

Abstract
Systemic identification of deterministic genes for different phenotypes is a primary application of high-throughput expression profiles. However, gene expression differences cannot be used when the differences between groups are not significant. Therefore, novel methods incorporating features other than expression differences are required. We developed a promising method using transcriptional response as an operational feature, which is quantified as the correlation between expression levels of pathway genes and target genes of the pathway. We applied this method to identify causative genes associated with chemo-sensitivity to tamoxifen and epirubicin. Genes whose transcriptional response was dysregulated only in the drug-resistant patient group were chosen for in vitro validation in human breast cancer cells. Finally, we discovered two genes responsible for tamoxifen sensitivity and three genes associated with epirubicin sensitivity. The method we propose here can be widely applied to identify deterministic genes for different phenotypes with only minor differences in gene expression levels.
Specific phenotypes are generally attributed to different gene expression levels. Since high-throughput measurement of gene expression levels has become possible, several studies have identified genes showing differential expression between two or more phenotypic groups with hope that these genes are responsible for the phenotypic differences. There are several successful examples1,2,3,4,5,6, however, this approach has not been successfully applied to clinical studies because of the inconsistency of gene expression profiling using microarrays7,8,9. Typically, gene expression levels do not show significant differences between groups. For example, few genes show differential expression between primary tumors that are metastasis-prone and those that are metastasis-free after tamoxifen treatment. Moreover, there are many resultant passenger genes that have no causative power for phenotypes10. This indicates that analysis of expression level alone is not sufficient.
Abnormal genes that do not show changes in expression level can result in phenotypic changes. For example, gain-of-function oncogenes can transform normal cells into neoplastic cells such as B-Raf in skin cancer. Conventional approaches that depend only on gene expression levels are not applicable to such cases. Instead, evaluation of functional outcomes is required to identify genes contributing to phenotypes. Therefore, operational relationship between gene expression levels and functional outcomes should be assessed to find phenotype deterministic genes. Among diverse functional outcomes, we used transcriptional response, which is related to how well target genes of transcriptional factors are regulated. Malfunctioning genes can deregulate transcriptional responses against cytotoxic drugs, sometimes triggering drug resistance11,12. To capture such an aberration, we compared correlation patterns regarding expression levels of pathway genes and their target genes in drug-sensitive and drug-resistant patients to identify genes with significant differences in transcriptional responses, instead of comparing gene expression levels in the two patient groups.
There are several previous reports in which correlation is evaluated in each phenotype. Hu et al. checked correlation difference with all genes between two conditions13. For a gene, however, not all the other genes should have correlation with it. Considering all other genes is likely to make noise. Hwang et al. also examined correlation, but focused on differentially expressed protein-protein interaction sub-network14. It can identify differential outcomes, but not the cause for them. Unlike these previous studies, we developed a simple, but powerful method for systemic identification of deterministic genes for phenotypes using transcriptional response, and identified genes that lost their transcriptional response in tamoxifen-resistant and epirubicin-resistant patients. We hypothesized that inhibition of these genes suppresses abnormal transcriptional responses, sensitizing cancer cells to tamoxifen or epirubicin. Computational prediction was confirmed by in vitro cell viablity assays.
