZ polymer sedimentation assay.
FtsZ polymers (P) were generated in a reaction mixture containing 50 mM HEPES, 50 mM KCl, and 5 mM MgCl2 in addition to 1 mM GTP during 3 min at 30°C and collected by ultracentrifugation (11 min, 80,000 rpm) at 4°C (TL-100 Ultracentrifuge; Beckman). The reactive buffer was then discarded and replaced by a reaction mixture containing 50 mM HEPES, 50 mM KCl, and 5 mM MgCl2 in addition to 1 mM ATP and 0.5 µg/ml of purified ZapE-H6 when indicated in a final volume of 100 µl. The reaction was stopped after 0, 1, or 3 min, as indicated. FtsZ polymers containing the pellet fraction (P) were separated from the soluble fraction (S) by ultracentrifugation (11 min, 80,000 rpm) at 4°C (TL-100 Ultracentrifuge; Beckman). Samples were resuspended in a Laemli buffer (1× final concentration) and subsequently subjected to SDS-PAGE gel analysis and transferred onto a nitrocellulose membrane. FtsZ and ZapE-H6 were detected in both fractions by Western blotting using rabbit polyclonal antibodies.
Bioinformatics.
Identification of ZapE homologous proteins among other organisms was performed using BlastP. The ZapE sequence comparisons were performed using ClustalW software. Length measurement of bacteria was performed with MicrobeTracker suite software (version 0.937) (15, 17, 31), and the data mining was performed using a MatLab computing system (R2012 version with Image Processing Toolbox and Statistics Toolbox). Statistical analyses were performed using GraphPad Prism 5 software.
Fluorescent protein fusion imaging.
In order to localize FtsZ-GFP and ZapE-mCherry and mutated versions of protein fusions in bacteria, the corresponding expression plasmids were transformed in E. coli K-12 MG1655 wild-type or ΔZapE strains, as indicated. The localization was performed on either fixed or living bacteria. The fixation of bacteria was performed by adding 4% paraformaldehyde (PFA) followed by a washing in PBS. The observation was performed using a Nikon Eclipse 80i or an epifluorescence microscope. The live observation of FtsZ-GFP and ZapE-mCherry during the cell division process was performed on an LB agarose (1%) pad using a 200 M Axiovert epifluorescence microscope (Zeiss) equipped with a Lambda LS 300W Xenon lamp and a CoolSnapHQ charge-coupled-device (CCD) camera.
In order to localize FtsZ-mCherry (pAKF133) in K-12 and K12::ΔzapE strains, bacteria were grown in an M9 minimum media at 37°C until an OD600 of 0.5 was reached and were further incubated on a minimal medium agar pad with 0.01% arabinose. The cell division process was observed using an inverted microscope (Nikon Ti) equipped with a 100× 1.4 numerical aperture (NA) PL-APO objective lens. Image stacks were acquired using Metamorph software (MDS) and a CCD camera (Photometrics Coolsnap HQ). A similar procedure was applied to observe the effect of ZapE-H6 overexpression on FtsZ-mCherry localization, through the addition of IPTG (1 mM) within the minimal medium agar pad with 0.01% arabinose (no IPTG was added during the liquid culture growth).
Image reconstruction (see Movie S3 in the supplemental material) was performed using Imaris software (Bitplane).
