

ABSTRACT
We have recently reported the isolation of a novel virus, provisionally designated C/swine/Oklahoma/1334/2011 (C/OK), with 50% overall homology to human influenza C viruses (ICV), from a pig in Oklahoma. Deep RNA sequencing of C/OK virus found a matrix 1 (M1) protein expression strategy that differed from that of ICV. The novelty of C/OK virus prompted us to investigate whether C/OK virus could exist in a nonswine species. Significantly, we found that C/OK virus was widespread in U.S. bovine herds, as demonstrated by reverse transcription (RT)-PCR and serological assays. Genome sequencing of three bovine viruses isolated from two herds in different states further confirmed these findings. To determine whether swine/bovine C/OK viruses can undergo reassortment with human ICV, and to clarify the taxonomic status of C/OK, in vitro reassortment and serological typing by agar gel immunodiffusion (AGID) were conducted. In vitro reassortment using two human ICV and two swine and bovine C/OK viruses demonstrated that human ICV and C/OK viruses were unable to reassort and produce viable progeny. Antigenically, no cross-recognition of detergent split virions was observed in AGID between human and nonhuman viruses by using polyclonal antibodies that were reactive to cognate antigens. Taken together, these results demonstrate that C/OK virus is genetically and antigenically distinct from ICV. The classification of the new virus in a separate genus of the Orthomyxoviridae family is proposed. The finding of C/OK virus in swine and bovine indicates that this new virus may spread and establish infection in other mammals, including humans.
IMPORTANCE
Influenza C viruses (ICV) are common human pathogens, infecting most people during childhood and adolescence, and typically cause mild respiratory symptoms. While ICV have been isolated from both pigs and dogs, humans are thought to be the natural viral reservoir. Previously, we characterized an ICV-like virus isolated from pigs exhibiting symptoms of influenza virus-like illness. Here, we show molecular and serological data demonstrating widespread circulation of similar viruses in bovines. Deep RNA sequencing, phylogenetic analysis, and in vitro reassortment experiments demonstrate that animal ICV-like viruses are genetically distinct from human ICV. Antigenically, we show that ICV-like viruses are not recognized by ICV antibodies. En masse, these results suggest that bovine influenza virus warrants classification as a new genus of influenza virus. The finding of this novel virus that can infect multiple mammalian species warrants further research into its role in human health.
INTRODUCTION
The Orthomyxoviridae family contains three genera of influenza viruses, influenza A, B, and C viruses, that are classified according to antigenic differences between their nucleoprotein (NP) and matrix 1 (M1) proteins (1–3). Such a classification is supported by low intergenic (20 to 30%) and high intragenic (>85%) homology of their M1 and NP proteins. Additionally, viral reassortment has been used to classify influenza viruses, as by definition, intragenic reassortment yields viable progeny (4).
Influenza C virus (ICV) is a ubiquitous pathogen of humans. Compared to influenza A and B viruses, it causes a mild respiratory illness or remains an asymptomatic infection. Most humans are seropositive to ICV by adolescence, and ICV is most commonly isolated from children less than 6 years of age (5, 6). Clinically, fever and respiratory symptoms are common and typically mild. Aside from humans, ICV has been isolated from pigs in China in 1981 and in the United States in 2011 (7, 8). Genetic analysis of the Chinese swine ICV found high similarities to contemporary human ICV, suggestive of interspecies transmission (9). In contrast, the ICV-like C/swine/Oklahoma/1334/2011 (C/OK) virus was only moderately related to previously characterized ICV, with approximately 50% overall homology. These results, in addition to a lack of cross-reactivity between antibodies against C/OK virus and human ICV in hemagglutination inhibition (HI) assays, suggested that C/OK virus conservatively represents a new but distant subtype of ICV. Previously, only influenza A viruses were thought to possess multiple subtypes (10). Cocirculation of multiple influenza virus subtypes allows for antigenic shift, the process of rapid evolution mediated by reassortment of the genomic RNA segments of two parent viruses.
Serological studies found that 19% and 9.9% of pigs in the United Kingdom and Japan, respectively, were seropositive to human ICV (11, 12). Similarly, 9.5% of pigs had measurable titers in the HI assay to C/OK virus (8). Together, these results suggest that pigs are frequently infected by ICV. Despite its ability to infect ferrets, a surrogate for human influenza virus studies, only 1.3% of human sera had a measurable titer to C/OK virus. Additionally, the relatively low geometric mean titer (GMT) in swine sera in the HI assay to C/OK virus (20.7) led us to speculate that neither swine nor humans are the natural reservoir of C/OK virus.
Using a reverse transcription (RT)-PCR primer and probe set targeting the PB1 gene of swine C/OK virus, we identified C/OK virus in samples from bovine respiratory disease diagnostic submissions. Detailed genetic, biological, and antigenic characterization suggests that C/OK viruses in cattle and swine are distinct from human ICV and likely represent a new genus in the Orthomyxoviridae family, with C/OK virus as the type species of influenza D virus. Additionally, widespread and high antibody titers to C/OK virus and relative ease of virus isolation in cattle indicate that bovines may represent the reservoir to this novel influenza virus. The finding of C/OK virus in both swine and bovine indicates that this new virus may spread and establish infection in other mammals, including humans.
RESULTS
Analysis of viral transcriptome by next-generation sequencing (NGS).
To extend the understanding of swine C/OK virus genome expression, we used Illumina-based deep RNA sequencing to study viral transcription in swine testicle (ST) cells, focusing on examining and comparing the transcription and splicing of swine C/OK virus with those reported previously for human ICV. Total cellular RNA was isolated from ST cells infected with swine C/OK at a multiplicity of infection (MOI) of 0.1 at 0, 18, and 36 h after infection. Corresponding cDNA libraries were prepared and loaded onto an Illumina Genome Analyzer IIx (GA IIx) sequencer for sequencing. The DNA sequence data sets were then assembled against the C/OK genome, which has seven single-stranded, negative-sense RNA segments. For each sample, we obtained on average over 9 million 100-nucleotide (nt) reads mapping to either C/OK virus or pig genomes. At 18 h postinfection, approximately 5% of the total reads mapped to the viral genomic segments, which was similar to that obtained at 36 h postinfection (data not shown). Read coverage for each of seven segments had significant overlap at both time points, demonstrating the active replication of the C/OK genome with a similar rate at both time points (Fig. 1). The pattern of read coverage clearly discriminated seven segments into three distinctive groups (Fig. 1). Group 1 included three viral polymerase genes (P3, PB1, and PB2 genes) that all had a large proportion of reads mapping to the 5′ and 3′ ends of segments, with a significant reduction in the number of reads mapping to the internal regions. The overrepresentation of sequencing reads in the 5′ and 3′ ends of the segments suggested the presence of internal deletions in copies of these polymerase genes. The subgenomic segments with the internal deletions appeared to range in size from 300 to 700 nucleotides, which have been previously found to be associated with defective interfering RNA in replicating influenza viruses, including human ICV (13–16). Group 2, represented by NP and hemagglutinin-esterase fusion (HEF) segments, had sequencing reads relatively evenly distributed across the complete segment compared to group 1. Interestingly, a reduction of read coverage was noted in the 5′-terminal half of the NP segment. The coverage reduction may indicate the presence of heterogeneous species of the NP genes with some sequences deleted within this region, which is similar to those observed for polymerase genes (PB1, PB2, and P3 genes). Group 3 (M and NS segments) displayed a large reduction in the number of reads mapping in certain regions of the segments, indicative of the presence of internal introns. Subsequent analysis of segment-derived sequencing reads identified potential splice junctions and introns present in both segments (Fig. 1; see Table S1 and S2 in the supplemental material).