Results
Overview of the approach
We defined a transcriptional response as a relationship between the activities of transcription factor (TF) modulators and expression levels of TF target genes, which can be calculated using several types of correlation or mutual information. We hypothesized that the transcriptional response (other than the expression level itself) can be used to differentiate between two phenotypic groups. For many signal transduction pathways, TFs are integration points of signals from proteins operating between TFs and receptors. Thus, we considered genes in the same pathway with TFs (pathway genes) as genes that can modulate transcriptional responses. A schematic diagram of the overall process is shown in Figure 1. To identify deterministic genes for specific phenotypes, we evaluated all signaling molecules in any pathways according to NetSlim, simply referred to as “pathway genes”. We considered target genes of TFs in the same pathway of each pathway gene to be controlled by a pathway gene (target genes of a pathway gene). Even when there are no expression differences in a pathway gene (Figure 1A) and a target gene (Figure 1B) between two phenotypes, expressional correlation between the two genes can differ (Figure 1C). Therefore, we considered the expressional correlation as representative for normality of the transcriptional regulation of pathway genes. In most datasets, for example, EGFR target genes have significant expressional correlation with EGFR only in tamoxifen-sensitive patients (Figure 1D). Expression of EGFR and its target gene (NUDT11), and the correlation between them is shown in Supplementary Fig. S1 online. To identify pathway genes that lose significant transcriptional regulation on their target genes in drug-resistant patients, we compared the number of target genes which have significant expressional correlation with a pathway gene in drug-resistant and drug-sensitive patients. We defined the significance rate (SR) of a pathway gene as the proportion of target genes which have significant expressional correlations with the pathway gene, and the significance score (SS) as the ratio of small SR over large SR after restriction to range [0, 1]. According to defined formula, higher SS indicates more deterministic genes for given phenotype (Figure 1E). We used SS as the numeric for normality of the transcriptional regulation of each pathway gene.
Figure 1. Overview of the study approach.

For a gene involved in a pathway (pathway gene), target genes of transcription factors involved in the same pathway were considered to be target genes of the pathway genes. Even if there are no expression differences between the pathway gene (A) and target gene (B) in two phenotypic groups, the correlation between them (C) can still differ. (D) EGFR target genes were significantly correlated only in drug-sensitive patients. Each rectangle represents EGFR target genes, and color indicates the portion of datasets in which the target gene had a Spearman's rank order correlation coefficient (P < 0.05) with EGFR in terms of mRNA expression level. (E) Target genes of a pathway gene were defined as target genes of transcription factors involved in the same pathways of the pathway gene. For each phenotypic group, we calculated the ratio of target genes with a significant expressional correlation with the pathway gene over all target genes, which is termed as the significance rate (SR) of the pathway gene. The ratio of small SR over large SR is restricted to range [0, 1], and termed as the significance score (SS) of the pathway gene.
Difference in transcriptional response with minor expression differences
Differentially expressed genes (DEGs) between tamoxifen-resistant and tamoxifen-sensitive patients in the datasets were identified. Among the eight datasets, there were no DEGs in the four datasets using a false discovery rate (FDR) < 0.05. GSE6532B had the largest number of DEGs, which accounted for only 5% of the tested genes (Figure 2A). On the other hand, SR was significantly increased in tamoxifen-sensitive patients compared to tamoxifen-resistant patients in all datasets (Figure 2B, all datasets had a paired t-test P-value < 0.0001). The SR distribution of all pathway genes confirmed this result (Figure 2C, the distributions were not normal in Kolmogorov-smirnove, D'agostino & Pearson omnibus, and Shapiro-Wilk normality tests). Based on SR, we calculated SS for all pathway genes (Figure 2D). The number of target genes of pathway genes was not associated with SS (based on the P-value of the Spearman's rank order correlation coefficient of target gene number and SS). We visualized the difference in SR between the two groups (SR of tamoxifen-sensitive patients - SR of tamoxifen-resistant patients) for all pathway genes over several datasets (Figure 3A). Of the top-ranked genes, we selected five that had no differences in expression level between the two groups (Figure 3B), and performed in vitro assays to examine the association between these genes and tamoxifen sensitivity.
Figure 2. Tamoxifen sensitivity is more strongly associated with SS than DEGs.

(A) The number of differentially expressed genes (DEGs) between tamoxifen-sensitive and tamoxifen-resistant patient groups over eight datasets. There were no DEGs in four datasets with FDR < 0.05. (B) SR in tamoxifen-sensitive and tamoxifen-resistant patients. All datasets have different SR distributions with P < 0.0001. The 95% confidence intervals of SR are (0.073, 0.075) and (0.144, 0.149) in tamoxifen-resistant and tamoxifen-sensitive groups, respectively. Distribution of SR (C) and SS (D) for tamoxifen sensitivity were averaged over all datasets.