SUPPLEMENTAL MATERIAL
Live observation of E. coli K-12 FtsZ-GFP and ZapE-mCherry time-dependent expression and localization during a cell division process. Time-lapse observation was performed on an E. coli K12::ΔzapE/pDSW230/pzapE-mCherry strain grown on an LB-agar pad at 30°C, using a 200 M Axiovert epifluorescence microscope (Zeiss). Image acquisition was performed every 3 min, and the movie is shown at 5 frames per second (fps). This result is representative of five individual observations from three independent experiments. Download
FtsZ-GFP (pDSW231) time-dependent expression and localization in a K12::ΔzapE strain during a cell division process. Time-lapse observation was performed on strains grown on an LB-agar pad at 30°C in the absence of IPTG, using a 200 M Axiovert epifluorescence microscope (Zeiss). Image acquisition was performed every 3 min, and movies are shown at 5 fps. These data are representative of the results of at least three independent observations. Bars are 2 µm. Download
Three-dimensional (3D) view of FtsZ/FtsZ-GFP helical polymerization in the presence of ZapE-H6 (1:1) and the absence of ATP at t = 0 min (see Fig. 5B). Image reconstruction was performed using Imaris software. 3D Z stack determinations were performed using 40 input images with resolution of z = 0.2 μm. Download
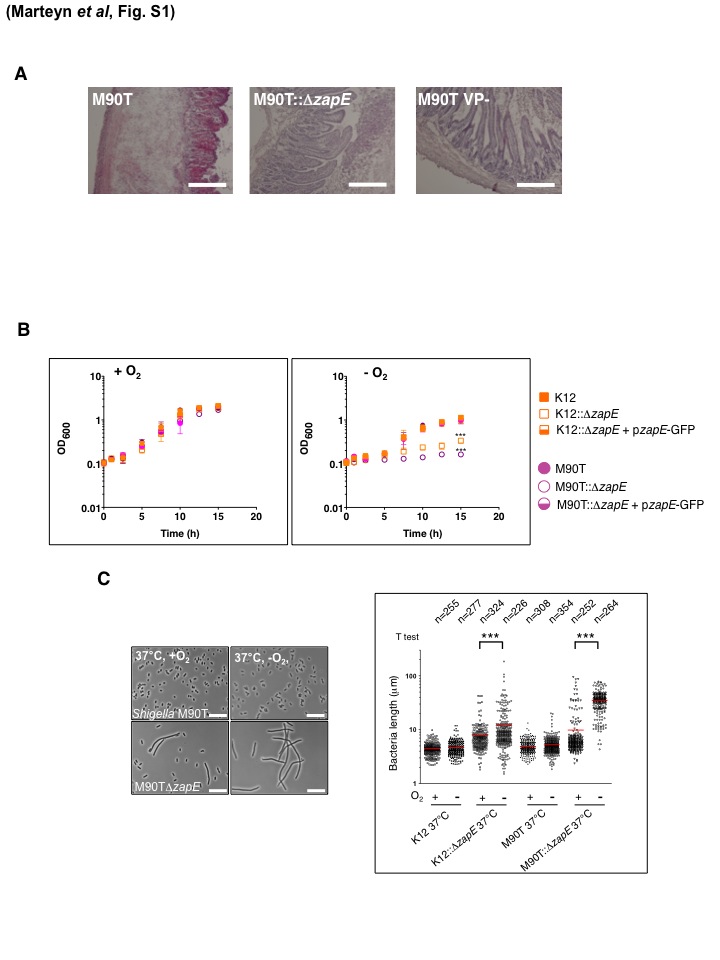
(A) Histopathological analysis of rabbit ileal loops infected by M90T, M90T::ΔzapE, and M90T VP- (BS176) strains. Paraffin-embedded tissues were stained using hematoxylin-eosin. Bars are 50 µm. Data represent K-12 ZapE homologous sequence comparisons. Growth of strain M90TΔzapE is attenuated in vivo. (B) Growth defect of K12::ΔzapE and M90T::ΔzapE strains under low-oxygen conditions. K-12, M90T, K12::ΔzapE, and M90T::ΔzapE strains were grown in minimum media in the presence (+O2) or absence (−O2) of oxygen at 37°C. Scale bars are 10 µm. (C) Phenotypic characterization of M90T and M90T::ΔzapE grown in minimum media in the presence (+O2) or absence (−O2) of oxygen at 37°C. Scale bars are 10 µm. Length measurements of bacteria were performed on each strain using MicrobeTracker software (1, 2); conditions were as indicated on the left and in the Fig. 2A legend. Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of bacteria measured per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). Download
