

Abstract
The nuclear factor erythroid 2 related factor 2 (NRF2) is a key regulator of endogenous inducible defense systems in the body. Under physiological conditions NRF2 is mainly located in the cytoplasm. However, in response to oxidative stress, NRF2 translocates to the nucleus and binds to specific DNA sites termed “anti-oxidant response elements” or “electrophile response elements” to initiate transcription of cytoprotective genes. Acute oxidative stress to the brain, such as stroke and traumatic brain injury is increased in animals that are deficient in NRF2. Insufficient NRF2 activation in humans has been linked to chronic diseases such as Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis.
New findings have also linked activation of the NRF2 system to anti-inflammatory effects via interactions with NF-κB. Here we review literature on cellular mechanisms of NRF2 regulation, how to maintain and restore NRF2 function and the relationship between NRF2 regulation and brain damage. We bring forward the hypothesis that inflammation via prolonged activation of key kinases (p38 and GSK-3β) and activation of histone deacetylases gives rise to dysregulation of the NRF2 system in the brain, which contributes to oxidative stress and injury.
Keywords: neuroinflammation, NRF2, anti-oxidants
1. Regulation of the NRF2 system
During evolution, cells have developed inducible defense systems against harmful endogenous and exogenous substances. Several transcription factors are involved in boosting the cell’s defenses. The major regulator of the so-called phase II and some phase III genes is the nuclear factor erythroid 2 related factor 2 (NRF2) (1, 2). The principle of the NRF2 system is to keep NRF2 protein low under normal conditions with the possibility of rapid induction in case of a sudden increase in oxidation status in the cell. This is achieved by constitutive synthesis and degradation of NRF2 with the possibility of rapid redirection of NRF2 to the nucleus.
NRF2 is a member of the cap’n’collar (CNC) family of transcription factors, which also include NRF1, NRF3 and p45 NF-E2. NRF2 is a basic leucine zipper (bZIP) protein that in the nucleus heterodimerizes with small MAF or JUN proteins followed by binding to specific DNA sites termed anti-oxidant response elements (ARE) or electrophile response elements (EpRE) (1, 3). NRF2-ARE binding can initiate transcription of hundreds of cytoprotective genes including enzymes in the glutathione defense system and proteasome subunits (4). New findings connect NRF2 not only to an elevated anti-oxidant capacity but also to expression of other types of protective proteins such as brain derived neurotrophic factor (5), the anti-apoptotic B-cell lymphoma 2 (6), the anti-inflammatory interleukin (IL)-10, the mitochondrial transcription (co)-factors NRF-1 and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (7), the iron exporter ferroportin 1 (8), and the autophagic protein p62 (9).
1.1 Regulation of NRF2 by KEAP1
The NRF2 molecule contains six functional domains named NRF2-ECH homologies (Neh 1-6). In the unstressed cell NRF2 is bound to Kelch-like ECH associated protein 1 (KEAP1) in the cytoplasm (10) via its KEAP1 binding domain. KEAP1 is a homodimer with three major domains. Each C-terminal in the KEAP1 dimer, the so called DRG domain, binds to NRF2, while the N-terminal part of KEAP1 binds to a E3 ubiquitin ligase complex (Rbx-1) via an adaptor, cullin-3 (11). This directs NRF2 to ubiqutination and constitutive degradation by the 26S proteasome (12).
There are several theories on how NRF2 is released from KEAP1 in response to cellular stress. NRF2 binds to the DRG sites of each KEAP1 subunit via two different binding sites, one high affinity (hinge) and one low affinity (latch) (13). One hypothesis is that NRF2 can still move freely when bound to the hinge site (high affinity), while the latch site restricts the movement of NRF2 and places the NRF2 lysins in the Neh2 regions for poly-ubiquitination via the cullin-3 E3 ligase. In this model, oxidative/electrophilic molecules modify cysteine residues of KEAP1, which activates NRF2 (14). The KEAP1 protein contains relatively many cysteines, which are in the vicinity of basic amino acids. This configuration lowers the pKa values for many of the thiols and makes them extra reactive to oxidants/electrophiles. Modifications of (some of) these cysteines result in a different conformation of KEAP1, which then can dissociate from the latch (low-affinity) site. The result is that KEAP1 is bound to NRF2 at the hinge site and that “new” non-bound NRF2 can be phosphorylated by protein kinase C (PKC) (15) and possibly other kinases (see below), translocated to the nucleus and initiate transcription of cytoprotective genes. Alternative hypotheses on how NRF2 becomes free from KEAP1 include that oxidants/electrophiles can dissociate the NRF2-KEAP1 complex directly (16) or that oxidants/electrophiles dissociate cullin-3 from KEAP1, which leads to blocking of further ubiquitination (17) and degradation of KEAP1 (18). Recent studies also show that NRF2 activation can be achieved by autophagic degradation of KEAP1 (see more below).
The abundance of the proteins in the NRF2 system discussed above appears to be regulated by an autoregulatory loop. Thus NRF2 regulates the transcription of KEAP1, cullin-3 and Rbx-1 and, in turn, KEAP1/cullin-3/Rbx-1 degrades NRF2 (19). The KEAP1 complex can also be imported into the nucleus and participate in the degradation of nuclear NRF2 (15). Further, feed back systems appear to exist where NRF2 activation induces an increase in proteasome expression and activity. In response to NRF2 activators, increased expression of genes coding for the proteasome subunits 20S and 19S have been observed (4, 20, 21). The NRF2-mediated induction of proteasome genes has been linked to increased resistance to oxidative stress (22, 23).
Although KEAP1 is the main regulator of NRF2 activation others exist, including the transcription factor Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), as discussed below. Another regulator of NRF2 is Broad-complex, tramtrack and bric-a-brac and CNC homology 1 (BACH1). This is a transcription factor that competes with NRF2 for binding to ARE sites and thus functions as a repressor of NRF2 activation (24). Interestingly BACH1 appears to serve as a negative feed-back regulator of NRF2 activation as it is regulated by NRF2-activating agents in an NRF2-dependent manner (25).
1.2. Phosphorylation of NRF2
The transport to and from the nucleus is influenced by factors such as the phosphorylation of NRF2. Several kinases have been shown to affect NRF2 transport differently via specific phosphorylation sites. One of the best characterized include phosphorylation by PKC (26), which is necessary for liberating NRF2 from KEAP1 (27) and thus promoting transport to the nucleus. Protein kinase CK2 can also phosphorylate NRF2, which promotes transport of NRF2 into the nucleus (28). Similar activating effects on NRF2 translocation to the nucleus have been observed for phosphatidylinositide 3-kinases (PI3K) (29), c-Jun N-terminal kinase (JNK), extracellular regulated kinase (ERK) (30) and PERK (31). However, it has been suggested that mitogen-activated protein kinases (MAPKs)-induced phosphorylation of NRF2 does not directly affect KEAP1-NRF2 protein interaction, but rather acts through indirect mechanisms (32).