Figure 3. Significant scores of pathway genes over all datasets.

(A) Significance scores of pathway genes over all datasets. Each column represents a dataset and each row represents a pathway gene. Color indicates differences in SR between the two groups (SR of tamoxifen-sensitive patients - SR of tamoxifen-resistant patients) instead of SS for clear visualization. Gray cells indicate the pathway genes that were not available on microarray chips used in the dataset. (B) Significance of expression level differences of the five top-ranked genes. For each gene, a dot indicates the q-value of differential expression in each dataset. For the five top-ranked genes, there were no genes that showed differential expression with FDR < 0.05 (yellow region) in any of datasets.
Deterministic genes for tamoxifen sensitivity
To verify the accuracy of the computational predictions, we evaluated the cytotoxic effect of tamoxifen after knockdown of the five top-ranked genes, namely, SNF1LK, TRAP1, JAK2, SOCS2, and FOSB. Titration of tamoxifen showed that cell death occurred in a dose-dependent manner in two breast cancer cell lines: MCF-7 and MDA-MB-231 cells. To examine the effects of each gene on cell viability, cells were treated with tamoxifen at a concentration of 1 μM. At 48 h post-transfection of siRNAs specific for each gene, cells were incubated in the presence or absence of tamoxifen for 24 h, and then cell viability was measured using WST-1 assay. Tamoxifen-induced cell death was significantly increased in cells transfected with siJAK2 or siSOCS2 (Figure 4A). Transfection of siRNAs without tamoxifen treatment did not induce significant level of cell death. These results were confirmed by flow cytometric analysis after staining with TMRE. Tamoxifen-induced cell death was remarkably increased after siRNA knockdown of JAK2 and SOCS2 (Figure 4B). These data validate our computational method and suggest that JAK2 and SOCS2 are deterministic genes for tamoxifen sensitivity in breast cancer. In accordance with these results, JAK2/STAT5 inhibition has been shown to be important to restore efficacy of dual PI3K/mTOR inhibitor in metastatic breast cancer15.
Figure 4. Tamoxifen-induced cell death with siRNA transfection.
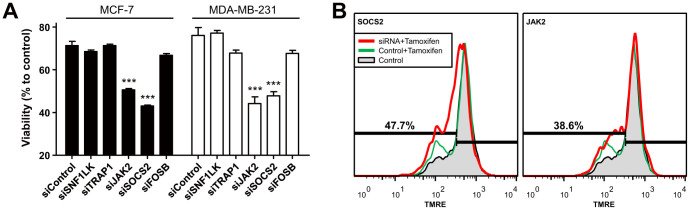
(A) Knockdown of SNF1LK, TRAP1, JAK2, SOCS2, and FOSB was enabled by siRNA transfection as described in Materials and Methods. At 48 h post-transfection, cells were treated with tamoxifen (1 μM) for 24 h, and then cell viability was measured using the WST-1 assay (mean ± SEM; Tukey's post hoc test was applied to significant group effects in ANOVA, P < 0.0001; ***P < 0.001, compared with non-treated control). (B) Cell viability was evaluated by flow cytometric analysis after TMRE staining.
Consistent loss of transcriptional response by JAK2 and SOCS2 in drug-resistant patients
Because JAK2 and SOCS2 were associated with tamoxifen sensitivity in the in vitro assays, we examined whether their target genes would have significant transcriptional responses in the tamoxifen-sensitive patients over several datasets. For most JAK2 (Figure 5A) and SOCS2 (Figure 5B) target genes, the transcriptional response was consistently lost in drug-resistant patients for all datasets. For SOCS2 target genes, two datasets (GSE1378, GSE1379) showed different sign of correlations with mRNA expression levels of SOCS2 compared to other datasets, but their correlations were still significant (Figure 5B). These data suggest that the status of transcriptional regulation is more consistent with drug-sensitivity than are gene expression levels, where only five DEGs were common to two datasets among the eight datasets.