{kind=link}
(A) Temperature-dependent phenotype of Shigella wild-type, ΔzapE mutant, and complemented strains. Bacteria were grown in rich media at the indicated temperature until an OD600 = 0.5 was reached. Scale bars are 10 µm. (B) Length measurements of bacteria were performed on each strain under the conditions described in the panel A legend and the Fig. 2B legend using MicrobeTracker software (2, 3). Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of measured bacteria per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). Download
{kind=link}
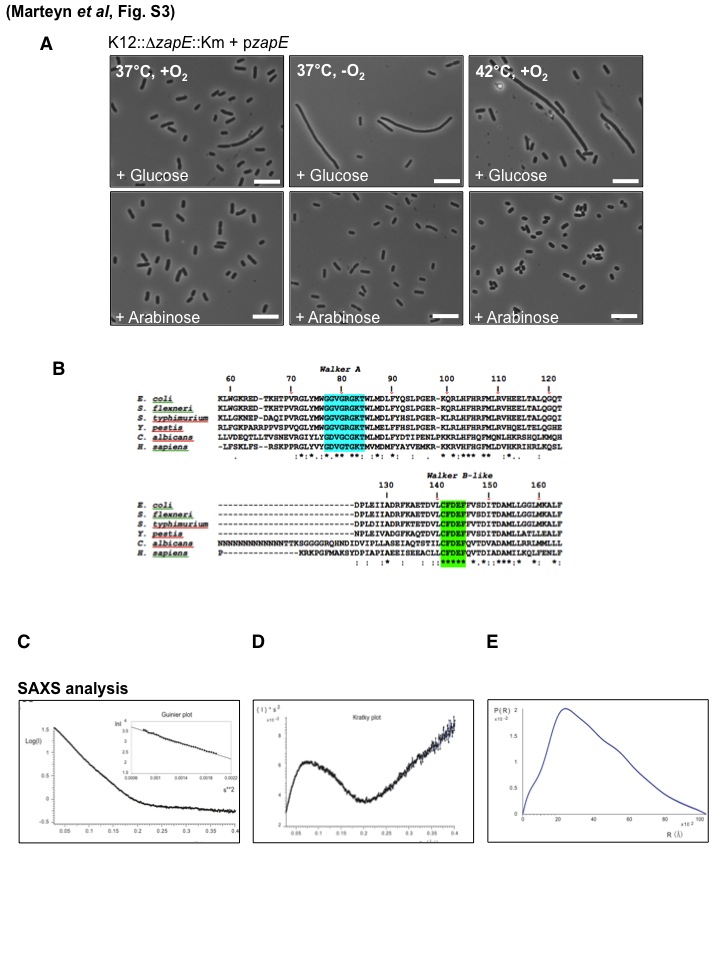
(A) Phenotype of strain K12::ΔzapE::Km/pzapE in the presence of glucose or arabinose. Bacteria were grown in a rich liquid media under the indicated conditions in the presence of glucose (0.2%) or arabinose (0.2%) until an OD600 = 0.5 was reached. Scale bars are 5 µm. (B) Partial primary sequence alignment of ZapE proteins from different species. Alignment was made using the ClustalW software, and identifications of putative Walker A and Walker B consensus sequences are highlighted. Alignment is shown in ZapE homologues identified in E. coli K-12 MG1655 (B3232, YhcM), Shigella flexneri 5A M90T (S3487), Salmonella enterica serovar Typhi (STY3526), Yersinia pestis (YPO3564), Candida albicans (AFG11), and Homo sapiens (LACE1). Sequence ID, percent homology, and percent identity and the P-loop sequence are detailed in Table S1C in the supplemental material. ZapE SAXS structure models are shown. (C, D, and E) SAXS properties of the ZapE full-length protein. In panel C, the final processed SAXS curve after subtraction of the buffer, extrapolation to zero concentration, and merging and scaling of curves coming from various concentrations is shown. In the inset, the Guinier plot in the small-angle range is almost linear, which is indicative of ZapE particle monodispersity. In panel D, the Kratky representation of the final processed SAXS curve in panel A provides the information of a globular particle, as there