Phosphorylation catalyzed by kinases can also increase NRF2 breakdown. The MAPK p38 has been shown to stabilize the interaction between KEAP1 and NRF2 thereby elevating the breakdown of NRF2 (33). Activation of glycogen synthase kinase 3-beta (GSK-3β) can cause degradation of NRF2 by the proteasome that is independent of KEAP1. Phosphorylation of the Neh6 domain in NRF2 by GSK-3β leads to recognition by a E3 ubiquitin ligase (beta-TrCP) (34). Binding of beta-TrCP couples NRF2 to the cullin-3/Rbx1 ubiqutination complex (35), directing NRF2 to proteasomal degradation. GSK3β activation acts upstream and phosphorylates Src kinases, leading to their nuclear localization and NRF2 phosphorylation (36, 37). Ultimately this will lead to export, with or without KEAP1, followed by proteasomal degradation of NRF2. Several Src kinases (FYN, SRC, YES, and FGR) in the nucleus can regulate NRF2 via phosphorylation of NRF2 Tyr568 that triggers nuclear export and degradation of NRF2. Thus, kinase activation is of utmost importance in regulation of the NRF2-system.
1.3 Autophagic regulation of NRF2
The protein p62, which represents both a selective autophagy substrate and a cargo receptor for autophagic degradation of misfolded proteins, has been reported to activate NRF2 in response to oxidative stress (9, 38–40). The promoter/enhancer region of the p62 gene contains ARE sites (41) and autophagy defects induce an excessive accumulation of p62 and oxidative stress (39, 42). An increase in endogenous p62, either due to a defect in autophagy or as a result of its ectopic expression, sequesters KEAP1 into aggregates, hence causing inhibition of KEAP1-mediated NRF2 ubiquitination and degradation (43). Thus, when autophagy is impaired, p62 accumulates and activates NRF2 by sequestering KEAP1 in inclusion bodies. In turn NRF2 promotes new p62 products creating a positive loop of NRF2 activation. With regard to the brain, recent findings demonstrate that interactions between p62 and the KEAP1-NRF2 signalling pathway play a key role in preventing oxidative injury and alleviate endoplasmic reticulum stress during cerebral ischemia/reperfusion (44).
1.4 Epigenetic regulation of NRF2
Histone acetylation and phosphorylation, methylation of CpG islands and synthesis of specific miRNAs are additional means by which cells can regulate the levels of NRF2. This field of NRF2 regulation is just beginning to be explored but it contains interesting targets for future therapeutic activation of the NRF2 system, including in the brain. We have shown that factors released from activated microglia activate histone deacetylases (HDACs), reduce the overall acetylation level of histone 3 and decrease the activation of NRF2 in cultured astrocytes. Restoring the acetylation level by HDAC inhibitors also repaired the NRF2 system indicating that histone acetylation may be one epigenetic mechanism to regulate NRF2 function (45) (see more below). In support of this idea, the HDAC inhibitor trichostatin A (TSA) increased cell viability in cortical neurons after oxygen-glucose deprivation and reduced infarct volume in wild-type mice subjected to permanent middle cerebral artery occlusion (MCAO) in an NRF2/KEAP1-dependent manner (46). TSA reduced the expression of KEAP1, activated NRF2 nuclear translocation, NRF2–ARE binding and increased expression of the NRF2 regulated proteins heme oxygenase (HO1), NAD(P)H:quinone oxidoreductase 1 (NQO1), and glutamate-cysteine ligase (GCL) catalytic subunit. It should be noted that direct acetylation of NRF2 by p300/CBP has been shown to stimulate NRF2 binding to DNA, an effect that could also contribute to the positive effects of HDAC inhibitors on NRF2 activation (47).
Elevated methylation of CpG islands in the promoter of the Nrf2 gene was shown to suppress expression of NRF2 in mice with prostate cancer. Interestingly the NRF2 function could be restored by phytochemicals that reduced the methylation level (48). To our knowledge CpG methylation of the Nrf2 promoter as a cause of low NRF2 function and oxidative stress in neurodegerative diseases and aging has not yet been explored (see more below). Similarly, several miRNAs (miR153, miR27a, miR142-5p, miR144) reduce NRF2 activation in neuronal SH-SY5Y cells (49), but there is overall very limited knowledge of the effects of miRNA modifications on the NRF2 system in the brain.
2. The NRF2 system and CNS disease
The NRF2 system is widely expressed in the CNS and is regulated in response to both acute cerebral insults and in neurodegenerative disease. In addition to its crucial regulatory role in the endogenous defense to various cellular stresses, NRF2 is recognized as an important regulator of inflammation in the brain. Dysregulation of these mechanisms has been suggested to contribute to brain injury.
2.1. CNS effects in animals with genetic deletion of Nrf2
Suppression of the NRF2 system by genetic deletion in animals leads to elevated neurotoxicity resulting from endogenous and exogenous stresses. However, spontaneous pathological effects in brains from Nrf2-deficient mice are less clear, but do exist. The predominant general findings in animals with genetic deletion of Nrf2, compared with age-matched control animals, are white matter leukoencephalopathy with widespread astrogliosis (50), but with no overall difference in microglial activation and overt signs of neurodegeneration. However, a predominant pro-inflammatory microglial phenotype was reported in dopamine metabolizing brain areas (striatum and ventral midbrain) of Nrf2 deficient animals. Markers characteristic of classical microglial activation, such as cyclooxygenase-2 (COX-2), inducible nitric oxide synthases (iNOS), IL-6, and tumor necrosis factor-α (TNF-α) were increased, while at the same time anti-inflammatory markers attributable to alternative microglial activation, including Found in inflammatory zone protein (FIZZ-1), YM-1, Arginase-1, and IL-4 were decreased (51). These studies show that Nrf2 deficient mice indeed demonstrate signs of neurodegeneration, but mainly in areas with high inherent oxidative reactions, i.e. dopamine-metabolizing areas.
On the other hand, different types of cellular stresses superimposed on an Nrf2-deficient background have significant detrimental effects on neuronal systems in the brain. Loss of NRF2-mediated transcription exacerbates vulnerability to the neurotoxin 6-hydroxydopamine (6-OHDA), both in cultured neurons and in Nrf2 deficient mice in vivo (52). In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) animal model of Parkinson’s disease there is greater loss of the dopamine transporter in the striatum (53), and exacerbated gliosis and dopaminergic nigrostriatal degeneration (54) in Nrf2 deficient mice compared to wild type controls. Also, in animal models of acute brain injury such as stroke and traumatic brain injury, NRF2 deficient mice show more severe injuries and more intense immunological reactions (55–57).