Figure 5. Correlation between target genes of JAK2 and SOCS2.

Each row represents a JAK2 (A) and SOCS2 (B) target gene, and each column represents a dataset. Color indicates the Spearman's rank order correlation coefficient between JAK2 or SOCS2 and their target genes. Gray cells mean that there is no probe for the target genes on the microarray chips used in the datasets.
Validation in another case: deterministic genes for epirubicin sensitivity
Similarly with the case of tamoxifen sensitivity, we applied our method in epirubicin-treated breast cancer samples (GSE16446). We evaluated efficacy of epirubicin in MDA-MB-231 cells after knockdown of the six top-ranked genes, namely, NOTCH4, HES5, IL6, BIRC2, RING1, and SPEN. At 48 h post-transfection of siRNAs specific for each gene, cells were incubated in the presence or absence of epirubicin for 24 h, and then cell viability was evaluated by trypan blue exclusion. Epirubicin-induced cell death was significantly increased in cells transfected with siNOTCH4, siHES5, or siBIRC2, suggesting that they are deterministic genes for epirubicin sensitivity. However, DEGs could not select deterministic genes for epirubicin sensitivity, because there were no DEGs with FDR < 0.05 including NOTCH4, HES5, and BIRC2 (Supplementary Fig. S2 online).
Discussion
Evaluation of transcriptional response is required to identify deterministic genes for different phenotypes. In the context of drug-resistance, pathway genes can convey defective signals to TFs and affect the transcription of target genes, eventually inducing drug-resistance. As in this case, operational phenotypes such as the response to growth factors or drugs depend not only on the quantity of proteins but also on their functional normality. Thus, we incorporated stimuli-response concepts into transcriptional control, and developed a method to identify deterministic genes that cause specific phenotypes. Our method was used to identify genes responsible for tamoxifen sensitivity in breast cancer. Notably, most genes showed non-significant differential expression between tamoxifen-sensitive and tamoxifen-resistant groups (Figure 2A). Even using re-sampling analysis by Li et al.16, only one gene was common DEG that has FDR < 0.05 for more than 50% of randomly selected microarray data in two of eight datasets. This suggests that traditional methods that depend on the quantity of gene expression levels would have been unsuccessful in this case.
Several studies have identified genes that represent prognosis1,2,17 after tamoxifen treatment, but such studies have focused on differences in gene expression between drug-resistant and drug-sensitive patients. The ratio of two genes (HOXB13:IL17BR) is one of prognostic factors18. However, the results of several studies have been inconsistent7,8,9 mainly due to the inaccuracy of expression profiling by microarrays. In contrast, the rank of expression level has been relatively well-conserved over several studies compared to expression level itself19. We speculated that our method would successfully identify causative genes for drug resistance because we used rank of correlations of genes. Moreover, the correlation is not affected by shift and scale, implying that the normalization method of expression levels, which is an important issue in handling gene expression profiles, has only a minor effect on the final results. Using our method, we revealed that JAK2 and SOCS2 are deterministic factors for tamoxifen sensitivity in breast cancer. In addition, we showed that NOTCH4, HES5, and BIRC2 and determine epirubicin sensitivity of breast cancer cells.
Our proposed method is not limited to drug resistance, but rather is applicable to any case where two different phenotypes are of interest, even when few genes are showing significant differential expression. Therefore, it is important to establish definite criteria by which phenotypes can be determined. For example, it is clear that cell death is the most important phenotypic criterion for drug-resistance. On the other hands, there is no clear standard for phenotypic differentiation regarding metastasis, even though anoikis, migration, and invasion are important contributing factors. To overcome these weaknesses of examining phenotypes, we incorporated transcriptional response as a functional output of phenotypes, and successfully identified causative genes for different phenotypes.