FIG 1 .

Transcriptome maps of C/OK virus segments. Numbers of normalized sequence reads (100 nucleotides each read) were mapped to each nucleotide position across the respective segments of C/OK virus genome. The x axis represents the length of each segment, while the y axis represents the number of sequencing reads mapping to each genomic segment. Arrow signs indicate intron regions present in NS and M segments that were identified by next-generation sequencing (NGS) analysis.
Follow-up RT-PCR and Sanger sequencing experiments confirmed the splice junctions for both NS and M segments and their introns determined in NGS analysis (see Fig. S2 to S4 and Tables S1 and S2 in the supplemental material). Further analysis showed that both splicing events utilized a consensus dinucleotide motif GT/AG: GT present at the extreme 5′ (donor) with AG detected at the extreme 3′ (acceptor) ends of the intron. The splicing pattern for the C/OK NS segment was similar to that of human influenza C virus (Fig. 2A) (17). In contrast, M segment splicing encoding mRNA for the M1 protein was different from that of the human ICV M1 mRNA (Fig. 2B). Rather than introduction of a termination codon to the M1 open reading frame, splicing of the C/OK M segment added a second exon encoding a 4-residue peptide to the primary M1 transcript (M1 mRNA, nucleotides 29 to 756 joined to 1050 to 1062). Variability in the length of the second exon of M1 observed between the two viruses adds to the novelty of the C/OK virus. In addition to NS2 and M1 expressed through splicing, NS and M segments of both human ICV and C/OK viruses also encode NS1 and M2 proteins, respectively, through the corresponding unspliced mRNA transcripts. These viruses likely utilize a similar mechanism to produce these proteins, although further experimental validation is required.
FIG 2 .
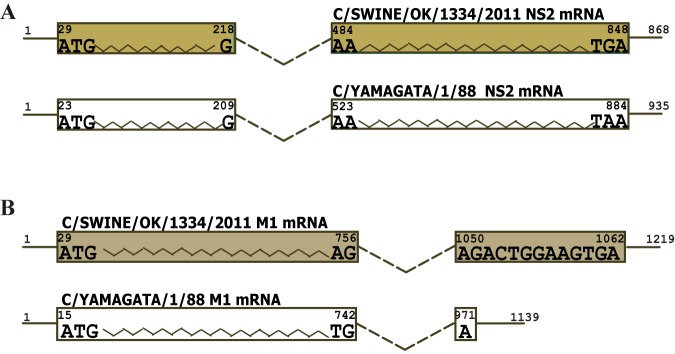
Splicing strategies of C/OK virus for the NS segment (A) and M segment (B). Panel A schematically illustrates a splicing strategy of the C/OK virus NS segment to produce NS2 protein in comparison to its counterpart in human influenza C viruses (ICV), while panel B describes a splicing strategy of the M segment to produce M1 protein in comparison to human ICV’s M1 protein synthesis.
FIG 3 .

Phylogenetic trees of the seven segments of bovine C/OK viruses. Maximum likelihood analysis in combination with 500 bootstrap replicates was used to derive trees based on the nucleotide sequences of the genomic segments. Bootstrap values are shown above and to the left of the major nodes.
FIG 4 .

Mean hemagglutination inhibition titers for eight bovine herds (11 to 27 animals per herd) to bovine C/660 and swine C/OK. Bovine sera from eight herds located in five different states were screened individually (11 to 27 animals per herd) in the HI assay using C/OK and C/660 as antigens. The x axis represents the sample size of each herd tested, while the y axis indicates the average HI titer and standard error from each herd.
Identification of swine C/OK virus in bovines.
The novelty of C/OK virus demonstrated above and biological distinctness from human ICV reported in our previous work (8) prompted us to investigate whether C/OK virus could exist in a nonswine species. Nasal swabs were submitted to Newport Laboratories for diagnostic testing from cattle with respiratory disease. A total of 45 samples were tested by relative real-time reverse transcription-PCR (rRT-PCR) for C/OK virus. Samples were collected in the late winter and spring of 2013 and originated from 6 different states. Eight samples (18%) were positive, all from Minnesota and Oklahoma. Virus isolation was attempted on PCR-positive samples using multiple cell lines, and viral replication was determined by rRT-PCR and hemagglutination (HA) assays. Virus was initially isolated from five different samples on human rectal tumor (HRT-18G) cells. The virus was noncytopathic but replicated to a high titer as determined by rRT-PCR (threshold cycle [CT] values of ~12 to 14) and hemagglutination assays (HA = 1,280 to 5,120 hemagglutinating units [HAU]/ml). Following the initial isolation, viruses were passaged on ST cells and displayed typical influenza virus cytopathic effects. Four of the isolated viruses all originated from the same herd in Minnesota. Two were randomly chosen for further study, C/bovine/Minnesota/628/2013 (C/628) and C/bovine/Minnesota/729/2013 (C/729), along with the remaining viral isolate from a bovine in Oklahoma, C/bovine/Oklahoma/660/2013 (C/660).
Viral genome sequencing.
The genome sequences of C/628, C/729, and C/660 were determined using an Ion Torrent sequencer. Primers designed to amplify the whole genome of IAV were modified with the 5′ and 3′ noncoding regions of C/OK virus (8, 18). Using viral genomic RNA as a template, the seven-segmented genome was amplified from the three viruses using a high-fidelity polymerase, and the resulting amplicons were sequenced. Sequences were assembled using C/OK virus as a template. As amplicons were produced using primers homologous to the 5′ and 3′ noncoding regions of C/OK virus, the sequence encompassed the segment between these regions. The full-length segments were sequenced with the exception of 5′ and 3′ noncoding regions. All segments had greater than 96% identity to C/OK virus. Segments with the highest homology to C/OK virus were PB1 (98.9 to 99.1% identity) and NS (98.8 to 99.2% identity), while the most divergent segments were HEF (96.7 to 99.0% identity) and P42 (96.9 to 99.2% identity).