2.2. NRF2 and neurodegenerative disease in humans
A dysfunctional NRF2 system has been linked to several chronic diseases in humans, including chronic kidney disease and chronic obstructive pulmonary disease (COPD) although the exact role for the NRF2 decline is yet to be characterized (58). The NRF2 system is compromised by increased age and aging is a strong risk factor for almost all neurological diseases involving oxidative stress. If suppression of NRF2 activity occurs in human brains the implications are elevated vulnerability to acute insults that involve oxidative stress, prolonged and more severe inflammation and likely an elevated risk to develop chronic diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS). Indeed, brains from AD patients have decreased amounts of NRF2 in astrocytes of the hippocampus, one of the brain areas where neurodegeneration starts in AD (59). The NRF2 target protein p62 is significantly reduced in the frontal cortex of AD patients (60). As discussed above, p62 has been linked to autophagy and it was recently suggested that interaction between p62 and KEAP1 provides a molecular basis for the formation of cytoplasmic inclusions observed in several neurodegenerative diseases (61). In post mortem brains from ALS patients NRF2 is reduced in the motor neurons in spinal cord and cortex (62).
Brains from individuals with PD are characterized by a prominent decrease in glutathione in the substantia nigra. In the neurons that survive in substantia nigra, NRF2 is localized to the nucleus indicating that in these cells NRF2 transport is functional. However, it is not known if the NRF2 transcription machinery is deficient even though NRF2 is transported to the nucleus (59). In agreement with a malfunctioning NRF2 system in PD, olfactory neurosphere-derived cells from patients with sporadic PD had lower glutathione levels, which could be restored by an NRF2 activating agent (63). The study indicates that therapy aiming for NRF2 activation could stop neurodegeration in PD. Further, genetic variation in the Nrf2 gene has been linked to both AD and PD progression (64, 65). Taken together, although there are only a few studies in humans, these do indicate that the NRF2 system is dysregulated in brains of individuals suffering from neurodegenerative diseases.
The underlying mechanisms behind the apparent poor NRF2 functioning in chronic neurodegenerative diseases are not known. The number of theoretical possibilities behind dysregulation is overwhelming in view of the complex regulation of the NRF2 system described above. However, one possible link between the NRF2 system and neurodegenerative diseases is that many of them are proteinopathies, as described below. It also appears clear, if it is correct to extrapolate from mice to humans, that lack of NRF2 does not induce general neurodegeneration per se. Rather it is when cells are stressed that absence of NRF2 will become detrimental to neurons. Thus in the following sections, we will also review data that supports the idea that inflammation and prolonged activation of key kinases are factors that lead to the paradoxical decrease in NRF2 observed in some neurodegenerative diseases.
2.3 Proteinopathies and NRF2
Several of the neurodegerative dieseases are proteinopathies where specific proteins become aggregated in the brain. In view of the role of NRF2 in upregulation of proteasomal enzymes, a link between proteopathies and NRF2 dysfunction is possible, and potentially upregulation of the NRF2 system could be beneficial under such circumstances. AD is characterized by brain accumulation of amyloid beta-protein (Aβ) plaques. Activation of NRF2, by tert-butylhydroquinone (TBHQ) treatment or adenoviral Nrf2 gene transfer, protected against Aβ toxicity in a transgenic mouse model of AD (66). Recently, it was also shown that the NRF2 activator carnosic acid suppressed the production of Aβ 1–42 by in SH-SY5Y human neuroblastoma cells (67).
In, PD the protein α-synuclein aggregates to form so called “Lewy bodies”, and dopaminergic neurons in the substantia nigra pars compacta degenerate. Several studies indicate a relationship between NRF2 and α-synuclein. Mutation and deletion of the protein DJ-1 can cause a variant of autosomal recessive familial PD. Overexpression of DJ-1 stabilizes NRF2 and reduces aggregation of α-synuclein (68, 69). The NRF2 activator triterpenoid 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) methyl amide reduces neurodegeneration of dopamine neurons, diminishes α-synuclein accumulation and oxidative stress in a experimental model of PD (70). It has also been shown that mice that lack NRF2 and have increased expression of human α-synuclein, have aggravated dopaminergic neuron loss, increased neuroinflammation and elevated protein aggregation (71). Interestingly, in a mutant α-synuclein transgenic mouse model, it appears sufficient to increase NRF2 expression in astrocytes to achieve protection against neurotoxicity. It was suggested that the protection was mediated via elevated degradation of mutant α-synuclein through the autophagy-lysosome pathway (72).
Huntington disease (HD) is an inherited neurodegenerative disease caused by an expansion of polyglutamine in the protein huntingtin. Expression of mutant huntingtin caused reduced activity of NRF2, and lowered the activation of the NRF2 pathway by the oxidant TBHQ in cultured cells (73). In a transgenic mouse model of HD triterpenoid derivatives in the diet upregulated NRF2/ARE induced genes in the brain, reduced oxidative stress, improved motor impairment and increased longevity (74).
In familial ALS, 20 % of the cases are due to mutations in the Cu/Zn superoxide dismutase gene (SOD1). In a cellular model of this disease it was found that NRF2 and activation of NRF2 driven genes were down-regulated by mutated SOD1 (75). In support, over-expression of NRF2, specifically in astrocytes, delayed onset and extended survival in a mouse model of familial ALS (76).
3. Neuroinflammation and the NRF2 system
The transcription factor NF-κB is a key regulator of inflammation, which also suppresses the transcription of ARE-dependent genes (77). Under physiological conditions, NF-κB is inactive and sequestered in the cytoplasm by binding to the inhibitory kinase, Inhibitor of nuclear factor kappa-B kinase subunit beta (IKK-β) protein. The relation between NRF2 and NF-κB is not well characterized but the identification of NF-κB binding cites in the promoter region of the Nrf2 gene suggests cross-talk between these two regulators of inflammatory processes (77). The NF-κB subunit p65 has been shown to function as a negative regulator of NRF2 activation either by depriving CREB binding protein (CBP) from NRF2 or by recruitment of histone deacetylase 3 (HDAC3), a corepressor, to ARE via interaction of and CBP or MafK, causing local histone hypoacetylation and down-regulation of NRF2-ARE signaling (78). In another study it was shown that p65 decreased NRF2 binding to its cognate DNA sequences and enhanced NRF2 ubiquitination. Nuclear translocation of KEAP1 was augmented by p65 and the studies suggested that NF-κB signaling inhibits the NRF2-ARE pathway through the interaction of p65 and KEAP1 (79). It has also been shown that KEAP1 is a negative regulator of NF-κB signaling via inhibition of IKK-β phosphorylation and by mediating IKK-β degradation by autophagocytosis (80). Further, activation of NRF2 in response to lipopolysaccharide (LPS) has been suggested to be dependent on the key innate immunity-regulating adaptor protein Myeloid differentiation primary response gene 88 (MyD88) (81). In NRF2-deficient mice, the inflammatory response in the brain and the microglia activation is much more pronounced in response to LPS when compared to normal animals (82). Thus a functional NRF2-system is important to regulate neuroinflammation in response to oxidative stress in the brain. However, emerging evidence indicates that although neuroinflammation, i.e. activation of microglia, is implicated in both acute and chronic brain diseases, it is not fully clear under which circumstances inflammation may be beneficial, deleterious or both (83).
