

Abstract
Recent studies have identified the human genes involved in the biosynthesis of the molybdenum cofactor. The human MOCS3 protein contains an N-terminal domain similar to the Escherichia coli MoeB protein and a C-terminal segment displaying similarities to the sulfurtransferase rhodanese. The MOCS3 protein is believed to catalyze both the adenylation and the subsequent generation of a thiocarboxylate group at the C terminus of the smaller subunit of molybdopterin (MPT) synthase. The MOCS3 rhodanese-like domain (MOCS3-RLD) was purified after heterologous expression in E. coli and was shown to catalyze the transfer of sulfur from thiosulfate to cyanide. In a defined in vitro system for the generation of MPT from precursor Z, the sulfurated form of MOCS3-RLD was able to provide the sulfur for the thiocarboxylation of MOCS2A, the small MPT synthase subunit in humans. Mutation of the putative persulfide-forming active-site cysteine residue C412 abolished the sulfurtransferase activity of MOCS3-RLD completely, showing the importance of this cysteine residue for catalysis. In contrast to other mammalian rhodaneses, which are mostly localized within mitochondria, MOCS3 in addition to the subunits of MPT synthase are localized in the cytosol.
Sulfur-containing biomolecules are primary cell compounds that are essential for life in all organisms, and their formation involves the introduction of sulfur atoms into metabolic precursors. The biosynthesis of many sulfur-containing biomolecules occurs by complex processes that are yet to be completely delineated. Among the metabolic pathways requiring sulfur transfer are those leading to the formation of FeS clusters, biotin, thiamine, lipoic acid, molybdopterin (MPT), and sulfur-containing bases in RNA (1). MPT, the basic component of the molybdenum cofactor (Moco), is a tricyclic pterin derivative that bears the cis-dithiolene group essential for molybdenum ligation (2). The biosynthesis of Moco is an ancient and ubiquitous pathway, because highly homologous proteins involved in Moco formation have been found in archea, bacteria, higher plants, Drosophila, and higher animals including human (3). Moco is essential for the activity of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase in humans (4). Human Moco deficiency leads to the pleiotropic loss of all these molybdoenzymes and usually progresses to death at an early age (4). The biosynthetic pathway of Moco can be divided into three steps: (i) formation of precursor Z, (ii) formation of MPT from precursor Z, and (iii) insertion of molybdenum to form Moco. Recent studies have identified the human genes involved in the biosynthesis of Moco (5). The MOCS2 locus encodes the two subunits of MPT synthase and has been shown to be bicistronic with overlapping reading frames encoding MOCS2A and MOCS2B, the congeners of Escherichia coli MoaD and MoaE (6). Human MPT synthase, like the E. coli enzyme, is a heterotetramer and is composed of two MOCS2A (≈9,700 Da) and two MOCS2B (≈20,800 Da) subunits (7). The sulfur used to generate the dithiolene moiety of MPT is carried on the MOCS2A subunit in the form of a C-terminal thiocarboxylate that must be regenerated during each catalytic cycle. So far, nothing is known about the sulfur donor or the mechanism of sulfur transfer to MPT synthase in humans. In contrast, the reaction mechanism of resulfuration of E. coli MPT synthase has been described in detail (8–11). Similar to ubiquitin-activating enzymes (E1), E. coli MoeB, the MPT synthase sulfurase, activates the C terminus of MoaD to form an acyl adenylate. Subsequently the MoaD acyl adenylate is converted to a thiocarboxylate by action of any of several NifS-like proteins using l-cysteine as the sulfur source. Sequence alignments of the human MoeB homologue MOCS3 showed that the N-terminal domain is homologous to E. coli MoeB, but an additional C-terminal domain is present in MOCS3 with homologies to rhodaneses (3). Thus, the sulfur transfer reaction in humans appears to involve different protein components compared to E. coli. Rhodaneses (thiosulfate: cyanide sulfurtransferases, E.C. 2.8.1.1) are widespread enzymes that catalyze in vitro the transfer of a sulfane sulfur atom from thiosulfate to cyanide. The biological role of rhodaneses is largely speculative, because the identification of their in vivo substrates has thus far proven unsuccessful. The information available at present points to a role for a catalytic active cysteine residue of rhodanese in sulfur ransfer. The cysteine is the first residue of a 6-aa active-site loop defining the ridge of the catalytic pocket that is expected to play a key role in substrate recognition and catalytic activity (12).