RNA sequencing was performed to determine the conserved 5′ and 3′ noncoding region sequences for C/628, C/729, and C/660. The terminal conserved noncoding regions for each segment were identical to those of C/OK (data not shown).
Phylogenetic analysis.
To further understand viral evolution, we performed phylogenetic analysis of bovine C/OK virus and representative influenza A, B, and C viruses as well as other members of the Orthomyxoviridae family, Thogoto virus and infectious salmon anemia virus (Fig. 3). All seven segments were analyzed individually using maximum likelihood analysis. All segments for C/628, C/660, and C/729 were closely related to C/OK. C/OK, C/628, C/660, and C/729 clustered most closely to human ICV, suggesting that these viruses diverged from human ICV after the divergence from ancestral IAV and IBV. Despite reports of frequent reassortment between ICV (19), all segments of the bovine ICV were highly homologous to C/OK, and there was no evidence of reassortment with human ICV. These findings led us to hypothesize that the C/OK clade of viruses is unable to reassort with human ICV.
In vitro reassortment.
To test our hypothesis and determine whether C/OK and the bovine ICV viruses are able to reassort with human ICV, an in vitro reassortment experiment was performed. Elucidation of reassortment between C/OK and human ICV enabled us determine whether this novel virus utilizes reassortment as a potential mechanism for viral evolution and production of new antigenic variants that may pose a threat to human and animal health. Another purpose of this experiment was to gain more evidence toward understanding the taxonomic status of C/OK virus, because, by definition, all members of an influenza virus genus can reassort with each other and give viable progeny (4).
Two representative human ICV (C/Taylor/1947 [C/Tay] and C/Johannesburg/66 [C/JHB]), the swine C/OK, and the bovine C/660 were chosen. Selection of bovine C/660 over other bovine C/OK isolates is due to considerations that this virus encodes a slightly divergent HEF protein. ST cells were coinfected with a multiplicity of infection of 0.1 for each virus with all pairwise combinations of virus, with all four viruses, or, as a control, cells were also inoculated singly with each virus (Table 1). Ten plaques from each well containing multiple viruses were analyzed by full genome sequencing to allow determination of segment parentage. The donor virus for each segment was determined for each plaque-purified virus using criteria of at least 99.5% identity. Due to the high similarity between the two human and also the two nonhuman viruses, the parent donor virus for every segment could not be determined.
TABLE 1 .
Genotypes identified in plaque-purified viruses isolated following infection of cells with influenza C viruses
| Virusesa | Parentage (no. of plaques)b |
||||||
|---|---|---|---|---|---|---|---|
| PB2 | PB1 | P3 | HEF | NP | P42 | NS | |
| C/JHB, C/Tay, C/OK, C/660 |
C/OK (8); C/660 (2) |
C/660 (9); NI (1) |
C/OK (6); C/660 (4) |
C/OK (7); C/660 (1); NI (2) | C/OK (4); C/660 (4); NI (2) | C/OK (1); C/660 (4); NI (5) | C/OK (7); C/660 (3) |
| C/JHB, C/Tay | NI (10) | NI (10) | C/JHB (7); C/Tay (3) |
C/Tay (4); NI (6) | NI (10) | C/Tay (10) | NI (10) |
| C/JHB, C/OK | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) |
| C/JHB, C/660 | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) |
| C/Tay, C/OK | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) | C/OK (10) |
| C/Tay, C/660 | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) | C/660 (10) |
| C/OK, C/660 | C/OK (10) | C/OK (3); C/660 (7) |
C/OK (8); C/660 (2) |
C/OK (10) | C/OK (6); C/660 (4) |
C/OK (4); C/660 (5); NI (1) | C/OK (6); C/660 (1); NI (3) |
Viruses used for coinfection.
Parentage of viral genome segments present in virus plaques from coinfected cells. Ten plaque-purified viruses were analyzed from each coinfection experiment. The number of plaques from each donor is indicated in parentheses. Some viral segment donors could not be identified (NI).
Reassortant viruses were identified following coinfection with the two human ICV, C/JHB and C/Tay, despite our inability to assign a parent virus for five of the 7 segments due to high sequence identity. Only the P3 and P42 segments were divergent enough to conclusively determine lineage. All P42 segments were derived from C/Tay, while only 3/10 viruses had P3 segments derived from C/Tay. Reassortant viruses were also identified following coinfection of cells with the two nonhuman ICV, C/OK and C/660, representing at least 7 distinct genotypes (see Fig. S1A in the supplemental material). In contrast, coinfection of cells with pairwise combinations of human and nonhuman ICV failed to yield reassortant viruses. In all four instances, all segments from all viruses were derived from the nonhuman ICV (C/OK or C/660). Consistent with these results, coinfection of cells with all four viruses gave reassortant viruses with segments derived from C/OK and C/660 (see Fig. S1B).
A second reassortment experiment was performed with C/JHB and C/OK using increased MOIs and an increased ratio of C/JHB to C/OK. Cells were coinfected with both C/JHB and C/OK with an MOI of 1.0:0.1, 1.0:1.0, or 10.0:1.0; as in the first reassortment experiment, all segments from all plaques isolated from cells coinfected with C/JHB and C/OK were derived from C/OK. Eighteen, 16, and 18 plaques, respectively, were analyzed by genome sequencing. Similar to the previous reassortment experiment, all segments were derived from C/OK.
In vitro growth kinetics were determined on ST cells for C/OK and C/JHB for the MOIs used in the second reassortment experiment (see Fig. S5 in the supplemental material). Cell inoculation was performed as in the reassortment experiment. Both C/JHB and C/OK had similar growth curves, with C/JHB consistently replicating at higher titers, approximately 1.0 log10 50% tissue culture infectious dose (TCID50)/ml, than C/OK at 24, 60, and 72 h postinfection with the MOIs used in the second reassortment experiment. Despite these results, all segments from the second reassortment experiment were derived from C/OK, and no reassortant viruses were identified.
The finding of reassortant viruses between the human viruses C/JHB and C/Tay as well as between the animal viruses C/OK and C/660 and the lack of reassortment between human and animal ICV suggests that human and animal ICV are unable to reassort and generate viable progeny. The failure to identify viable reassortant viruses provides the first functional evidence that this new group of viruses is genetically distinct from human ICV and represents a new genus of influenza virus in the Orthomyxoviridae family.
Antibody cross-reactivity.