3.1 Context dependent NRF2 regulation
We have shown that LPS-induced systemic inflammation can both reduce and aggravate neuronal damage induced by postnatal hypoxia-ischemia (84). Neuropathological outcome was assessed in neonatal rats that were subjected to LPS prior to an episode of hypoxia-ischemia. It was found that the neuropathological effects were time dependent so that an interval of 24 h between the LPS injection and hypoxia-ischemia protected the brain, whereas a 72 h interval caused enhanced brain damage. The deleterious effect was inhibited by the anti-oxidant and glutathione precursor N-acetylcysteine. Our interpretation of this study was that neonatal infection can reduce the anti-oxidant capacity via pro-inflammatory cytokines, a hypothesis which we have further explored mechanistically in vitro. Treatment of astrocytes with the pro-inflammatory cytokine TNF-α (10 ng/ml) or medium from microglia exposed to LPS (10 ng/ml) activated the NRF2-system after 24 h. This protective up-regulation of the NRF2-system was mediated by activation of several kinases including AKT, ERK and JNK (45, 85, 86) (Fig 1a). In contrast, treatment of astrocytes with medium from activated microglia or TNF-α for 72 h down-regulated proteins in the NRF2-system and prevented induction of glutathione synthesis via NRF2 (Fig 1b). Inhibitors of GSK-3β and p38 kinases blocked the detrimental effects induced by prolonged inflammation. Also HDAC activity was elevated after 72 h and the level of acetylated histone 3 was decreased in astrocytes treated with medium from activated microglia for 72 h (45). The mechanisms behind these epigenetic effects were also related to activation of GSK-3β and p38 as inhibitors of each of these kinases blocked histone deacetylation. The negative effects on NRF2 activation by these two kinases is well established by earlier work in which it was shown that the HDAC blocker TSA and the less selective blocker valproate reversed the acetylation levels, restored the inducibility of the NRF2-system and protected astrocytes from oxidative stress (87, 88) The data clearly demonstrate that prolonged inflammation can induce HDAC activation, which in turn down-regulates the NRF2-activation likely via the kinases p38 and GSK3-β. There are few previous reports on such a link, but a recent study suggests that GSK3-β can directly phosphorylate and activate one HDAC subtype, which then exerts neurotoxic effects (89). Similar, but less pronounced, effects on the NRF2 system in brains were observed in vivo in LPS injected 8 day old rats (90).
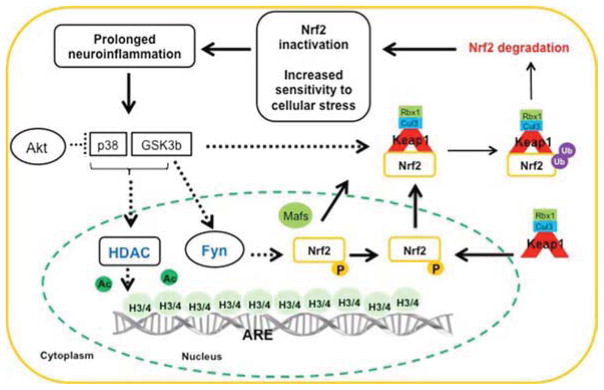
Figure 1. Regulation of NRF2 activation following acute (A) and prolonged (B) inflammation.
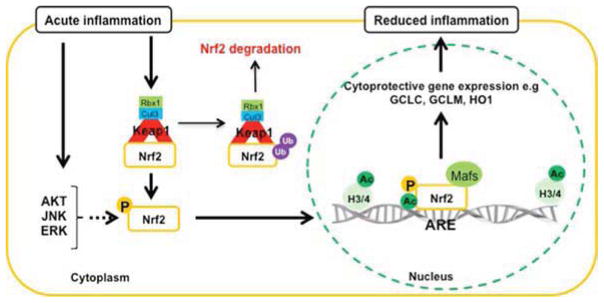
(A) Based on our studies (45, 85, 86, 90) we suggest that acute inflammation can oxidize/modify certain thiols in the KEAP1 dimer and activate kinases such as AKT, ERK and JNK. This will lead to that NRF2 escapes from being ubiquinated by Cullin-3 (Cul3) and then transported to the nucleus, a process that is promoted by phosphorylation. Inside the nucleus, NRF2 combines with proteins such as MAFs. The complex binds to certain DNA motifs in promoters, so called anti-oxidant response elements (ARE), of hundreds of protective genes. The NRF2-mediated transcription is enhanced by acetylation of histones (H3/4). The effect of acute inflammation on NRF2 activation is reduced oxidative stress by elevated levels of cytoprotective proteins and anti-oxidants such as glutathione, creating a cellular environment that will reduce inflammation. For a more complete description of established (solid arrows) and putative (dashed arrows) kinases involved in NRF2 activation during acute inflammation see main text.
(B) Prolonged neuroinflammation, i.e. overactivation of microglia and elevated levels of proinflammatory cytokines, leads to sustained activation of GSK-3β and p38 (45, 85, 86, 90). We propose that the activation of GSK-3β and p38 leads to phosphorylation of NRF2 at sites that enhance the binding to KEAP1, which will elevate the ubiquitination of NRF2 and thereby enhance degradation. GSK-3β can also phosphorylate FYN, which is transported to the nucleus. Inside the nucleus phosphorylation of NRF2 by FYN will lead to export of NRF2 out of the nucleus. Intranuclear KEAP1 can compete with binding of NRF2 to ARE, and the complex NRF2-KEAP1 can be exported to the cytosol where NRF2 is degraded. Further, decreased histone acetylation caused by elevated HDAC activity will reduce NRF2 mediated transcription. The increase in HDAC activity can be partly caused by the prolonged activation of GSK-3β and p38. The inactivation of the NRF2 system will lead to sustained and elevated activation of microglia and increased vulnerability to oxidative stress. Note the key importance of activation of AKT as a mean to reduce GSK-3β activation. For a more complete description of established (solid arrows) and putative (dashed arrows) kinases involved in NRF2 activation during prolonged inflammation see main text.
3.2 Effect of peripheral inflammation on NRF2 regulation in CNS
There is evidence to suggest that inflammation around birth may have long-term detrimental effects on the NRF2 system. A dysfunctional NRF2 system in adulthood after prenatal inflammation was indicated by a decrease in the activity of GCL at four and seventeen months after prenatal injection of LPS (91). A lower level of the modulatory subunit of GCL paralleled the decrease in glutathione. As this subunit is a target of NRF2 activation it is possible that the reason for the low glutathione levels is related to poor NRF2 function. However, further studies are needed to support this as several other enzymes in the glutathione system were unchanged or elevated. The work of Cai and coworkers show that systemic neonatal infection, induced by LPS, leads to prolonged changes in neuroinflammation and enhanced vulnerability of the substantia nigra to environmental toxins such as rotenone (92). The mechanisms are yet to be resolved but one possibility is that the NRF2 system is down-regulated after postnatal inflammation similar to that observed with prenatal inflammation. In support of this idea, we have found that neonatal LPS, at least in the short term, suppresses NRF2 target genes in cells at the blood-cerebral spinal fluid barrier (93).