Here we describe the purification and characterization of human MOCS3 and the separate MOCS3 rhodanese-like domain (RLD) after heterologous expression in E. coli. In vitro, both proteins displayed thiosulfate sulfurtransferase activity using cyanide as sulfur acceptor. Because the N-terminal MoeB-domain of MOCS3 was found to be inactive for MPT biosynthesis, the activity of the separately purified MOCS3-RLD was characterized in detail. The sulfurated form of MOCS3-RLD was shown to be able to transfer the sulfur to MOCS2A in a defined in vitro system containing MOCS2A, MOCS2B, MoeB, and Mg-ATP. This observation provides evidence for a physiological substrate of a rhodanese-like protein in humans. Site-directed mutagenesis of cysteine residues in MOCS3-RLD showed that the active-site loop cysteine residue C412 is essential for sulfurtransferase activity.
Cellular localization of MOCS3, MOCS2A, and MOCS2B in HeLa cells as fusions to the GFP showed a cytosolic localization for all three proteins.
Materials and Methods
Bacterial Strains, Media, and Growth Conditions. The E. coli moeB(DE3) mutant strain was described in refs. 13 and 14. E. coli BL21(DE3) cells and pET15b were obtained from Novagen. pGEX-6P-1, glutathione-Sepharose resin, and PreScission protease were obtained from Amersham Pharmacia. Cell strains containing expression plasmids were grown aerobically at either 30°C (pET15b-RLD) or 22°C (pAM17) in LB medium containing 150 μg/ml ampicillin. The human cervical adenocarcinoma cell line (HeLa) were maintained in a Steri-Cult 200 (Forma Scientific) with 10% CO2, 95% humidity in DMEM (PAA Laboratories) plus 10% FCS.
Cloning, Expression, and Purification of MOCS3, MOCS3-MoeBD, MOCS3-RLD, and MOCS3-RLD Variants. The genes encoding MOCS3, the MOCS3 MoeB-like domain (MOCS3-MoeBD), and the 158 C-terminal amino acids of MOCS3 (MOCS3-RLD) were amplified by PCR. The oligonucleotide primers used were as follows: (i) 5′-GGATCCATGGCTTCCCGGGAGG and 3′-GTCGACTCAGTACTGTGGAAATGTTCC primers were used cloning MOCS3 into the BamHI and SalI site of the multiple cloning site of pGEX-6P-1, which results in an N-terminal fusion protein of MOCS3 to the GST, designated pAM17. (ii) 5′-GGTCGCCATGGCTTCCCGGG and 3′CTATACCAGGATCCTCAGTAC primers were used for cloning MOCS3 into the NcoI and BamHI sites of pET15b, resulting in pSL171. (iii) 5′-CATATGGCTTCCCGGGAGGAGGTACTCG and 3′-GGATCCTCATTCCCCGCAAGCTGCACAGTCG primers were used to clone the gene sequence of the 302 N-terminal MoeB-like domain of MOCS3 into the NdeI and BamHI sites of pET15b, resulting in an N-terminal His-tag fusion of MOCS3-MoeBD, designated pAM21. (iv) 5′-CATATGCGGCCCACTGTGACTGATCTGC and 3′-GGATCCTCAGTACTGTGGAAATGTTCC primers were used to clone the gene sequence of the 158 C-terminal amino acids of MOCS3 (MOCS3-RLD) into the NdeI and BamHI sites of pET15b, resulting in an N-terminal His-tag fusion of MOCS3-RLD, designated pET15b-RLD. By using PCR mutagenesis, amino acid exchanges C239A and C412A were introduced into MOCS3, and amino acid exchanges C316A, C324A, C365A, and C412A were introduced into MOCS3-RLD.