The inability of swine C/OK and bovine C/660 to reassort in vitro with human ICV plus the observed novelty of C/OK virus prompted us to employ serological typing by agar gel immunodiffusion (AGID) to probe their antigenic relatedness. Antigenic differences between NP and M1 proteins detected in AGID or other similar assays allow division of influenza viruses into three genera (3, 20). AGID has been widely and routinely used to type influenza viruses, especially prior to the advent of RT-PCR and sequencing technologies. For the AGID experiment, similar amounts of split viral antigens (12 log2 HAU) were used with normal uninfected cell-derived mock antigens as negative controls. Antisera generated against representative IAV, IBV, and ICV recognized intragenus, not intergenus, virus antigens in the AGID (Table 2). No cross-reactivity between genera and no recognition of mock antigens were observed, indicating the specificity of the AGID. Significantly, polyclonal antibody against C/Tay virus did not recognize detergent-treated swine C/OK and bovine C/660 virus antigens but was reactive to C/JHB virus antigen, in addition to its own antigen. Conversely, both swine C/OK and bovine influenza C virus antigens, not human influenza C virus antigens (C/JHB and C/Taylor), were recognized by C/OK virus-specific polyclonal antibody. Considering that NP and M1 are major structural proteins of influenza virus, we concluded that the major differences of NP and M1 are responsible for the failure in cross-recognition of viral antigens between these two groups of viruses in the AGID. This result is similar to what we observed for reference influenza A, B, and C viruses in the AGID. As such, based on this finding together with results from in vitro reassortment and NGS analysis, we propose that C/OK virus may be considered a representative strain of a new genus in the Orthomyxoviridae family.
TABLE 2 .
Results of AGID assay
| Antigen (virus or mock control) |
Presence or absence of visible white precipitation linea
|
|||
|---|---|---|---|---|
| A/NWS/34(H1)- A/Equine/Prague/1/56(N7) |
B/Hong Kong/8/73(Matrix) | C/Taylor/1233/1947 | C/swine/OK/1334/2011 | |
| A/WSN/1933 | + | − | − | − |
| B/Brisbane/60/2008 | − | + | − | − |
| C/Taylor/1233/1947 | − | − | + | − |
| C/Johannesburg/1/1966 | − | − | + | − |
| C/swine/OK/1334/2011 | − | − | − | + |
| C/bovine/OK/660/2013 | − | − | − | + |
| MDCK mockb | − | − | − | − |
| HRT-18G mock | − | − | − | − |
| PBS | − | − | − | − |
+, presence of a visible white precipitation line between antigen and antiserum wells; −, absence of a visible white precipitation line between antigen and antiserum wells.
The same protocol to produce viral antigens was employed to prepare supernatants collected from uninfected cells.
Seroprevalence of C/OK virus in bovines.
Given the relatively high percentage of bovine respiratory disease samples positive for C/OK-like virus by rRT-PCR, HI assays were performed on bovine sera to determine if antibodies to this virus were similarly prevalent. Our previous study of seroprevalence of C/OK virus in swine found 9.5% of pigs were positive, with a GMT of 20.7 (8). Bovine sera from eight herds located in five different states were screened individually (11 to 27 animals per herd) in the HI assay using C/OK and C/660 as antigens (Fig. 4). With the exception of one herd, all herds had geometric mean titers greater than 40 to both viruses, suggesting that C/OK virus infection is widespread in bovines and that antibodies generated against C/OK and C/660 are cross-reactive in the HI assay. The widespread presence of C/OK virus in bovines suggests that they likely represent a reservoir of this new group of viruses.
DISCUSSION
Until recently, ICV was thought to consist of a single subtype and possess the lowest rate of evolution among the various genera of influenza viruses (10). ICV, unlike IAV, is thought to exist in a human reservoir and has previously only rarely been shown to infect alternative species, such as pigs and dogs (7, 9, 21). Despite the presence of multiple cocirculating lineages, ICV are relatively genetically homogenous and thought to be near evolutionary equilibrium in humans (22). A recently isolated influenza C-like virus raised many questions about whether our understanding of ICV was correct. This virus, C/OK, was isolated from a pig in Oklahoma displaying influenza virus-like illness (8). Genetic analysis found only moderate (~53%) similarity to human influenza C viruses. Additionally, serological analysis found that antibodies against C/OK failed to recognize IAV, IBV, or human ICV. This virus also displayed phenotypes of broad cell tropism and had permissive growth at 37°C, which further distinguish this novel virus from human ICV. C/OK was provisionally identified as a subtype of ICV primarily due to its overall genetic organization (7 RNA segments) and both coding and noncoding region similarities to ICV. Despite these, the numerous incongruities observed with C/OK warranted further investigation.
Previous serological results using both human and swine sera found that approximately 1 and 10%, respectively, were positive for antibodies to C/OK, with low titers (HI range of 10 to 40). Infrequent isolation of C/OK virus from pigs plus a lower percentage of RT-PCR samples positive to C/OK virus (<0.1%) during the routine testing of nasal swabs from pigs challenged a theory that that swine is the primary host of C/OK virus. Based on these results, we suspected an alternate reservoir for this virus. Screening of a bovine serum panel representing animals from multiple states and production conditions (cow-calf and dairy) identified widespread, high antibody titers to C/OK viruses, suggesting that bovines may represent the reservoir to this virus. Serologically, the magnitude and percent positive for HI titers were similar to values reported for wild waterfowl, the established reservoir of IAV (23). Previous studies have shown that bovines are susceptible to IAV infection, and multiple subtypes of IAV have been isolated from cattle (24). Correlations have also been shown between IAV antibodies and respiratory disease and reduced milk yield (25, 26). Antibodies to IAV, IBV, and ICV have all been reported in cattle (27); however, the percentage of infected animals and titers were considerably lower than reported here for C/OK virus.
ICV differs from IAV and IBV in that it codes for a hemagglutinin esterase fusion protein that catalyzes receptor binding, cleavage, and membrane fusion. As opposed to IAV and IBV, which use α-2,3- and α-2,6-linked sialic acids on cell membrane proteins as receptors, ICV binds to 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2) as a receptor (28, 29). Another bovine pathogen, bovine respiratory coronavirus (BCV), encodes an evolutionarily related hemagglutinin esterase that also utilizes Neu5,9Ac2 as a receptor (30, 31). BCV can infect both the upper and lower respiratory tract and cause respiratory disease, implying that the receptors required for ICV are present in the bovine respiratory tract (31). Pathogenesis studies are required to evaluate whether C/OK virus utilizes Neu5,9Ac2 as a functional receptor and can replicate throughout the respiratory tract and establish whether C/OK virus causes disease in cattle.