Systemic inflammation is often observed in AD patients and increased serum TNF-α levels are associated with a two-fold increase in the rate of cognitive decline (94). Other studies that support the concept that peripheral inflammation causes increased vulnerability to the brain include observations that patients with PD have inflammatory reactions within the enteric nervous system (95) and in models of PD increased vulnerability of the substantia nigra is observed in rats with ulcerative colitis (96). Thus, there are indications that peripheral inflammation can sensitize the brain to become vulnerable and increase the risk of development of neurodegenerative disease.
4. Prophylactic activation of the NRF2 system
For humans it appears that adopting a diet rich in phytochemicals, such as the Mediterranean diet, decreases the risk of developing stroke, dementia and AD (97, 98). A diet rich in strawberries, blueberries and spirulina has also been shown to reduce inflammation and oxidative stress (99, 100). Whether this is due to increased activation of NRF2 in the brain is not known. It is unclear whether it is even necessary that the phytochemicals enter the brain. As described above, brain inflammation may be partly driven from peripheral inflammation and reducing peripheral inflammation by dietary means (101) may reduce inflammation also in brain. It should be noted also that moderate exercise can activate NRF2, for example in the aging heart (102). This type of endogenous activation of the NRF2 system is attractive. It is interesting to speculate that such endogenous activation could be used in combination with an NRF2 promoting diet to reduce inflammatory reactions. In brain, activation of NRF2 could also be achieved via the increase in peripheral IGF-1 that enters the brain after exercise (103). IGF-1 may be one beneficial mediator of exercise on brain health, via activation of NRF2 by promoting increased AKT (104) and decreased GSK-3β activity (88). Another more direct link between neuronal activity and NRF2 is that activation of the muscarinic receptor M1 activates NRF2 partly by inhibition of GSK-3β (105).
5. Restoring decreased NRF2 function
Designing therapies that aim to elevate NRF2 function are likely to be more beneficial if the underlying mechanisms for dysregulation can be deciphered. There is even a potential risk that NRF2 activators can be harmful if used in high concentrations in tissues that are deficient in NRF2. The basis for such detrimental effects is hormesis in which a low concentration of a substance such as phytochemicals present in fruit and vegetables results in activation and protection but a high dose can result in toxicity (106). The dose response curve could be shifted in diseased tissue so that a normally beneficial concentration of phytochemical becomes toxic. An example of this is the effect of the NRF2 activator sulphoraphane that has been described for naïve and TNF-α treated synoviocytes. The naïve cells were protected, but the TNF-α exposed cells underwent apoptosis (107). A similar type of argument concerning context biology of NRF2 in cancer was recently discussed (108). Activation of NRF2 can reduce tumorigenesis in its early stages but in later stages of tumorigenesis NRF2 activation can develop malignant cancer cells resistant to treatment.
From the discussions above one reason for a decrease in the NRF2 system by inflammation in brain appears to be related to prolonged activation of p38 and GSK-3β. These kinases are activated in chronic neurodegenerative diseases and have been suggested earlier as potential targets in AD and PD (109, 110). Therefore, a therapy that is directed to increase activation of NRF2 via release from KEAP1 only may be inadequate if GSK-3β is still activated. We speculate that under such circumstances NRF2 could be exported out of the nucleus via GSK-3β, may not enter the nucleus due to p38 activity or may not be able to activate/bind to promoter sites because of deacetylated histones. Thus a combination of HDAC, p38 and/or GSK-3β inhibition in combination with NRF2 activation may be the most effective way to restore a dormant/inadequate NRF2 system and reduce inflammation in neurodegenerative diseases. As GSK-3β is inhibited by AKT phosphorylation it follows that therapies/diets/conditions that activate PI3K (upstream of AKT) may be as effective as inhibitors of GSK-3β. Interestingly, several established NRF2 activators such as curcumin, sulphoraphane, spirulina and ketonic diet, appear to have multiple positive effects on the regulatory machinery of NRF2. For example, the turmeric ingredient curcumin has been shown to inactivate GSK-3β, inhibit DNA methyltransferase, modulate HDACs and miRNAs (111, 112). Likewise, the broccoli component sulforaphane is a HDAC inhibitor (113) and a spirulina extract reduces the expression of HDACs (114). A neuroprotective ketogenic diet activates NRF2 nuclear accumulation in both the hippocampus and liver (115) and produces ketone bodies that act as HDACs (116). This is in stark contrast to a high lard diet, which reduces NRF2 in hippocampus (117). Some formulas used in traditional medicine activate NRF2-mediated antioxidant signaling pathways and thus contain potential substances for more refined NRF2 activation (118). Further research on the active substances, their safety, ability to cross the blood brain barrier and action on the key kinases and epigenetic regulators in NRF2 activation is warranted.
6. NRF2 as a pharmaceutical target
There is an increasing clinical interest in using NRF2 activators for therapeutic purposes. One approach to activate/restore the NRF2 system is to use drugs that are already used clinically and have been found to activate the NRF2 system. Examples of this group include melatonin which activates NRF2 (119) and is used in the clinic against sleep disorders in children and adolescents (120), valproate, an anti-epileptic which restores NRF2 via HDAC inhibition, and celecoxib, a COX-2 inhibitor used in arthritic patients (121). To our knowledge, none of these have been used as therapeutics with the aim to restore NRF2 levels in brain disorders. However, there are a number of NRF2 inducers that have been tested clinically. Sulforaphane-rich broccoli sprout extract is undergoing clinical evaluation in schizophrenia and autism disorders (www.ClinicalTrials.gov; NCT01716858; NCT01474993). Experimental studies have shown that the fumaric acid ester Dimethyl fumarate (DMF) protects the brain from oxidative stress via NRF2-dependent mechanisms (122). The use of DMF is approved for psoriasis and was found to have beneficial effects in relapsing-remitting multiple sclerosis in recent placebo-controlled phase II and III studies (123, 124). Further, clinical trials in the treatment of MS using DMF are ongoing (www.ClinicalTrials.gov; NCT01930708). Triterpenoids are strong inducers of NRF2-regulated genes (125), and new synthetic triterpenoids penetrate the blood-brain barrier well and have been shown to reduce both the number of CD11b positive microglia and the transcription of proinflammatory cytokines in an MPTP-model of PD, suggesting beneficial effects on the brain (126). Previous phase II clinical trials reported beneficial effects of the triterpenoid bardoxolone methyl (CDDO-Me) in ameliorating kidney function in patients with less severe clinical conditions (127). However, lately questions have arisen about the therapeutic use of CDDO-Me and a phase III clinical trial in patients at the late stage of chronic kidney disease, associated with severe renal dysfunction or cardiovascular failure, has been terminated due to safety concerns (www.ClinicalTrials.gov; NCT01351675; 99).