is a maximum apparent in the low “s” area. Still, this maximum is not completely apparent. (E) The pair distribution function. The indirect Fourier transform of the final processed SAXS curve corresponding to Fig. 2D provides the first geometric characteristics of the ZapE particle in real space. The maximum diameter of the particle is estimated at 103 Å. Download
{kind=link}
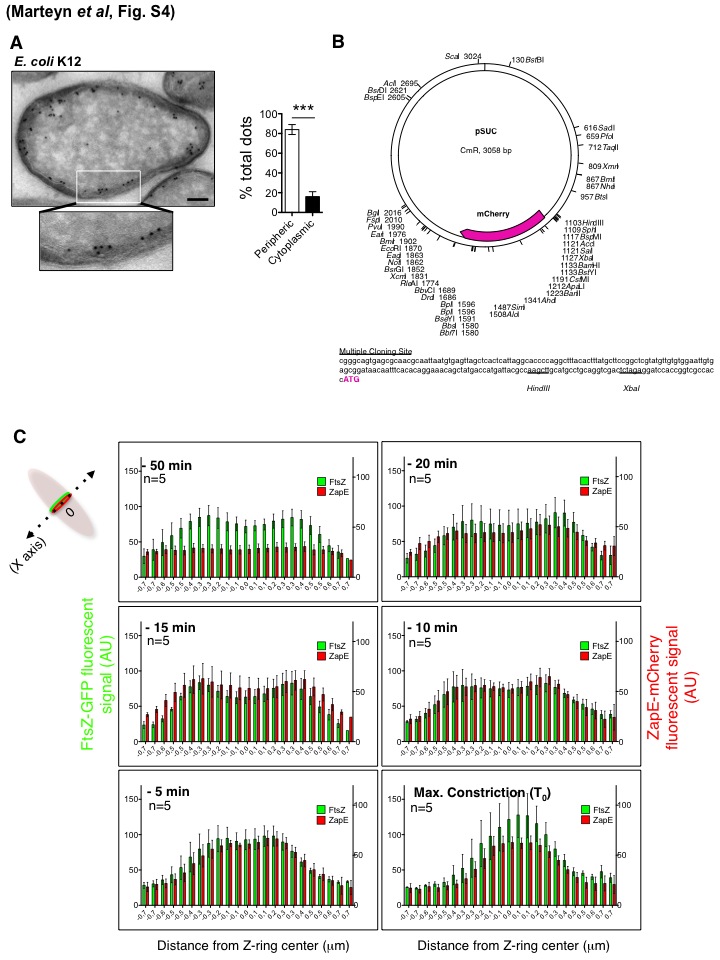
ZapE localization. (A) ZapE localization was performed in K-12 bacteria by electron microscopy using immunogold staining with a polyclonal α-ZapE antibody. The bar is 200 nm. Localization quantification is indicated, and error bars show the SD. *** indicates P < 0.001 (Student’s t test). (B) pSUC plasmid description. A schematic representation of the pSUC plasmid map in addition to its multiple-cloning-site sequence (HindIII and XbaI restriction sites are underlined) is shown. (C) Colocalization of FtsZ-GFP and ZapE-mCherry signals during Z-ring constriction and FtsZ-GFp localization in an E. coli ΔzapE mutant. FtsZ-GFP and ZapE-mCherry fluorescent signals (AU) from Movie S1 in the supplemental material and Fig. 3C were quantified in relation to the distance from the Z-ring center (as indicated on the top scheme). Measurements were performed on images acquired at the time of maximal constriction (Max. Constriction) and at 50 min, 20 min, 15 min, 10 min, and 5 min before. The x axis indicates the distances from the Z-ring center (−0.7 to 0.7 µm). n = 5 independent observations; error bars show the SD. Download
{kind=link}
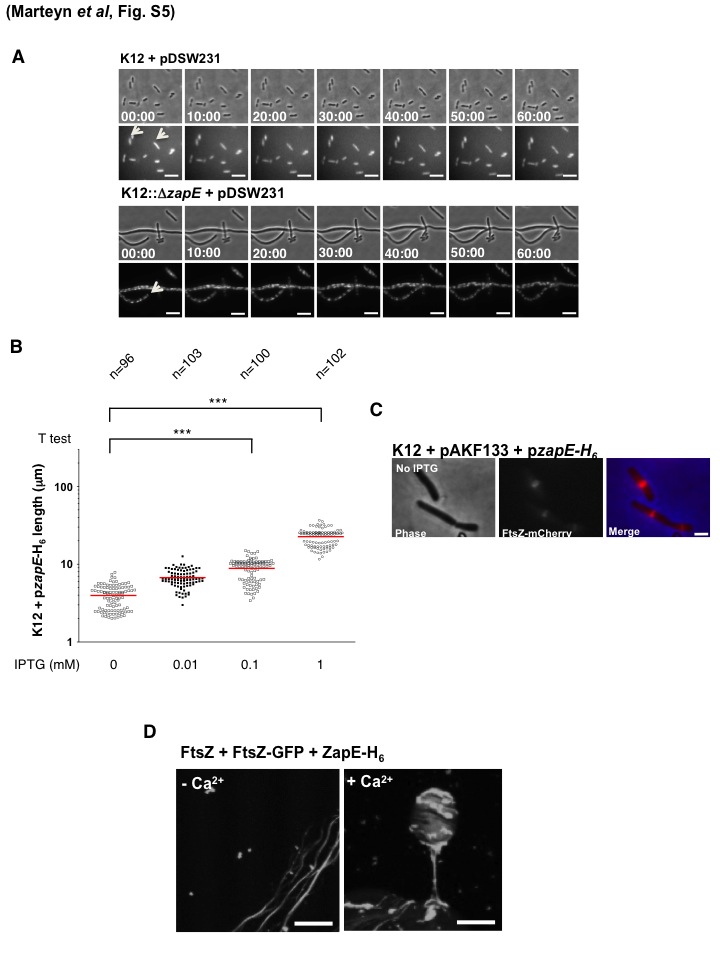
Phenotypic consequences of ZapE inactivation or FtsZ inactivation and overexpression and role of Ca2+ in purified FtsZ/FtsZ-GFP polymerization in vitro. (A) FtsZ-GFP localization in K-12 and K12::ΔzapE strains. Data represent the stability of the altered Z-rings in the K12::ΔzapE strain. FtsZ-GFP (pDSW231) time-dependent expression and localization in E. coli K-12 and E. coli K12::ΔzapE strains (see Movie S2 in the supplemental material) during a cell division process are shown. Time-lapse observation was performed on strains grown on an LB-agar pad at 30°C in the absence of IPTG, using a 200 M Axiovert epifluorescence microscope (Zeiss). (B) Length measurements of bacteria were performed on each of the strains under the conditions described for Fig. 5C using MicrobeTracker software (2, 4). Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of measured bacteria per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). (C) As described for the control represented in Fig. 5A, the effect of the ZapE basal level of expression (pzapE-H6) on Z-ring stability (FtsZ-mCherry, pAKF133) in the absence of IPTG was determined. Bacteria were grown in minimum media at 37°C until an OD600 of 0.5 was reached in the presence of arabinose (0.01%). (D) Ca2+ effect on ZapE modulation of FtsZ polymerization in vitro. Polymerization of FtsZ (10 µM) and FtsZ-GFP (5 µM) with 1 mM GTP was performed after addition of ZapE-H6 (10 µM) without ATP during 3 min, in the absence or presence of 10 mM CaCl2, as described in references 5 and 6. Bars are 15 µm. Download
{kind=link}
(A) Strains and plasmids used in this study. (B) Oligonucleotides used in this study. (C) E. coli K-12 ZapE (B3232, YhcM) homologue identification in prokaryotic and eukaryotic organisms using ClustalW.
Supplemental methods. Download
ACKNOWLEDGMENTS
We acknowledge access to the Protein Science Facility, Karolinska Institutet. We gratefully thank Jeff Errington for his initial interest in this project. We thank William Margolin for biochemistry advice. We thank David Weiss for supplying the various Fts protein fusions. We thank Arnaud Echard for scientific discussions and advice. We gratefully acknowledge the synchrotron beamline and excellent user support at the ESRF BioSAXS BM29 beamline. We thank Carmen R. Beuzón López and Spyridoula Charova for offering part of their beam time for the ZapE measurements.
B.S.M. and P.S.J. were supported by EU ERC Homeoepith grant no. 272398 and EU FP7 Tornado grant no. 222720.
Footnotes
Citation Marteyn BS, Karimova G, Fenton AK, Gazi AD, West N, Touqui L, Prevost M-C, Betton J-M, Poyraz O, Ladant D, Gerdes K, Sansonetti PJ, Tang CM. 2014. ZapE is a novel cell division protein interacting with FtsZ and modulating the Z-ring dynamics. mBio 5(2):e00022-14. doi:10.1128/mBio.00022-14.