The genome sequences of three bovine viruses were very similar to those of the recently described swine C/OK virus (8). Phylogenetic analysis showed that this clade of viruses was most closely related to human ICV; however, the distance between animal C/OK viruses and human ICV was similar to the differences between IAV and IBV for many segments. Additionally, there was no evidence of reassortment between human ICV and nonhuman C/OK virus. While our sample set was limited to three bovine viruses, we suspected that C/OK virus might be unable to reassort with human ICV. The abilities to reassort and yield viable progeny are necessary criteria for inclusion of a virus within a genus of influenza virus (4). An in vitro experiment was designed to determine if C/OK virus could generate viable reassortants with human ICV. Not unexpectedly, no reassortment between swine/bovine C/OK viruses and human ICV was observed despite the methodologies’ success in detecting reassortment separately between human and nonhuman ICV. These results strongly suggest that C/OK viruses are unable to reassort with human ICV and by definition that C/OK viruses represent a new genus of influenza virus. Previous work found that the 5′ and 3′ noncoding regions of swine C/OK virus were similar to those of human ICV but did contain one difference (position 5 from the 3′ terminus) and a polymorphism at position 1 from the 3′ terminus (8). The conserved 5′ and 3′ noncoding regions for the three bovine viruses presented in this work were identical to those of C/OK. The noncoding regions are highly conserved within each influenza virus genus and form panhandle structures due to inverted complementarity, which is critical for genome replication and packaging (32–35). The inability of C/OK viruses to reassort with human ICV may be due to these minor differences in noncoding regions.
Influenza viruses have long been classified into three genera on the basis of antigenic differences among the conserved NP and M proteins (4, 20, 36). Despite molecular tools such as RT-PCR and sequencing, the serological assay is still an important and irreplaceable method to type influenza viruses, especially for newly identified influenza viruses, such as C/OK virus in this case. Homologies of NP and M1 between IAV and IBV are approximately 30 to 40%, which are slightly higher than those observed between IAV and ICV and between IBV and ICV (10 to 20%). The approximate 40% similarity between C/OK and human ICV M1 and NP proteins further confounds the taxonomic position of C/OK virus. Results of our AGID experiments suggested that the new group of viruses (swine C/OK and bovine C/660) is antigenically distinct from human ICV, which is in good agreement with results obtained from the in vitro reassortment experiment.
Influenza viruses exploit various splicing strategies to express their viral proteins to maximize the genome coding capacity. To date, splicing has been identified only in the two smallest segments, M and NS. It is generally believed that all influenza viruses possess a similar mechanism to generate spliced mRNA from the NS segment to code for the NS2 protein (17, 37). Different mechanisms for M segment splicing toward the production of M1 or M2 protein are reported in IAV and ICV, while the M segment of IBV is not spliced. IAV utilizes an alternative splicing mechanism to synthesize its M2 protein, while translation of M1 protein is made from an unspliced colinear mRNA (38). In contrast, the M1 protein of ICV is produced from a spliced mRNA, while its M2 protein is derived from a protein (p42) generated from an unspliced colinear mRNA followed by proteolytic cleavage by a signal peptidase at an internal signal sequence (2, 39, 40). Using deep RNA sequencing coupled with conventional RT-PCR and Sanger sequencing for confirmation, alternatively spliced mRNA for both NS and M segments in C/OK-infected cells was identified. C/OK virus is similar to other influenza viruses in NS segment splicing to generate the NS2 protein. In contrast to the ICV M segment that produces the M1 protein through splicing that solely introduces a termination codon (consisting of nucleotides 752, 753, and 982) into a spliced mRNA, splicing of the C/OK M segment adds an additional 4-amino-acid peptide into the preceding exon. Variability in the length of the second exon of M1 adds to the novelty of this newly identified virus in swine and cattle.
It is unknown if C/OK virus has an impact on human health. The ability to infect and transmit in ferrets, a model for human influenza virus pathogenesis studies, suggests this pathogen has the potential to cause disease in humans. Limited serology on a subset of human serum samples showing a 1.3% positive rate also supports this hypothesis. The prevalence of C/OK virus in cattle and presumptive spillover to swine, both of which live in close proximity to humans, further highlights its potential threats to human health, which merit further studies.
In summary, several lines of evidence reported here present a compelling argument that C/OK virus is sufficiently distinct from human ICV to warrant classification as a new genus of influenza virus. Phylogenetic analysis found that the divergence between swine/bovine C/OK viruses and human ICV is similar to that between IAV and IBV. Serologically, C/OK virus is antigenically unrelated to human ICV. C/OK virus also encodes a novel mechanism for generating the M1 protein and, importantly, is unable to reassort with human ICV and generate viable progeny. In ecology, this new group of viruses differs from human ICV in that it resides in bovine. Furthermore, a serological survey suggested that C/OK virus frequently replicates and establishes its infection in bovines, with both percent positive and geometric mean titers considerably higher than those observed for humans and pigs, suggestive that bovines represent the reservoir of C/OK-like viruses. In conclusion, we propose that C/OK virus warrants creation of a new influenza virus genus, influenza D virus, with D/OK (formerly C/OK) as the type species.
MATERIALS AND METHODS
Virus isolation.
Nasal swabs or lung tissues were collected from cattle exhibiting respiratory disease and submitted to Newport Laboratories for diagnostic investigations during the late winter and spring of 2013. Samples originated from Oklahoma, Minnesota, Iowa, Indiana, Texas, Nebraska, and Tennessee and originated from animals of different ages. Virus isolation was performed on both ST and HRT-18G cells. Virus growth was determined by cytopathic effects, in addition to rRT-PCR and hemagglutination assays, using turkey red blood cells. rRT-PCR was performed as previously described (8).
Genome sequencing and analysis.
Viral RNA was isolated using a MagMAX viral RNA isolation kit. Full-genome amplification was performed as previously described except that the primers were modified to match the noncoding regions of C/OK: 3′ end, 5′ ACGCGTGATCGCATAAGCAG 3,′ and 5′ end, 5′ ACGCGTGATCAGCAGTAGCAAG 3′ (18). Amplicons were used for library preparation using the NEBNext Fast DNA library prep set for Ion Torrent. Libraries were sequenced using an Ion Torrent personal genome machine with the manufacturer’s reagents and protocols. Contigs were assembled using C/OK (accession numbers NC_922305 to NC_922311) and C/Ann Arbor/1/50 (accession numbers NC_006306 to NC_006312) as the templates and utilizing the SeqMan NGen module from DNAStar. Phylogenetic analysis was performed using Mega 5.1 software using maximum likelihood analysis with 500 bootstrap replicates to verify tree topology. The terminal segment sequences of C/660, C/628, and C/729 were determined using an RNA sequencing kit (Ion Total RNA-Seq kit v2) by following the manufacturer’s instructions with viral RNA as described above.
Next-generation sequencing of the viral transcriptome.
Swine testicle cells were infected with swine C/OK at an MOI of 0.1. Total cellular RNA was isolated from mock-infected cells (0 h) and infected cells at 18 h and 36 h postinfection, respectively, with TRIZOL followed by RNA purification with ethanol. RNA quality was tested using a Bioanalyzer 2100 (Agilent, Palo Alto, CA) and then processed for cDNA library construction by using a cDNA library prep kit (catalog number FC-122-1001; Illumina Inc.) according to the manufacturer’s instruction. cDNA library construction and all other procedures were conducted in Genomics Core Research Facility (GCRF) at the University of Nebraska-Lincoln (UNL). Briefly, mRNAs were purified from the total RNA using oligo(dT) magnetic beads followed by fragmentation. The resultant mRNAs were reverse transcribed to cDNAs that were subjected to an end repair process followed by ligation to the adapters. After separation in agarose gel through electrophoresis, cDNA fragments with a size of about 200 bp were excised, extracted, and amplified by PCR using two primers that match the ends of adaptors. PCR-enriched samples were then sequenced by an Illumina Genome Analyzer IIx (GA IIx) sequencer in the GCRF at UNL.