For expression of MOCS3, pAM17 was transformed into E. coli BL21(DE3) cells. The cells were grown in 6-liter cultures at 22°C, and expression was induced at A600 = 0.6 with 100 μM isopropyl β-d-thiogalactopyranoside. Growth was continued for 16 h, and the cells were harvested by centrifugation. The cells were resuspended in 50 ml of 10 mM sodium phosphate, 140 mM NaCl, and 2.7 mM KCl (pH 7.5) and lysed by three passages through a French pressure cell. After centrifugation, the supernatant was combined with 10 ml of glutathione-Sepharose equilibrated in the same buffer, and the slurry was stirred for 2 h at 4°C. The resin was then poured into a column and washed with 150 ml of 10 mM sodium phosphate, 140 mM NaCl, and 2.7 mM KCl (pH 8.0). After elution of the GST-MOCS3 fusion protein with 50 mM Tris·HCl and 20 mM glutathione (reduced) (pH 8.0), the buffer was changed with a PD10 column to 50 mM Tris·HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT (pH 7.5), and the GST tag was cleaved by treatment with 20 units of PreScission protease at 4°C. MOCS3 was separated from cleaved GST by chromatography on a Superose 12 gel filtration column equilibrated in 50 mM Hepes, 200 mM NaCl, and 2.5 mM DTT (pH 7.5).
For expression of MOCS3-RLD and variants, the corresponding plasmids were transformed into E. coli BL21(DE3) cells. The cells were grown in 6-liter cultures at 30°C, and expression was induced at A600 = 0.6 with 50 μM isopropyl β-d-thiogalactopyranoside. Growth was continued for 16 h, and the cells were harvested by centrifugation. The cells were resuspended in 50 ml of 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 2 mM 2-mercaptoethanol, and 1 mM thiosulfate (pH 8.0) and lysed by three passages through a French pressure cell, and, after centrifugation, the supernatant was combined with 15 ml of nickel nitrilotriacetate resin equilibrated in the same buffer and stirred for 2 h at 4°C. The resin was then poured into a column and washed with 150 ml of 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 2 mM 2-mercaptoethanol, and 1 mM thiosulfate (pH 8.0) and 150 ml of 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 2 mM 2-mercaptoethanol, and 1 mM thiosulfate (pH 8.0). Proteins were eluted with buffer containing 50 mM NaH2PO4, 300 mM NaCl, 500 mM imidazole, 2 mM 2-mercaptoethanol, and 1 mM thiosulfate (pH 8.0). Final purification of MOCS3-RLD and MOCS3-RLD-C412A was achieved by chromatography on a Superose 12 gel filtration column equilibrated in 50 mM Hepes, 200 mM NaCl, 2 mM 2-mercaptoethanol, and 1 mM thiosulfate (pH 8.0).
MOCS2A, MOCS2B, and MoeB were purified as described in refs. 7 and 10. Protein concentrations were determined by the method of Bradford or by using their calculated extinction coefficients at 280 nm.
Enzyme Assays. Nitrate reductase activity was assayed in extracts at room temperature with benzyl viologen as electron donor as described by Jones and Garland (15). One unit of nitrate reductase activity is described as the production of 1 μmol of nitrite per min per mg of protein. Human sulfite oxidase was purified after the method described in ref. 16, and activity was assayed by monitoring the reduction of cytochrome c at 550 nm.
Sulfurtransferase activity was measured by the method of Westley (17). Reaction mixtures contained 100 mM Tris-acetate, 50 mM ammonium thiosulfate, and 50 mM KCN (pH 8.6) in a volume of 500 μl. After 5–30 min, formaldehyde (250 μl, 15%) was added to quench the reaction, and color was developed by the addition of 750 μl of ferric nitrate reagent [100 g of Fe(NO3)3·9H2O and 200 ml of 65% HNO3 per 1,500 ml]. Thiocyanate (complexed with iron) was quantitated by A460 nm using ε = 4,200 M–1·cm–1.