NRF2- based therapy is still an open field of discussion, however, the failure of CDDO-Me highlights important issues concerning the clinical use of NRF2 activators. So far results indicate that activation of NRF2 may be more successful in preventing oxidation-related pathologies than in reversing advanced pathological conditions. Thus introduction of novel NRF2-based therapies should perhaps consider a more preventive approach in the future (128).
7. Concluding remarks
NRF2 has a well-established role as the regulator of key endogenous defenses against oxidative stress in the body. Emerging evidence suggests that NRF2, in addition to its anti-oxidant functions, may also play an important role in regulating inflammation in the brain. Thus, human neurodegenerative diseases have been associated with inflammation and dysregulation of the NRF2 system, in particular sensitizing vulnerable brain regions to additional stresses. We propose that prolonged inflammation induces activation of specific kinases and epigenetic modifications, which block the NRF2 system and thereby contribute to neurological injury. Diets or behavioural changes (e.g., exercise) that boost the NRF2 system may be important avenues to reduce the risk of neurodegenerative disease.
Highlights.
Prolonged inflammation induces key kinases, p38 and GSK-3β
Prolonged inflammation activates histone deacetylases
Prolonged inflammation contributes to oxidative damage via dysregulation of Nrf2
Acknowledgments
This research received financial assistance from the Swedish Research Council (VR2009-2630, 2012-2992), a Government grant to a researcher in Public Health Service at the Sahlgrenska University Hospital (ALFGBG-142881), European Union grant FP7, (Neurobid, HEALTHF2-2009-241778), the Leducq foundation (DSRR_P34404), Åhlén-stiftelsen, Wilhelm and Martina Lundgren Foundation, Swedish Brain Foundation (FO2013-095) and the National Institutes of Health (R01 GM044842). Barbara D’Angelo was supported by Foundation Olle Engkvist Byggmästare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 2.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 4.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakata H, Niizuma K, Yoshioka H, Kim GS, Jung JE, Katsu M, Narasimhan P, Maier CM, Nishiyama Y, Chan PH. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J Neurosci. 2012;32:3462–3473. doi: 10.1523/JNEUROSCI.5686-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287:9873–9886. doi: 10.1074/jbc.M111.312694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harada N, Kanayama M, Maruyama A, Yoshida A, Tazumi K, Hosoya T, Mimura J, Toki T, Maher JM, Yamamoto M, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys. 2011;508:101–109. doi: 10.1016/j.abb.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 10.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20:3906–3917. doi: 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. 2009;122:4452–4464. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 19.Kaspar JW, Jaiswal AK. An autoregulatory loop between Nrf2 and Cul3-Rbx1 controls their cellular abundance. J Biol Chem. 2010;285:21349–21358. doi: 10.1074/jbc.M110.121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 21.Kapeta S, Chondrogianni N, Gonos ES. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem. 2010;285:8171–8184. doi: 10.1074/jbc.M109.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwak MK, Cho JM, Huang B, Shin S, Kensler TW. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic Biol Med. 2007;43:809–817. doi: 10.1016/j.freeradbiomed.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 23.Pickering AM, Linder RA, Zhang H, Forman HJ, Davies KJ. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J Biol Chem. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 25.Jyrkkanen HK, Kuosmanen S, Heinaniemi M, Laitinen H, Kansanen E, Mella-Aho E, Leinonen H, Yla-Herttuala S, Levonen AL. Novel insights into the regulation of antioxidant-response-element-mediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by Nrf2. Biochem J. 2011;440:167–174. doi: 10.1042/BJ20110526. [DOI] [PubMed] [Google Scholar]
- 26.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 28.Pi J, Bai Y, Reece JM, Williams J, Liu D, Freeman ML, Fahl WE, Shugar D, Liu J, Qu W, et al. Molecular mechanism of human Nrf2 activation and degradation: role of sequential phosphorylation by protein kinase CK2. Free Radic Biol Med. 2007;42:1797–1806. doi: 10.1016/j.freeradbiomed.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003;546:181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- 30.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, Khor TO, Li W, Ma J, Kong AN. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther. 2006;5:1918–1926. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- 31.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Z, Huang Z, Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One. 2009;4:e6588. doi: 10.1371/journal.pone.0006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keum YS, Yu S, Chang PP, Yuan X, Kim JH, Xu C, Han J, Agarwal A, Kong AN. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006;66:8804–8813. doi: 10.1158/0008-5472.CAN-05-3513. [DOI] [PubMed] [Google Scholar]
- 34.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012;32:3486–3499. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niture SK, Jain AK, Shelton PM, Jaiswal AK. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J Biol Chem. 2011;286:28821–28832. doi: 10.1074/jbc.M111.255042. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]
- 38.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 40.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8:1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 41.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212–217. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Kang J, Li H, Su J, Wu J, Xu Y, Yu H, Xiang X, Yi H, Lu Y, et al. Regulation of endoplasmic reticulum stress in rat cortex by p62/ZIP through the Keap1-Nrf2-ARE signalling pathway after transient focal cerebral ischaemia. Brain Inj. 2013;27:924–933. doi: 10.3109/02699052.2013.793397. [DOI] [PubMed] [Google Scholar]
- 45.Correa F, Mallard C, Nilsson M, Sandberg M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3beta. Neurobiol Dis. 2011;44:142–151. doi: 10.1016/j.nbd.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012;52:928–936. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su ZY, Khor TO, Shu L, Lee JH, Saw CL, Wu TY, Huang Y, Suh N, Yang CS, Conney AH, et al. Epigenetic reactivation of Nrf2 in murine prostate cancer TRAMP C1 cells by natural phytochemicals Z-ligustilide and Radix angelica sinensis via promoter CpG demethylation. Chem Res Toxicol. 2013;26:477–485. doi: 10.1021/tx300524p. [DOI] [PubMed] [Google Scholar]