An infection experiment consisting of three time points (0, 18 h, and 36 h) was duplicated, and derived RNA samples were sequenced in deep RNA sequencing analysis. Three samples representative of three time points from each experiment were combined and loaded into one lane for sequencing. Each of the 6 samples submitted had approximately 9 million 100-nucleotide single-end reads, and the sequencing results passed quality control.
Bioinformatic analysis of the NGS data set.
Reads from each duplicate were merged and mapped to C/OK reference genomes using the GSNAP program (41). The resulting SAM files were converted to BAM files, and read coverage at each position of the C/OK genome segments was calculated using SAMtools (42). Sequencing reads were normalized according to total number of reads to viral and pig genomes obtained at each time point. Integrative Genomics Viewer (IGV) was used for visualization and mapping quality assurance (43). Reads mapped to C/OK genome segments with splicing were further analyzed with bash scripts. The accession numbers for the SRA database of NCBI are as follows: SRX471882 for the two repeats of 0 h, SRX471884 for 18 h, and SRX471846 for 36 h following infection.
Cell culture and in vitro reassortment.
ST and human rectal tumor (HRT-18G) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 5% fetal bovine serum. Influenza C/Taylor/1233/1947 (C/Tay) virus was provided by BEI Resources, while C/Johannesburg/1/66 (C/JHB) virus was a gift from P. Palese (Mt. Sinai Medical School, New York). For routine cultivation, cell medium was replaced by DMEM and virus was inoculated with a multiplicity of infection (MOI) of 0.001. Nonhuman ICV were cultivated at 33°C or 37°C, while human ICV were cultivated at 33°C. For in vitro reassortment, all viruses were propagated at 33°C in ST cells and titrated to determine 50% tissue culture infectious dose per milliliter (TCID50/ml). A single well of a 6-well plate with a monolayer of ST cells was coinfected with viruses as shown in Table 1 with an MOI of 0.1. Viruses were adsorbed for 1 h, and then the monolayer was washed with phosphate-buffered saline (PBS) and refed with DMEM. After 3 days, cell culture supernatants were serially diluted and plated onto a monolayer of ST cells with an agar overlay, and after an additional 1 to 3 days, individual plaques were picked and expanded on ST cells. Ten plaques were expanded for each reassortment experiment, and a single plaque was chosen from cultures of parental viruses.
A second reassortment experiment was performed under conditions designed to optimize reassortment between C/JHB and C/OK. Cells were coinfected with both C/JHB and C/OK with either an MOI of 1.0:0.1, 1.0:1.0, or 10.0:1.0, as described above. After 3 days, cell culture supernatants were serially diluted and plated onto a monolayer of ST cells with an agar overlay, and after an additional 1 to 3 days, individual plaques were picked and expanded on ST cells. Twenty plaques were expanded for each reassortment experiment.
In vitro growth kinetics were determined on ST cells for C/JHB and C/OK with the MOIs used in the second reassortment experiment. Cell inoculation was performed as described above using the same viral stocks used in the reassortment experiments. Samples were collected at 24, 60, and 72 h postinfection and analyzed by titration on ST cells.
Serology.
Bovine sera were treated with receptor-destroying enzyme overnight at 37°C and then heat inactivated at 56°C for 30 min. Hemagglutination inhibition (HI) assays were run with 0.5% packed turkey red blood cells as described in the WHO’s Manual on Animal Influenza Diagnosis and Surveillance (36). Bovine sera were a gift from the SD State University Veterinary Diagnostic Laboratory and represented samples from five states (South Dakota, Vermont, Pennsylvania, Idaho, and California) submitted for unrelated diagnostic testing.
All influenza A, B, and C reference virus antigens and antisera were obtained from Biodefense and Emerging Infections Research Resources Repository (BEI Resources). AGID antigens for swine C/OK virus, bovine influenza C-like virus, human influenza virus C/JHB, and human influenza virus C/Taylor were prepared according to the protocol of WHO and the World Organization for Animal Health (OIE). Supernatants from uninfected cell cultures (MDCK and HRT-18G) were prepared as mock controls using the same procedures as those used for production of viral antigens. Rabbit polyclonal antibody against swine C/OK virus was prepared using purified C/OK virus antigens. AGID plates were prepared according to the standard procedure, and assays were performed in triplicate according to WHO/OIE protocol (36). Similar concentrations of viral antigens for all tested strains (approximately 12 log2 HA units per 0.025 ml) were used in parallel with mock controls for antigen recognition by antisera in AGID with two different dilutions (neat and 1:2).
Nucleotide sequence accession numbers.
Genome sequences were entered into GenBank under accession numbers KF425652 to KF425658 (C/628), KF425659 to KF425665 (C/660), and KF425666 to KF425672 (C/729).
SUPPLEMENTAL MATERIAL
Supplemental Materials and Methods. Download
Genotypes in plaque-purified viruses following coinfection of swine testicle (ST) cells with C/OK and C/660 (A) and with C/OK, C/660, C/JHB, and C/Tay (B). Different-colored squares denote the identities of viral segments. Download
RT-PCR amplification of swine C/OK M, NS, and NP segments from infected ST cells with C/OK virus at an MOI of 0.01 at 18 h postinfection. Download
Full-length of C/OK virus M1 mRNA (5′-to-3′ direction) amplified by RT-PCR and confirmed by sequencing. The blue letters indicate sequence of the 5′ noncoding region and the 3′ noncoding region of M1 mRNA; the black letters with a gray highlight represent the sequence of primary M1 mRNA transcript; the black letters highlighted in yellow indicate the additional sequence introduced to primary M1 mRNA transcript following splicing. Download
Full-length of C/OK NS2 mRNA (5′-to-3′ direction) amplified by RT-PCR and confirmed by sequencing. The blue letters indicate the sequence of the 5′ noncoding region and 3′ noncoding region of NS2 mRNA; the black letters with a gray highlight represent the sequence of the first exon of NS2 mRNA transcript; the black letters highlighted in yellow indicated the sequence introduced to the first exon of NS2 following splicing. Download
Growth of influenza C/swine/Oklahoma/1334/2011 and influenza C/Johannesburg/1/66 virses in ST cell cultures. Cells were inoculated with a multiplicity of infection of 0.1 and 1.0 for C/OK and 1.0 and 10.0 for C/JHB and incubated at 33°C. Virus was titrated at the indicated time points on ST cells. Titers of viruses used for inoculation were 6.9 TCID50/ml for C/OK virus and 7.9 TCID50/ml for C/JHB virus. Download
Location of the internal intron within the C/OK M segment identified by NGS at 18 h (A) and 36 h (B) postinfection of ST cells.