For single turnover experiments, MOCS3-RLD was preincubated for 30 min with 50 mM thiosulfate, and excess thiosulfate was removed by gel filtration before KCN was added to the mixture.
MPT Synthase Reactions. MPT synthase reactions were performed at room temperature in a total volume of 400 μl of 100 mM Tris·HCl, pH 7.2 (7). MOCS2A-OH (18.6 μM), 3.1 μM MOCS2B, 10 μM MoeB, 10 μM MOCS3-RLD-SH or MOCS3-RLD variants, 2.5 mM MgCl2, and 2.5 mM ATP were incubated for 15 min on ice, and the formation of MPT was started by addition of 3 nM precursor Z. After 2 h, the reaction was terminated by the addition of 50 μl of acidic iodine to convert MPT to form A, which was purified as described in refs. 7 and 18.
Cellular Localization Studies. GFP fusions of MOCS3, MOCS2A, and MOCS2B were generated by amplifying cDNAs by PCR and subcloning the products into the mammalian expression vectors pEGFP-C2 and pEGFP-N2 (Clontech). HeLa cells, 1.5 × 104 per 12-well chamber slide, were transiently transfected with 4 μg of GFP–plasmid DNA by using the method based on Ca3(PO4)2. At 18–28 h after transfection, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and placed in Mowiol. Fluorescence was detected with a conventional light microscope equipped with epifluorescence (Axiophot, Zeiss) and the software program metamorph (Visiton Systems).
Results
Purification of MOCS3 and MOCS3-RLD. Eukaryotic MoeB homologues have been identified in fungi (Aspergillus nidulans, CnxF) (19), plants (Arabidopsis thaliana, Cnx5) (3), humans (MOCS3) (3), and other organisms showing C- and N-terminal extensions in comparison to E. coli MoeB. Although the ≈50 aa or longer N-terminal extensions are not conserved, the ≈158 aa C-terminal extensions show homologies to RLDs with a conserved cysteine residue proposed to be involved in sulfur transfer by formation of a cysteine persulfide (Fig. 1A).
Fig. 1.

Domain structure and purification of MOCS3 and separate MOCS3-RLD. (A) Two-domain structure of MOCS3. The MOCS3 cysteine to alanine substitutions generated in this report are indicated. The putative 6-aa active-site loop characteristic for rhodanese-like proteins is shown. (B) SDS/polyacrylamide gel (15%) analysis after different purification stages of MOCS3. Lane 1, molecular mass marker; lane 2, 20 μg of protein after glutathione-Sepharose chromatography; lane 3, 15 μg of protein after cleavage with PreScission protease; lanes 4 and 5, 15-μg fractions after gel filtration chromatography of the PreScission protease-cleaved fusion protein. (C) SDS/polyacrylamide gel (15%) analysis of purification stages of MOCS3-RLD. Lane 1, molecular mass marker; lane 2, 30 μgof E. coli extract after cell lysis; lane 3, 15 μg of protein after nickel nitrilotriacetate chromatography; lane 4, 20 μg of MOCS3-RLD after gel filtration chromatography.
Human MOCS3 is encoded by an intronless gene located on chromosome 20, and, surprisingly, no mutations in MOCS3 have been identified in Moco-deficient patients to date.
For purification of MOCS3, the corresponding gene was cloned into the pGEX-6P-1 GST-fusion vector for heterologous expression in E. coli. For separate purification of the C-terminal MOCS3-RLD, the corresponding fragment encoding the 158 C-terminal amino acids of MOCS3 was cloned into the E. coli pET15b expression vector, resulting in an N-terminal His6-tagged recombinant protein.