- 49.Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS One. 2012;7:e51111. doi: 10.1371/journal.pone.0051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hubbs AF, Benkovic SA, Miller DB, O’Callaghan JP, Battelli L, Schwegler-Berry D, Ma Q. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 52.Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burton NC, Kensler TW, Guilarte TR. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 54.Innamorato NG, Jazwa A, Rojo AI, Garcia C, Fernandez-Ruiz J, Grochot-Przeczek A, Stachurska A, Jozkowicz A, Dulak J, Cuadrado A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS One. 2010;5:e11838. doi: 10.1371/journal.pone.0011838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin W, Wang H, Yan W, Zhu L, Hu Z, Ding Y, Tang K. Role of Nrf2 in protection against traumatic brain injury in mice. J Neurotrauma. 2009;26:131–139. doi: 10.1089/neu.2008.0655. [DOI] [PubMed] [Google Scholar]
- 58.Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol. 2010;298:F662–671. doi: 10.1152/ajprenal.00421.2009. [DOI] [PubMed] [Google Scholar]
- 59.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salminen A, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H, Alafuzoff I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2012;96:87–95. doi: 10.1016/j.pneurobio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 61.Tanji K, Maruyama A, Odagiri S, Mori F, Itoh K, Kakita A, Takahashi H, Wakabayashi K. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J Neuropathol Exp Neurol. 2013;72:18–28. doi: 10.1097/NEN.0b013e31827b5713. [DOI] [PubMed] [Google Scholar]
- 62.Sarlette A, Krampfl K, Grothe C, Neuhoff N, Dengler R, Petri S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2008;67:1055–1062. doi: 10.1097/NEN.0b013e31818b4906. [DOI] [PubMed] [Google Scholar]
- 63.Cook AL, Vitale AM, Ravishankar S, Matigian N, Sutherland GT, Shan J, Sutharsan R, Perry C, Silburn PA, Mellick GD, et al. NRF2 activation restores disease related metabolic deficiencies in olfactory neurosphere-derived cells from patients with sporadic Parkinson’s disease. PLoS One. 2011;6:e21907. doi: 10.1371/journal.pone.0021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergstrom P, Minthon L, Bogdanovic N, Andreasen N, Gustafson DR, et al. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech Ageing Dev. 2010;131:105–110. doi: 10.1016/j.mad.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 65.von Otter M, Landgren S, Nilsson S, Celojevic D, Bergstrom P, Hakansson A, Nissbrandt H, Drozdzik M, Bialecka M, Kurzawski M, et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson’s disease. BMC Med Genet. 2010;11:36. doi: 10.1186/1471-2350-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanninen K, Malm TM, Jyrkkanen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Yla-Herttuala S, Levonen AL, Koistinaho J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci. 2008;39:302–313. doi: 10.1016/j.mcn.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 67.Meng P, Yoshida H, Matsumiya T, Imaizumi T, Tanji K, Xing F, Hayakari R, Dempoya J, Tatsuta T, Aizawa-Yashiro T, et al. Carnosic acid suppresses the production of amyloid-beta 1-42 by inducing the metalloprotease gene TACE/ADAM17 in SH-SY5Y human neuroblastoma cells. Neurosci Res. 2013;75:94–102. doi: 10.1016/j.neures.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 68.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu F, Nguyen JL, Hulleman JD, Li L, Rochet JC. Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson’s disease. J Neurochem. 2008;105:2435–2453. doi: 10.1111/j.1471-4159.2008.05333.x. [DOI] [PubMed] [Google Scholar]
- 70.Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, Liby KT, Williams C, Royce D, Risingsong R, et al. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS One. 2009;4:e5757. doi: 10.1371/journal.pone.0005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rabano A, Kirik D, Cuadrado A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 72.Gan L, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci. 2012;32:17775–17787. doi: 10.1523/JNEUROSCI.3049-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin YN, Yu YV, Gundemir S, Jo C, Cui M, Tieu K, Johnson GV. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One. 2013;8:e57932. doi: 10.1371/journal.pone.0057932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M, Beal MF. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic Biol Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J, et al. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–1706. doi: 10.1093/brain/awh503. [DOI] [PubMed] [Google Scholar]
- 76.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nair S, Doh ST, Chan JY, Kong AN, Cai L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br J Cancer. 2008;99:2070–2082. doi: 10.1038/sj.bjc.6604703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta. 2008;1783:713–727. doi: 10.1016/j.bbamcr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 79.Yu M, Li H, Liu Q, Liu F, Tang L, Li C, Yuan Y, Zhan Y, Xu W, Li W, et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal. 2011;23:883–892. doi: 10.1016/j.cellsig.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 80.Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal. 2010;22:1645–1654. doi: 10.1016/j.cellsig.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 81.Kim KH, Lyu JH, Koo ST, Oh SR, Lee HK, Ahn KS, Sadikot RT, Joo M. MyD88 is a mediator for the activation of Nrf2. Biochem Biophys Res Commun. 2011;404:46–51. doi: 10.1016/j.bbrc.2010.11.051. [DOI] [PubMed] [Google Scholar]
- 82.Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 83.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 84.Wang X, Svedin P, Nie C, Lapatto R, Zhu C, Gustavsson M, Sandberg M, Karlsson JO, Romero R, Hagberg H, et al. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann Neurol. 2007;61:263–271. doi: 10.1002/ana.21066. [DOI] [PubMed] [Google Scholar]
- 85.Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M. The Nrf2-inducible antioxidant defense in astrocytes can be both up- and down-regulated by activated microglia:Involvement of p38 MAPK. Glia. 2011;59:785–799. doi: 10.1002/glia.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Correa F, Mallard C, Nilsson M, Sandberg M. Dual TNFalpha-induced effects on NRF2 mediated antioxidant defence in astrocyte-rich cultures: role of protein kinase activation. Neurochem Res. 2012;37:2842–2855. doi: 10.1007/s11064-012-0878-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naidu S, Vijayan V, Santoso S, Kietzmann T, Immenschuh S. Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J Immunol. 2009;182:7048–7057. doi: 10.4049/jimmunol.0900006. [DOI] [PubMed] [Google Scholar]