Location of the internal intron within the C/OK NS segment identified by NGS at 18 h (A) and 36 h (B) postinfection of ST cells.
ACKNOWLEDGMENTS
We thank Peter Palese (Mt. Sinai Medical School, New York) for providing the C/Johannesburg/1/66 virus. Special thanks to the SDSU Veterinary Diagnostic lab for providing bovine serum samples and to NIAID biodefense and Emerging Infections Research Resources Repository (BEI Resources) for providing a panel of viral antigens and antisera of influenza A, B, and C viruses as well as the C/Taylor/1947 virus. We thank the Molecular Diagnostic group at Newport Laboratories (Brent Wassman, Karen Schwartz, and Josh Elston) for assistance with PCR and the Genetic Analysis Group (Elyse Cooper and Danielle McKeown) for assistance with sequencing.
Work done in the Feng Li lab was supported in part by SDSU AES 3AH-203 and SD 2010 Research Center (Biological Control and Analysis of Applied Photonics [BCAAP]) Fund SJ163.
Footnotes
Citation Hause BM, Collin EA, Liu R, Huang B, Sheng Z, Lu W, Wang D, Nelson EA, Li F. 2014. Characterization of a novel influenza virus in cattle and swine: proposal for a new genus in the Orthomyxoviridae family. mBio 5(2):e00031-14. doi:10.1128/mBio.00031-14.
REFERENCES
- 1. Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1690 In Knipe D, Howley P. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Yamashita M, Krystal M, Palese P. 1988. Evidence that the matrix protein of influenza C virus is coded for by a spliced mRNA. J. Virol. 62:3348–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sugawara K, Nishimura H, Hongo S, Kitame F, Nakamura K. 1991. Antigenic characterization of the nucleoprotein and matrix protein of influenza C virus with monoclonal antibodies. J. Gen. Virol. 72:103–109. 10.1099/0022-1317-72-1-103 [DOI] [PubMed] [Google Scholar]
- 4. McCauley JW, Hongo S, Kaverin NV, Kochs G, Lamb RA, Matrosovich MN, Perez DR, Palese P, Presti RM, Rimstad E. 2012. Orthomyxoviridae, p 749–761 In King AMQ, Adams MJ, Carstens EB, Lefkowitz E. (ed), Virus taxonomy. Classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier, New York, NY. [Google Scholar]
- 5. Matsuzaki Y, Katsushima N, Nagai Y, Shoji M, Itagaki T, Sakamoto M, Kitaoka S, Mizuta K, Nishimura H. 2006. Clinical features of influenza C virus infection in children. J. Infect. Dis. 193:1229–1235. 10.1086/502973 [DOI] [PubMed] [Google Scholar]
- 6. Gouarin S, Vabret A, Dina J, Petitjean J, Brouard J, Cuvillon-Nimal D, Freymuth F. 2008. Study of influenza C virus infection in France. J. Med. Virol. 80:1441–1446. 10.1002/jmv.21218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuanji G, Fengen J, Ping W, Min W, Jiming Z. 1983. Isolation of influenza C virus from pigs and experimental infection of pigs with influenza C virus. J. Gen. Virol. 64:177–182. 10.1099/0022-1317-64-1-177 [DOI] [PubMed] [Google Scholar]
- 8. Hause BM, Ducatez M, Collin EA, Ran Z, Liu R, Sheng Z, Armien A, Kaplan B, Chakravarty S, Hoppe AD, Webby RJ, Simonson RR, Li F. 2013. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 9:e1003176. 10.1371/journal.ppat.1003176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuanji G, Desselberger U. 1984. Genome analysis of influenza C viruses isolated in 1981/82 from pigs in China. J. Gen. Virol. 65:1857–1872. 10.1099/0022-1317-65-11-1857 [DOI] [PubMed] [Google Scholar]
- 10. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown IH, Harris PA, Alexander DJ. 1995. Serological studies of influenza viruses in pigs in Great Britain 1991-2. Epidemiol. Infect. 114:511–520. 10.1017/S0950268800052225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamaoka M, Hotta H, Itoh M, Homma M. 1991. Prevalence of antibody to influenza C virus among pigs in Hyogo prefecture Japan. J. Gen. Virol. 72:711–714. 10.1099/0022-1317-72-3-711 [DOI] [PubMed] [Google Scholar]
- 13. Saira K, Lin X, Depasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, Dwyer DE, Freiberg M, Horban A, Losso M, Lynfield R, Wentworth DN, Holmes EC, Davey R, Wentworth DE, Ghedin E, INSIGHT FLU002 Study Group. INSIGHT FLU003 Study Group 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J. Virol. 87:8064–8074. 10.1128/JVI.00240-13 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis AR, Nayak DP. 1979. Sequence relationships among defective interfering influenza viral RNAs. Proc. Natl. Acad. Sci. U. S. A. 76:3092–3096. 10.1073/pnas.76.7.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nayak DP, Chambers TM, Akkina RK. 1985. Defective-interfering (DI) RNAs of influenza viruses: origin, structure, expression, and interference. Curr. Top. Microbiol. Immunol. 114:103–151 [DOI] [PubMed] [Google Scholar]
- 16. Davis AR, Hiti AL, Nayak DP. 1980. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc. Natl. Acad. Sci. U. S. A. 77:215–219. 10.1073/pnas.77.1.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakada S, Graves PN, Palese P. 1986. The influenza C virus NS gene: evidence for a spliced mRNA and a second NS gene product (NS2 protein). Virus Res. 4:263–273. 10.1016/0168-1702(86)90005-5 [DOI] [PubMed] [Google Scholar]
- 18. Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J. Virol. 83:10309–10313. 10.1128/JVI.01109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng G, Hongo S, Kimura H, Muraki Y, Sugawara K, Kitame F, Numazaki Y, Suzuki H, Nakamura K. 1996. Frequent occurrence of genetic reassortment between influenza C virus strains in nature. J. Gen. Virol. 77:1489–1492. 10.1099/0022-1317-77-7-1489 [DOI] [PubMed] [Google Scholar]
- 20. Dowdle WR, Galphin JC, Coleman MT, Schild GC. 1974. A simple double immunodiffusion test for typing influenza viruses. Bull. World Health Organ. 51:213–218 [PMC free article] [PubMed] [Google Scholar]
- 21. Ohwada K, Kitame F, Homma M. 1986. Experimental infections of dogs with type C influenza virus. Microbiol. Immunol. 30:451–460. 10.1111/j.1348-0421.1986.tb02971.x [DOI] [PubMed] [Google Scholar]
- 22. Buonagurio DA, Nakada S, Fitch WM, Palese P. 1986. Epidemiology of influenza C virus in man: multiple evolutionary lineages and low rate of change. Virology 153:12–21. 10.1016/0042-6822(86)90003-6 [DOI] [PubMed] [Google Scholar]