The soluble fraction of MOCS3 was purified on glutathione-Sepharose. After cleavage with PreScission protease and subsequent gel filtration on a Superose 12 column, two major bands were displayed on Coomassie brilliant blue R-stained SDS gels with sizes of 50 kDa and 60 kDa (Fig. 1B). Although the 50-kDa band corresponds to MOCS3 (calculated molecular mass of 49,665 Da), the 60-kDa band was identified as GroEL after N-terminal sequencing (calculated mass of 57,328 Da). All attempts to separate GroEL from MOCS3 failed, showing that the proteins form a tight complex. In general, after heterologous expression, the major MOCS3 fraction was present in inclusion bodies in the insoluble fraction. In contrast, expression of the MOCS3-RLD domain yielded a protein with an approximate monomeric mass of 18 kDa as the major soluble protein after cell lysis (Fig. 1C). This value corresponded closely to the calculated molecular mass of 19,890 Da for the His6-tagged MOCS3-RLD. The protein was purified by nickel nitrilotriacetate and Superose 12 chromatography and displayed a single band on Coomassie brilliant blue R-stained SDS gels (Fig. 1C). The elution position of MOCS3-RLD from a size exclusion column revealed that the protein exists as a monomer in solution (data not shown).
Test of Functional Complementation of an E. coli moeB Mutant Strain by MOCS3. To determine whether MOCS3 can perform the role of MoeB for Moco biosynthesis in E. coli cells, functional complementation studies of an E. coli moeB mutant strain were performed. Complementation of the E. coli mutant by the human homolog would result in the production of active nitrate reductase (15). As shown in Table 1, neither MOCS3 alone nor the N-terminal MoeB-like part of MOCS3 without the 158-aa C-terminal extension was able to complement the E. coli moeB mutant strain, because no nitrate reductase activity was detected. It was shown previously that complementation of an E. coli moaD mutant strain was only achieved when MOCS2A and MOCS2B were coexpressed with MOCS3, suggesting that E. coli MoeB is incapable of activating MOCS2A (7). To analyze whether MOCS3 is unable to activate E. coli MoaD, but is capable of activating coexpressed MOCS2A, a construct that coexpresses MOCS2A, MOCS2B, and MOCS3 (7) was tested for its ability to complement the E. coli moeB mutant strain. As shown in Table 1, using this coexpression construct nitrate reductase activity was reconstituted to 28% of that compared to the value after expression of E. coli MoeB in the mutant strain. This suggests that MOCS3 can activate MOCS2A but not endogenous E. coli MoaD, a failure that could be either due to the inability of the two proteins to favorably interact or to slight differences in the mechanism of activation and sulfur transfer to MoaD or MOCS2A.
Table 1. Nitrate reductase activity after functional complementation of an E. coli moeB– strain.
| E. coli strain | Expressed proteins | NR, units* |
|---|---|---|
| moeB(DE3) | — | ND |
| moeB(DE3)xpMW15eB† | E. coli MoeB | 1.54 |
| moeB(DE3)xpMW15eB-C187A† | E. coli MoeB-C187A | 1.12 |
| moeB(DE3)xpAM21‡ | MOCS3Δ303-460 | ND |
| moeB(DE3)xpAM17‡ | GST-MOCS3 | ND |
| moeB(DE3)xpSL171‡ | MOCS3 | ND |
| moeB(DE3)xpSL206§ | MOCS2A + MOCS2B + MOCS3 | 0.43 |
| moeB(DE3)xpSL206-C239A‡ | MOCS2A + MOCS2B + MOCS3-C239A | 0.08 |