Methods
Microarray datasets and pre-processing
We used four original microarray datasets from the Gene Expression Omnibus (GEO) database: GSE129093, GSE1378, GSE1379, and GSE6532. Because GSE6532 includes data from several hospitals, we split it into five datasets according to hospital and microarray platform. All datasets contained ER-positive patients. We classified patients as tamoxifen-resistant if there was annotation (in the GSM description) that metastasis had occurred; otherwise, they were classified as tamoxifen-sensitive. In all datasets, disease free survival (DFS) of tamoxifen-sensitive patients group was longer than that of tamoxifen-resistant patients group (all datasets had a paired t-test P-value < 0.001). We used GSE16446 in the case of epirubicin sensitivity. Total 38 arrays were used for drug sensitive and resistant groups since the other arrays have no information about occurrence of distant metastasis after therapy. The number of patients and other information are provided in Table 1.
Table 1. Overview of the study approach.
| GSE ID | GPL ID | The number of resistant patients | The number of sensitive patients | ER-STATE |
|---|---|---|---|---|
| GSE1378 | GPL1223 | 28 | 32 | Positive |
| GSE1379 | GPL1223 | 28 | 32 | Positive |
| GSE6532A | GPL96 | 12 | 54 | Positive |
| GSE6532B | GPL97 | 19 | 80 | Positive |
| GSE6532C | GPL570 | 28 | 59 | Positive |
| GSE6532D | GPL96 | 16 | 32 | Positive |
| GSE6532E | GPL97 | 21 | 48 | Positive |
| GSE12093 | GPL96 | 20 | 116 | Positive |
| GSE16446 | GPL570 | 19 | 19 | Negative |
All probe IDs in microarray chips were removed during processing if detection P-values were larger than 0.05 or tags for detection were absent. Probes that detected less than 75% in all sample groups were also removed in later analyses. Remaining probes were converted into HPRD20 ID where the average intensities of the probes for the same HPRD were assigned as expression intensity of the HPRD ID. Quantile-quantile normalization was applied to all chips.
Pre-defined pathways
We used NetSlim database21 whose pathways are one of the most well-organized forms, even though there are small number of pathways. NetSlim pathways start from external stimuli and end with transcription factors. For pathways containing several sub-pathways such as EGFR pathway, NetSlim pathways are neatly demarcated. Moreover, protein-protein and enzyme-substrate interactions in NetSlim are selected based on stringent criteria21. We manually removed genes denoted as transcriptionally induced in the pathways. Finally, we used 605 pathway genes, and 9063 target genes of pathway genes.
Cell cultures
MCF-7 and MDA-MB-231 cells were generously provided by Dr. Mi-Young Kim (KAIST, Daejeon, Korea), and maintained at 37°C in an atmosphere containing 5% CO2 in Dulbecco's modified Eagle's medium (Welgene, Seoul, Korea) supplemented with 10% fetal bovine serum (Gibco, Gaithersburg, MD), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). Tamoxifen was purchased from Sigma-Aldrich (St. Louis, MO).
Cell viability assay
WST-1 assays were performed to determine cell viability. MCF-7 and MDA-MB-231 cells were seeded in 24-well plates at a density of 4 × 104 cells/well in quadruplicate, and WST-1 reagent (Nalgene, Rochester, NY) was added to each well up to 5% of the media volume. After incubation for 2 h at 37°C in a 5% CO2 incubator, the absorbance at 450 nm was measured using a microplate reader (Bio-Rad, Richmond, CA). Cell death was confirmed by staining with 100 nM tetramethylrhodamine ethyl ester (TMRE, Sigma-Aldrich). Ten thousand cells were analyzed on a Caliber flow cytometer. Cell viability was also measured by trypan blue exclusion. Each cell suspension was mixed with the same volume of 0.4% trypan blue dye (Gibco), followed by counting with hemocytometer in quadruplicate. Cells that stained blue were scored as nonviable.
siRNA transfection
A small interfering RNA (siRNA) duplex targeted to SNF1LK, TRAP1, JAK2, SOCS2, FOSB, NOTCH4, HES5, IL6, BIRC2, RING1, SPEN, and an siRNA with a random sequence (negative control) was synthesized by Bioneer (Daejeon, Korea). Transient transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Statistical analysis
DEGs were identified by a q-value < 0.05 calculated with “qvality”22 for the P-value of the two-tailed Student's t-test. Correlations used in SRs were calculated using the Spearman's rank order correlation coefficient and their P-values were approximated using Student's distribution23. After treatment of anticancer drugs, each sample was compared by one-way analysis of variance (ANOVA) with Tukey's post hoc test being applied to significant main effects.