REFERENCES
- 1. Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354:161–164. 10.1038/354161a0 [DOI] [PubMed] [Google Scholar]
- 2. de Boer PA. 2010. Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13:730–737. 10.1016/j.mib.2010.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lutkenhaus J, Pichoff S, Du S. 2012. Bacterial cytokinesis: from Z ring to divisome. Cytoskeleton (Hoboken) 69:778–790. 10.1002/cm.21054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galli E, Gerdes K. 2010. Spatial resolution of two bacterial cell division proteins: ZapA recruits ZapB to the inner face of the Z-ring. Mol. Microbiol. 76:1514–1526. 10.1111/j.1365-2958.2010.07183.x [DOI] [PubMed] [Google Scholar]
- 5. Hale CA, Shiomi D, Liu B, Bernhardt TG, Margolin W, Niki H, de Boer PA. 2011. Identification of Escherichia coli ZapC (YcbW) as a component of the division apparatus that binds and bundles FtsZ polymers. J. Bacteriol. 193:1393–1404. 10.1128/JB.01245-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Durand-Heredia J, Rivkin E, Fan G, Morales J, Janakiraman A. 2012. Identification of ZapD as a cell division factor that promotes the assembly of FtsZ in Escherichia coli. J. Bacteriol. 194:3189–3198. 10.1128/JB.00176-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marteyn B, Scorza FB, Sansonetti PJ, Tang C. 2011. Breathing life into pathogens: the influence of oxygen on bacterial virulence and host responses in the gastrointestinal tract. Cell. Microbiol. 13:171–176. 10.1111/j.1462-5822.2010.01549.x [DOI] [PubMed] [Google Scholar]
- 8. Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration of Escherichia coli in the mouse intestine. Infect. Immun. 75:4891–4899. 10.1128/IAI.00484-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. West NP, Sansonetti P, Mounier J, Exley RM, Parsot C, Guadagnini S, Prévost MC, Prochnicka-Chalufour A, Delepierre M, Tanguy M, Tang CM. 2005. Optimization of virulence functions through glucosylation of Shigella LPS. Science 307:1313–1317. 10.1126/science.1108472 [DOI] [PubMed] [Google Scholar]
- 10. Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prévost MC, Sansonetti P, Tang CM. 2010. Modulation of Shigella virulence in response to available oxygen in vivo. Nature 465:355–358. 10.1038/nature08970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anilkumar G, Srinivasan R, Anand SP, Ajitkumar P. 2001. Bacterial cell division protein FtsZ is a specific substrate for the AAA family protease FtsH. Microbiology 147(Pt 3):516–517. 10.1002/jobm.200610236 [DOI] [PubMed] [Google Scholar]
- 12. Srinivasan R, Ajitkumar P. 2007. Bacterial cell division protein FtsZ is stable against degradation by AAA family protease FtsH in Escherichia coli cells. J. Basic Microbiol. 47:251–259. 10.1002/jobm.200610236 [DOI] [PubMed] [Google Scholar]
- 13. Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. U. S. A. 95:5752–5756. 10.1073/pnas.95.10.5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szwedziak P, Wang Q, Freund SM, Löwe J. 2012. FtsA forms actin-like protofilaments. EMBO J. 31:2249–2260. 10.1038/emboj.2012.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stouf M, Meile JC, Cornet F. 2013. FtsK actively segregates sister chromosomes in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 110:11157–11162. 10.1073/pnas.1304080110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187:2233–2243. 10.1128/JB.187.7.2233-2243.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181:508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thanedar S, Margolin W. 2004. FtsZ exhibits rapid movement and oscillation waves in helix-like patterns in Escherichia coli. Curr. Biol. 14:1167–1173. 10.1016/j.cub.2004.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu XC, Margolin W. 1997. Ca2+-mediated GTP-dependent dynamic assembly of bacterial cell division protein FtsZ into asters and polymer networks in vitro. EMBO J. 16:5455–5463. 10.1093/emboj/16.17.5455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu W, Park KT, Holyoak T, Lutkenhaus J. 2011. Determination of the structure of the MinD-ATP complex reveals the orientation of MinD on the membrane and the relative location of the binding sites for MinE and MinC. Mol. Microbiol. 79:1515–1528. 10.1111/j.1365-2958.2010.07536.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong G, Yang Q, Wang Q, Kim YI, Wood TL, Osteryoung KW, van Oudenaarden A, Golden SS. 2010. Elevated ATPase activity of KaiC applies a circadian checkpoint on cell division in Synechococcus elongatus. Cell 140:529–539. 10.1016/j.cell.2009.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Errington J, Daniel RA, Scheffers DJ. 2003. Cytokinesis in bacteria. Microbiol. Mol. Biol. Rev. 67:52–65. 10.1128/MMBR.67.1.52-65.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheffers D, Driessen AJ. 2001. The polymerization mechanism of the bacterial cell division protein FtsZ. FEBS Lett. 506:6–10. 10.1016/S0014-5793(01)02855-1 [DOI] [PubMed] [Google Scholar]