Analysis of the Sulfurtransferase-Activity of MOCS3 and MOCS3-RLD. In vitro, sulfurtransferase activity can be measured as thiosulfate sulfurtransferase activity by using thiosulfate as sulfur donor or as mercaptopyruvate sulfurtransferase (MST) activity by using β-mercaptopyruvate as sulfur donor (20). Cyanide can be used as sulfur acceptor in vitro, yielding thiocyanate (rhodanide) and sulfite or pyruvate, respectively. To analyze the substrate specificities of MOCS3, thiosulfate, β-mercaptopyruvate, and l-cysteine were tested as sulfur sources. As shown in Fig. 2A, MOCS3 exhibited sulfurtransferase activity only with thiosulfate, clearly identifying the enzyme as thiosulfate sulfurtransferase. The thiosulfate sulfurtransferase activity of the separated MOCS3-RLD protein was comparable to that of intact MOCS3 (Fig. 2B), showing that the MoeB domain of MOCS3 is not involved in sulfur transfer in vitro. We determined the Km for thiosulfate as 0.25 mM and the Km for cyanide as 0.28 mM. The Km for thiosulfate is 16 times lower and the Km for cyanide 4.6 times higher than the values reported for bovine rhodanese (Km thiosulfate, 3.7 mM; Km cyanide, 60 μM) (21). Single-turnover experiments with sulfur-substituted enzyme gave a value of 35.7% of active enzyme that is able to form the persulfide-intermediate (data not shown). However, time course experiments showed that the enzyme was able to perform multiple turnovers (Fig. 2C). To determine the release of sulfite during the reaction, we used human sulfite oxidase in a reaction mixture containing MOCS3-RLD and thiosulfate. We were able to determine the production of 0.35 mol sulfite per mol of MOCS3-RLD in the reaction mixture (data not shown), confirming the data that only 35% of MOCS3-RLD are active. The total amount of sulfite was increased after the addition of cyanide to the reaction mixture, which is required for the turnover of MOCS3-RLD (data not shown).
Fig. 2.

Rate of thiocyanate formation by MOCS3 and MOCS3-RLD with different sulfur sources. Dependence of the rate of thiocyanate formation on the concentration of MOCS3 (A) and MOCS3-RLD (B) with 50 mM thiosulfate (squares), 50 mM β-mercaptopyruvate (dots), and 10 mM l-cysteine (triangles) as sulfur sources. (C) Time dependence of the rate of thiocyanate formation catalyzed by MOCS3 (dots) and MOCS3-RLD (squares) with thiosulfate as sulfur source.
To obtain direct evidence for the persulfide group on MOCS3-RLD, thiosulfate-treated MOCS3-RLD was analyzed by mass spectrometry. Two definite peaks with a mass difference reflecting the presence of a persulfide group were detected for thiosulfate-treated MOCS3-RLD (data not shown). However, the majority of thiosulfate-treated MOCS3-RLD was determined to be present in its unsulfurated form, showing that a significant part of the enzyme exists in an inactive form, most likely because of an incorrect folding of the protein.
Site-Directed Mutagenesis of Cysteine Residues in MOCS3-RLD. In Fig. 1A, it can be seen that the C-terminal MOCS3-RLD contains a conserved cysteine residue that is part of a highly conserved 6-aa active-site loop characteristic for rhodaneses. To ascertain whether this cysteine residue is involved in the formation of a putative persulfide group before sulfur transfer reaction, site-directed mutagenesis was performed with MOCS3-RLD to replace the cysteine residue 412 with alanine. In addition, the remaining three cysteine residues C316, C324, and C365 were also exchanged to an alanine to analyze additional effects of these amino acid substitutions for enzyme activity. As shown in Table 2, analysis of the thiosulfate sulfurtransferase activity of the MOCS3-RLD variants showed that although C316A, C324A, and C365A variants displayed comparable activities to wild-type MOCS3-RLD, variant C412A was devoid of detectable activity. This clearly identified C412 as the catalytically active cysteine residue involved in formation of the covalent persulfide intermediate.
Table 2. Thiosulfate sulfurtransferase activity and in vitro formation of MPT by MOCS3-RLD and variants.