- 88.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- 89.Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Correa F, Ljunggren E, Patil J, Wang X, Hagberg H, Mallard C, Sandberg M. Time-Dependent Effects of Systemic Lipopolysaccharide Injection on Regulators of Antioxidant Defence Nrf2 and PGC-1alpha in the Neonatal Rat Brain. Neuroimmunomodulation. 2013;20:185–193. doi: 10.1159/000347161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu Y, Carvey PM, Ling Z. Altered glutathione homeostasis in animals prenatally exposed to lipopolysaccharide. Neurochem Int. 2007;50:671–680. doi: 10.1016/j.neuint.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cai Z, Fan LW, Kaizaki A, Tien LT, Ma T, Pang Y, Lin S, Lin RC, Simpson KL. Neonatal Systemic Exposure to Lipopolysaccharide Enhances Susceptibility of Nigrostriatal Dopaminergic Neurons to Rotenone Neurotoxicity in Later Life. Dev Neurosci. 2013 doi: 10.1159/000346156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D’Angelo B, Ek CJ, Sandberg M, Mallard C. Expression of the Nrf2-system at the blood-CSF barrier is modulated by neonatal inflammation and hypoxia-ischemia. J Inherit Metab Dis. 2013;36:479–490. doi: 10.1007/s10545-012-9551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–48. doi: 10.1016/j.nbd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 96.Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson’s disease. J Neurochem. 2010;114:1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 97.Lourida I, Soni M, Thompson-Coon J, Purandare N, Lang IA, Ukoumunne OC, Llewellyn DJ. Mediterranean diet, cognitive function, and dementia: a systematic review. Epidemiology. 2013;24:479–489. doi: 10.1097/EDE.0b013e3182944410. [DOI] [PubMed] [Google Scholar]
- 98.Psaltopoulou T, Sergentanis TN, Panagiotakos DB, Sergentanis IN, Kosti R, Scarmeas N. Mediterranean diet and stroke, cognitive impairment, depression: A meta-analysis. Ann Neurol. 2013 doi: 10.1002/ana.23944. [DOI] [PubMed] [Google Scholar]
- 99.Chen L, Xin X, Yuan Q, Su D, Liu W. Phytochemical properties and antioxidant capacities of various colored berries. J Sci Food Agric. 2013 doi: 10.1002/jsfa.6216. [DOI] [PubMed] [Google Scholar]
- 100.Gemma C, Mesches MH, Sepesi B, Choo K, Holmes DB, Bickford PC. Diets enriched in foods with high antioxidant activity reverse age-induced decreases in cerebellar beta-adrenergic function and increases in proinflammatory cytokines. J Neurosci. 2002;22:6114–6120. doi: 10.1523/JNEUROSCI.22-14-06114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Konstantinidou V, Covas MI, Sola R, Fito M. Up-to date knowledge on the in vivo transcriptomic effect of the Mediterranean diet in humans. Mol Nutr Food Res. 2013;57:772–783. doi: 10.1002/mnfr.201200613. [DOI] [PubMed] [Google Scholar]
- 102.Gounder SS, Kannan S, Devadoss D, Miller CJ, Whitehead KS, Odelberg SJ, Firpo MA, Paine R, 3rd, Hoidal JR, Abel ED, et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS One. 2012;7:e45697. doi: 10.1371/journal.pone.0045697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cotman CW, Berchtold NC, Christie LA. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30:464–472. doi: 10.1016/j.tins.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 104.Sequea DA, Sharma N, Arias EB, Cartee GD. Calorie restriction enhances insulin-stimulated glucose uptake and Akt phosphorylation in both fast-twitch and slow-twitch skeletal muscle of 24-month-old rats. J Gerontol A Biol Sci Med Sci. 2012;67:1279–1285. doi: 10.1093/gerona/gls085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Espada S, Rojo AI, Salinas M, Cuadrado A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J Neurochem. 2009;110:1107–1119. doi: 10.1111/j.1471-4159.2009.06208.x. [DOI] [PubMed] [Google Scholar]
- 106.Son TG, Camandola S, Mattson MP. Hormetic dietary phytochemicals. Neuromolecular Med. 2008;10:236–246. doi: 10.1007/s12017-008-8037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fragoulis A, Laufs J, Muller S, Soppa U, Siegl S, Reiss LK, Tohidnezhad M, Rosen C, Tenbrock K, Varoga D, et al. Sulforaphane has opposing effects on TNF-alpha stimulated and unstimulated synoviocytes. Arthritis Res Ther. 2012;14:R220. doi: 10.1186/ar4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yasuda S, Sugiura H, Tanaka H, Takigami S, Yamagata K. p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent Nerv Syst Agents Med Chem. 2011;11:45–59. doi: 10.2174/187152411794961040. [DOI] [PubMed] [Google Scholar]
- 110.Kanninen K, White AR, Koistinaho J, Malm T. Targeting Glycogen Synthase Kinase-3beta for Therapeutic Benefit against Oxidative Stress in Alzheimer’s Disease: Involvement of the Nrf2-ARE Pathway. Int J Alzheimers Dis. 2011;2011:985085. doi: 10.4061/2011/985085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang HC, Xu K, Jiang ZF. Curcumin-mediated neuroprotection against amyloid-beta-induced mitochondrial dysfunction involves the inhibition of GSK-3beta. J Alzheimers Dis. 2012;32:981–996. doi: 10.3233/JAD-2012-120688. [DOI] [PubMed] [Google Scholar]
- 112.Teiten MH, Dicato M, Diederich M. Curcumin as a regulator of epigenetic events. Mol Nutr Food Res. 2013 doi: 10.1002/mnfr.201300201. [DOI] [PubMed] [Google Scholar]
- 113.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 114.Ku CS, Pham TX, Park Y, Kim B, Shin MS, Kang I, Lee J. Edible blue-green algae reduce the production of pro-inflammatory cytokines by inhibiting NF-kappaB pathway in macrophages and splenocytes. Biochim Biophys Acta. 2013;1830:2981–2988. doi: 10.1016/j.bbagen.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Milder JB, Liang LP, Patel M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis. 2010;40:238–244. doi: 10.1016/j.nbd.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez-Kim SO, White CL, Purpera MN, Uranga RM, Bruce-Keller AJ, et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem. 2010;114:1581–1589. doi: 10.1111/j.1471-4159.2010.06865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang S, Penchala S, Prabhu S, Wang J, Huang Y. Molecular basis of traditional Chinese medicine in cancer chemoprevention. Curr Drug Discov Technol. 2010;7:67–75. doi: 10.2174/157016310791162794. [DOI] [PubMed] [Google Scholar]
- 119.Wang Z, Ma C, Meng CJ, Zhu GQ, Sun XB, Huo L, Zhang J, Liu HX, He WC, Shen XM, et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J Pineal Res. 2012;53:129–137. doi: 10.1111/j.1600-079X.2012.00978.x. [DOI] [PubMed] [Google Scholar]
- 120.Cummings C. Melatonin for the management of sleep disorders in children and adolescents. Paediatr Child Health. 2012;17:331–336. [PMC free article] [PubMed] [Google Scholar]
- 121.Wang JS, Ho FM, Kang HC, Lin WW, Huang KC. Celecoxib induces heme oxygenase-1 expression in macrophages and vascular smooth muscle cells via ROS-dependent signaling pathway. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:159–168. doi: 10.1007/s00210-010-0586-6. [DOI] [PubMed] [Google Scholar]
- 122.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 123.Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, Polman CH, Schmierer K, Yousry TA, Yang M, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 124.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 125.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 126.Kaidery NA, Banerjee R, Yang L, Smirnova NA, Hushpulian DM, Liby KT, Williams CR, Yamamoto M, Kensler TW, Ratan RR, et al. Targeting Nrf2-mediated gene transcription by extremely potent synthetic triterpenoids attenuate dopaminergic neurotoxicity in the MPTP mouse model of Parkinson’s disease. Antioxid Redox Signal. 2013;18:139–157. doi: 10.1089/ars.2011.4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 128.Zhang DD. Bardoxolone brings Nrf2-based therapies to light. Antioxid Redox Signal. 2013;19:517–518. doi: 10.1089/ars.2012.5118. [DOI] [PMC free article] [PubMed] [Google Scholar]