- 24. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller VL, Beer KB, Loomis WP, Olson JA, Miller SI. 1992. An unusual pagC::TnphoA mutation leads to an invasion- and virulence-defective phenotype in Salmonellae. Infect. Immun. 60:3763–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 27. Martinez E, Bartolomé B, de la Cruz F. 1988. pACYC184-derived cloning vectors containing the multiple cloning site and lacZ alpha reporter gene of pUC8/9 and pUC18/19 plasmids. Gene 68:159–162 [DOI] [PubMed] [Google Scholar]
- 28. Valdivia RH, Falkow S. 1996. Bacterial genetics by flow cytometry: rapid isolation of Salmonella typhimurium acid-inducible promoters by differential fluorescence induction. Mol. Microbiol. 22:367–378. 10.1046/j.1365-2958.1996.00120.x [DOI] [PubMed] [Google Scholar]
- 29. Sansonetti PJ, d’Hauteville H, Formal SB, Toucas M. 1982. Plasmid-mediated invasiveness of “Shigella-like” Escherichia coli. Ann. Microbiol. (Paris) 133:351–355 [PubMed] [Google Scholar]
- 30. Yu XC, Margolin W. 1997. Ca2+-mediated GTP-dependent dynamic assembly of bacterial cell division protein FtsZ into asters and polymer networks in vitro. EMBO J. 16:5455–5463. 10.1093/emboj/16.17.5455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80:612–627. 10.1111/j.1365-2958.2011.07579.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live observation of E. coli K-12 FtsZ-GFP and ZapE-mCherry time-dependent expression and localization during a cell division process. Time-lapse observation was performed on an E. coli K12::ΔzapE/pDSW230/pzapE-mCherry strain grown on an LB-agar pad at 30°C, using a 200 M Axiovert epifluorescence microscope (Zeiss). Image acquisition was performed every 3 min, and the movie is shown at 5 frames per second (fps). This result is representative of five individual observations from three independent experiments. Download
FtsZ-GFP (pDSW231) time-dependent expression and localization in a K12::ΔzapE strain during a cell division process. Time-lapse observation was performed on strains grown on an LB-agar pad at 30°C in the absence of IPTG, using a 200 M Axiovert epifluorescence microscope (Zeiss). Image acquisition was performed every 3 min, and movies are shown at 5 fps. These data are representative of the results of at least three independent observations. Bars are 2 µm. Download
Three-dimensional (3D) view of FtsZ/FtsZ-GFP helical polymerization in the presence of ZapE-H6 (1:1) and the absence of ATP at t = 0 min (see Fig. 5B). Image reconstruction was performed using Imaris software. 3D Z stack determinations were performed using 40 input images with resolution of z = 0.2 μm. Download
(A) Histopathological analysis of rabbit ileal loops infected by M90T, M90T::ΔzapE, and M90T VP- (BS176) strains. Paraffin-embedded tissues were stained using hematoxylin-eosin. Bars are 50 µm. Data represent K-12 ZapE homologous sequence comparisons. Growth of strain M90TΔzapE is attenuated in vivo. (B) Growth defect of K12::ΔzapE and M90T::ΔzapE strains under low-oxygen conditions. K-12, M90T, K12::ΔzapE, and M90T::ΔzapE strains were grown in minimum media in the presence (+O2) or absence (−O2) of oxygen at 37°C. Scale bars are 10 µm. (C) Phenotypic characterization of M90T and M90T::ΔzapE grown in minimum media in the presence (+O2) or absence (−O2) of oxygen at 37°C. Scale bars are 10 µm. Length measurements of bacteria were performed on each strain using MicrobeTracker software (1, 2); conditions were as indicated on the left and in the Fig. 2A legend. Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of bacteria measured per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). Download
(A) Temperature-dependent phenotype of Shigella wild-type, ΔzapE mutant, and complemented strains. Bacteria were grown in rich media at the indicated temperature until an OD600 = 0.5 was reached. Scale bars are 10 µm. (B) Length measurements of bacteria were performed on each strain under the conditions described in the panel A legend and the Fig. 2B legend using MicrobeTracker software (2, 3). Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of measured bacteria per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). Download