Author Contributions
J.L. and J.P. conceived basic concept. J.L. performed computational analysis. J.P. performed cell death assays. J.L. and J.P. wrote the manuscript. C.C. supervised and coordinated this research. All authors discussed results of this manuscript.
Supplementary Material
Supplementary Fig. S1 and S2
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2012R1A2A4A01007108).
References
- Chanrion M. et al. A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clin Cancer Res 14, 1744–52 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M. P. et al. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J Clin Oncol 23, 732–40 (2005). [DOI] [PubMed] [Google Scholar]
- van de Vijver M. J. et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347, 1999–2009 (2002). [DOI] [PubMed] [Google Scholar]
- van't Veer L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–6 (2002). [DOI] [PubMed] [Google Scholar]
- Cardoso F. et al. Clinical application of the 70-gene profile: the MINDACT trial. J Clin Oncol 26, 729–35 (2008). [DOI] [PubMed] [Google Scholar]
- Choi J. K., Yu U., Yoo O. J. & Kim S. Differential coexpression analysis using microarray data and its application to human cancer. Bioinformatics 21, 4348–55 (2005). [DOI] [PubMed] [Google Scholar]
- Ein-Dor L., Kela I., Getz G., Givol D. & Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics 21, 171–8 (2005). [DOI] [PubMed] [Google Scholar]
- Kothapalli R., Yoder S. J., Mane S. & Loughran T. P. Jr Microarray results: how accurate are they? BMC Bioinformatics 3, 22 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis J. P. Microarrays and molecular research: noise discovery? Lancet 365, 454–5 (2005). [DOI] [PubMed] [Google Scholar]
- Bozic I. et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 107, 18545–50 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K. et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 109, E2127–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove E. A. & Sutherland R. L. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9, 631–43 (2009). [DOI] [PubMed] [Google Scholar]
- Hu R., Qiu X., Glazko G., Klebanov L. & Yakovlev A. Detecting intergene correlation changes in microarray analysis: a new approach to gene selection. BMC Bioinformatics 10, 20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T. & Park T. Identification of differentially expressed subnetworks based on multivariate ANOVA. BMC Bioinformatics 10, 128 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi A. et al. JAK2/STAT5 Inhibition Circumvents Resistance to PI3K/mTOR Blockade: A Rationale for Cotargeting These Pathways in Metastatic Breast Cancer. Cancer Cell 22, 796–811 (2012). [DOI] [PubMed] [Google Scholar]
- Li J. et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun 1, 34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitroda S. P., Khodarev N. N., Beckett M. A., Kufe D. W. & Weichselbaum R. R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci U S A 106, 5837–41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X. J. et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 5, 607–16 (2004). [DOI] [PubMed] [Google Scholar]
- Eddy J. A., Hood L., Price N. D. & Geman D. Identifying tightly regulated and variably expressed networks by Differential Rank Conservation (DIRAC). PLoS Comput Biol 6, e1000792 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad T. S. et al. Human Protein Reference Database--2009 update. Nucleic Acids Res 37, D767–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju R. et al. NetSlim: high-confidence curated signaling maps. Database (Oxford) 2011, bar032 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Storey J. D. & Noble W. S. QVALITY: non-parametric estimation of q-values and posterior error probabilities. Bioinformatics 25, 964–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press W. H. Numerical recipes: the art of scientific computing. (Cambridge University Press, Cambridge, UK; New York, 2007).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1 and S2