- 23. Wilson HM, Hall JS, Flint PL, Franson JC, Ely CR, Schmutz JA, Samuel MD. 2013. High seroprevalence of antibodies to avian influenza viruses among wild waterfowl in Alaska: implications for surveillance. PLoS One 8:e58308. 10.1371/journal.pone.0058308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lopez JW, Woods GT. 1984. Influenza virus in ruminants: a review. Res. Commun. Chem. Pathol. Pharmacol. 45:445–462 [PubMed] [Google Scholar]
- 25. Brown IH, Crawshaw TR, Harris PA, Alexander DJ. 1998. Detection of antibodies to influenza A virus in cattle in association with respiratory disease and reduced milk yield. Vet. Rec. 143:637–638 [PubMed] [Google Scholar]
- 26. Crawshaw TR, Brown IH, Essen SC, Young SC. 2008. Significant rising antibody titres to influenza A are associated with an acute reduction in milk yield in cattle. Vet. J. 178:98–102. 10.1016/j.tvjl.2007.07.022 [DOI] [PubMed] [Google Scholar]
- 27. Kawano J, Onta T, Kida H, Yanagawa R. 1978. Distribution of antibodies in animals against influenza B and C viruses. Jpn. J. Vet. Res. 26:74–80 [PubMed] [Google Scholar]
- 28. Suzuki Y. 2005. Sialobiology of influenza: molecular mechanism of host range variation of influenza viruses. Biol. Pharm. Bull. 28:399–408. 10.1248/bpb.28.399 [DOI] [PubMed] [Google Scholar]
- 29. Herrler G, Szepanski S, Schultze B. 1991. 9-O-acetylated sialic acid, a receptor determinant for influenza C virus and coronaviruses. Behring Inst. Mitt. 89:177–184 [PubMed] [Google Scholar]
- 30. Zhang XM, Kousoulas KG, Storz J. 1992. The hemagglutinin/esterase gene of human coronavirus strain OC43: phylogenetic relationships to bovine and murine coronaviruses and influenza C virus. Virology 186:318–323. 10.1016/0042-6822(92)90089-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saif LJ. 2010. Bovine respiratory coronavirus. Vet. Clin. North Am. Food Anim. Pract. 26:349–364. 10.1016/j.cvfa.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Desselberger U, Racaniello VR, Zazra JJ, Palese P. 1980. The 3′ and 5′-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 8:315–328. 10.1016/0378-1119(80)90007-4 [DOI] [PubMed] [Google Scholar]
- 33. Zheng H, Palese P, García-Sastre A. 1996. Nonconserved nucleotides at the 3′ and 5′ ends of an influenza A virus RNA play an important role in viral RNA replication. Virology 217:242–251. 10.1006/viro.1996.0111 [DOI] [PubMed] [Google Scholar]
- 34. Lee YS, Seong BL. 1998. Nucleotides in the panhandle structure of the influenza B virus virion RNA are involved in the specificity between influenza A and B viruses. J. Gen. Virol. 79:673–681 [DOI] [PubMed] [Google Scholar]
- 35. Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. 1987. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. U. S. A. 84:8140–8144. 10.1073/pnas.84.22.8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. WHO Manual on animal influenza diagnosis and surveillance. 2002. World Health Organization, Geneva, Switzerland: http://whqlibdoc.who.int/hq/2002/WHO_CDS_CSR_NCS_2002.5.pdf [Google Scholar]
- 37. Briedis DJ, Lamb RA. 1982. Influenza B virus genome: sequences and structural organization of RNA segment 8 and the mRNAs coding for the NS1 and NS2 proteins. J. Virol. 42:186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lamb RA, Lai CJ, Choppin PW. 1981. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: colinear and interrupted mRNAs code for overlapping proteins. Proc. Natl. Acad. Sci. U. S. A. 78:4170–4174. 10.1073/pnas.78.7.4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pekosz A, Lamb RA. 1998. Influenza C virus CM2 integral membrane glycoprotein is produced from a polypeptide precursor by cleavage of an internal signal sequence. Proc. Natl. Acad. Sci. U. S. A. 95:13233–13238. 10.1073/pnas.95.22.13233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hongo S, Sugawara K, Muraki Y, Matsuzaki Y, Takashita E, Kitame F, Nakamura K. 1999. Influenza C virus CM2 protein is produced from a 374-amino acid protein (P42) by signal peptidase cleavage. J. Virol. 73:46–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu TD, Nacu S. 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26:873–881. 10.1093/bioinformatics/btq057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The sequence alignment/MAP format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14:178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods. Download
Genotypes in plaque-purified viruses following coinfection of swine testicle (ST) cells with C/OK and C/660 (A) and with C/OK, C/660, C/JHB, and C/Tay (B). Different-colored squares denote the identities of viral segments. Download
RT-PCR amplification of swine C/OK M, NS, and NP segments from infected ST cells with C/OK virus at an MOI of 0.01 at 18 h postinfection. Download
Full-length of C/OK virus M1 mRNA (5′-to-3′ direction) amplified by RT-PCR and confirmed by sequencing. The blue letters indicate sequence of the 5′ noncoding region and the 3′ noncoding region of M1 mRNA; the black letters with a gray highlight represent the sequence of primary M1 mRNA transcript; the black letters highlighted in yellow indicate the additional sequence introduced to primary M1 mRNA transcript following splicing. Download
Full-length of C/OK NS2 mRNA (5′-to-3′ direction) amplified by RT-PCR and confirmed by sequencing. The blue letters indicate the sequence of the 5′ noncoding region and 3′ noncoding region of NS2 mRNA; the black letters with a gray highlight represent the sequence of the first exon of NS2 mRNA transcript; the black letters highlighted in yellow indicated the sequence introduced to the first exon of NS2 following splicing. Download
Growth of influenza C/swine/Oklahoma/1334/2011 and influenza C/Johannesburg/1/66 virses in ST cell cultures. Cells were inoculated with a multiplicity of infection of 0.1 and 1.0 for C/OK and 1.0 and 10.0 for C/JHB and incubated at 33°C. Virus was titrated at the indicated time points on ST cells. Titers of viruses used for inoculation were 6.9 TCID50/ml for C/OK virus and 7.9 TCID50/ml for C/JHB virus. Download
Location of the internal intron within the C/OK M segment identified by NGS at 18 h (A) and 36 h (B) postinfection of ST cells.
Location of the internal intron within the C/OK NS segment identified by NGS at 18 h (A) and 36 h (B) postinfection of ST cells.