(A) Phenotype of strain K12::ΔzapE::Km/pzapE in the presence of glucose or arabinose. Bacteria were grown in a rich liquid media under the indicated conditions in the presence of glucose (0.2%) or arabinose (0.2%) until an OD600 = 0.5 was reached. Scale bars are 5 µm. (B) Partial primary sequence alignment of ZapE proteins from different species. Alignment was made using the ClustalW software, and identifications of putative Walker A and Walker B consensus sequences are highlighted. Alignment is shown in ZapE homologues identified in E. coli K-12 MG1655 (B3232, YhcM), Shigella flexneri 5A M90T (S3487), Salmonella enterica serovar Typhi (STY3526), Yersinia pestis (YPO3564), Candida albicans (AFG11), and Homo sapiens (LACE1). Sequence ID, percent homology, and percent identity and the P-loop sequence are detailed in Table S1C in the supplemental material. ZapE SAXS structure models are shown. (C, D, and E) SAXS properties of the ZapE full-length protein. In panel C, the final processed SAXS curve after subtraction of the buffer, extrapolation to zero concentration, and merging and scaling of curves coming from various concentrations is shown. In the inset, the Guinier plot in the small-angle range is almost linear, which is indicative of ZapE particle monodispersity. In panel D, the Kratky representation of the final processed SAXS curve in panel A provides the information of a globular particle, as there is a maximum apparent in the low “s” area. Still, this maximum is not completely apparent. (E) The pair distribution function. The indirect Fourier transform of the final processed SAXS curve corresponding to Fig. 2D provides the first geometric characteristics of the ZapE particle in real space. The maximum diameter of the particle is estimated at 103 Å. Download
ZapE localization. (A) ZapE localization was performed in K-12 bacteria by electron microscopy using immunogold staining with a polyclonal α-ZapE antibody. The bar is 200 nm. Localization quantification is indicated, and error bars show the SD. *** indicates P < 0.001 (Student’s t test). (B) pSUC plasmid description. A schematic representation of the pSUC plasmid map in addition to its multiple-cloning-site sequence (HindIII and XbaI restriction sites are underlined) is shown. (C) Colocalization of FtsZ-GFP and ZapE-mCherry signals during Z-ring constriction and FtsZ-GFp localization in an E. coli ΔzapE mutant. FtsZ-GFP and ZapE-mCherry fluorescent signals (AU) from Movie S1 in the supplemental material and Fig. 3C were quantified in relation to the distance from the Z-ring center (as indicated on the top scheme). Measurements were performed on images acquired at the time of maximal constriction (Max. Constriction) and at 50 min, 20 min, 15 min, 10 min, and 5 min before. The x axis indicates the distances from the Z-ring center (−0.7 to 0.7 µm). n = 5 independent observations; error bars show the SD. Download
Phenotypic consequences of ZapE inactivation or FtsZ inactivation and overexpression and role of Ca2+ in purified FtsZ/FtsZ-GFP polymerization in vitro. (A) FtsZ-GFP localization in K-12 and K12::ΔzapE strains. Data represent the stability of the altered Z-rings in the K12::ΔzapE strain. FtsZ-GFP (pDSW231) time-dependent expression and localization in E. coli K-12 and E. coli K12::ΔzapE strains (see Movie S2 in the supplemental material) during a cell division process are shown. Time-lapse observation was performed on strains grown on an LB-agar pad at 30°C in the absence of IPTG, using a 200 M Axiovert epifluorescence microscope (Zeiss). (B) Length measurements of bacteria were performed on each of the strains under the conditions described for Fig. 5C using MicrobeTracker software (2, 4). Three independent bacterial cultures were imaged for each strain under each set of growth conditions. “n” indicates the total number of measured bacteria per condition. *** indicates the statistical significance of the results of comparisons of the highlighted sets of conditions; P < 0.001 (Student’s t test). (C) As described for the control represented in Fig. 5A, the effect of the ZapE basal level of expression (pzapE-H6) on Z-ring stability (FtsZ-mCherry, pAKF133) in the absence of IPTG was determined. Bacteria were grown in minimum media at 37°C until an OD600 of 0.5 was reached in the presence of arabinose (0.01%). (D) Ca2+ effect on ZapE modulation of FtsZ polymerization in vitro. Polymerization of FtsZ (10 µM) and FtsZ-GFP (5 µM) with 1 mM GTP was performed after addition of ZapE-H6 (10 µM) without ATP during 3 min, in the absence or presence of 10 mM CaCl2, as described in references 5 and 6. Bars are 15 µm. Download
(A) Strains and plasmids used in this study. (B) Oligonucleotides used in this study. (C) E. coli K-12 ZapE (B3232, YhcM) homologue identification in prokaryotic and eukaryotic organisms using ClustalW.
Supplemental methods. Download