Analysis of Sulfurtransferase Activity of MOCS3-RLD and Cysteine to Alanine Variants in an in Vitro System for the Biosynthesis of MPT. With the finding that MOCS3-RLD has thiosulfate sulfurtransferase activity, it was of interest to determine whether the persulfide sulfur can be used for the thiocarboxylation of MOCS2A. A fully defined in vitro system containing MOCS2A, MOCS2B, Mg-ATP, and sulfurated MOCS3-RLD was used to analyze the ability of MOCS3-RLD-SH to provide the sulfur for MOCS2A. MOCS3 alone was shown to be inactive in the formation of the activated acyl-adenylate form of MOCS2A because of an inactive N-terminal MoeB domain (data not shown). In addition, it was not possible to carry out such studies on the MoeB-domain of MOCS3, because attempts to express the MOCS3 MoeB-domain separately (amino acid residues 1–302) resulted in an inactive protein that precipitated during purification. Thus, for the in vitro assay, we substituted MOCS3 with E. coli MoeB because in vitro activation studies showed that MoeB is able to adenylate MOCS2A (data not shown). The thiocarboxylation of MOCS2A was assayed by its ability to convert precursor Z to MPT in conjunction with MOCS2B. MPT formation can be monitored after acidic iodine treatment and its conversion to the oxidized fluorescent degradation product form A. In addition to MOCS3-RLD, the cysteine to alanine variants were tested for their sulfurtransferase activity in this system. As shown in Table 2, the addition of precursor Z gave rise to MPT formation in this system with MOCS3-RLD-SH as sulfur donor. When MOCS3-RLD was substituted with the C412A variant, no MPT was formed. This shows the importance of residues MOCS3-C412 in the sulfur transfer reaction for the thiocarboxylation of MOCS2A. In contrast, MOCS3-RLD variants C316A, C324A, and C365A expressed the same activity as wild-type MOCS3-RLD (Table 2).
Analysis of the Importance of Residue C239 in the MOCS3 MoeB-Domain for the Sulfur Transfer Reaction. Sequence alignments showed that residue C239 in the MOCS3–MoeB domain corresponds to residue C187 in E. coli MoeB (Fig. 1A), which in the MoeB crystal structure was shown to be located in a disordered loop region in close proximity to the adenylation site (8). This cysteine residue also corresponds to the highly conserved active site cysteine residue in the ubiquitin-activating enzyme E1 family, and there was shown to be involved in thioester formation with ubiquitin. To analyze the importance of MOCS3–C239 for the sulfur transfer reaction from the MOCS3-RLD domain to MOCS2A, we exchanged this residue to an alanine. For the analysis of the ability of the MOCS3–C239A variant to complement an E. coli moeB mutant strain, a construct coexpressing MOCS2A, MOCS2B, and MOCS3–C239A was transformed into E. coli moeB mutant cells, and nitrate reductase activity was determined. The results in Table 1 show that the C239A variant was able to restore nitrate reductase activity to a level of 19% compared to wild-type MOCS3 and 5% compared to E. coli MoeB. For comparison, we also analyzed the ability of the corresponding E. coli MoeB–C187A variant to restore nitrate reductase activity in this strain. The E. coli MoeB–C187A variant expressed an ability of 73% to restore nitrate reductase activity in the E. coli moeB– strain compared to wild-type MoeB. The results show that although cysteine residue C239 is important for the activity of MOCS3, mutation of the corresponding residue C187 in E. coli MoeB results only in a slightly reduced activity of the protein variant in vivo.
Cellular Localization of MOCS3, MOCS2A, and MOCS2B. Human sulfite oxidase is located in the intermembrane space of mitochondria, whereas xanthine dehydrogenase and aldehyde oxidase are located in the cytosol. To determine the cellular localization of MOCS2A, MOCS2B, and MOCS3 in human cells, we constructed N-terminal and C-terminal fusion proteins to GFP to visualize the in vivo cellular localization of all three proteins. All six GFP fusion plasmids were transiently transfected into HeLa cell lines, and subcellular localization was visualized by fluorescent microscopy. As shown in Fig. 3A, the C-terminal MOCS3–GFP fusion proteins are diffusely distributed throughout the cytoplasm. C-terminal fusions of MOCS2B or MOCS2A to GFP showed the same localization in the cytosol (Fig. 3 B and C). However, as shown in Fig. 3C, the MOCS2A-GFP fusion was also found to be localized in the cell nucleus. N-terminal fusions of all three proteins with GFP resulted in the same cellular localization (data not shown).
Fig. 3.
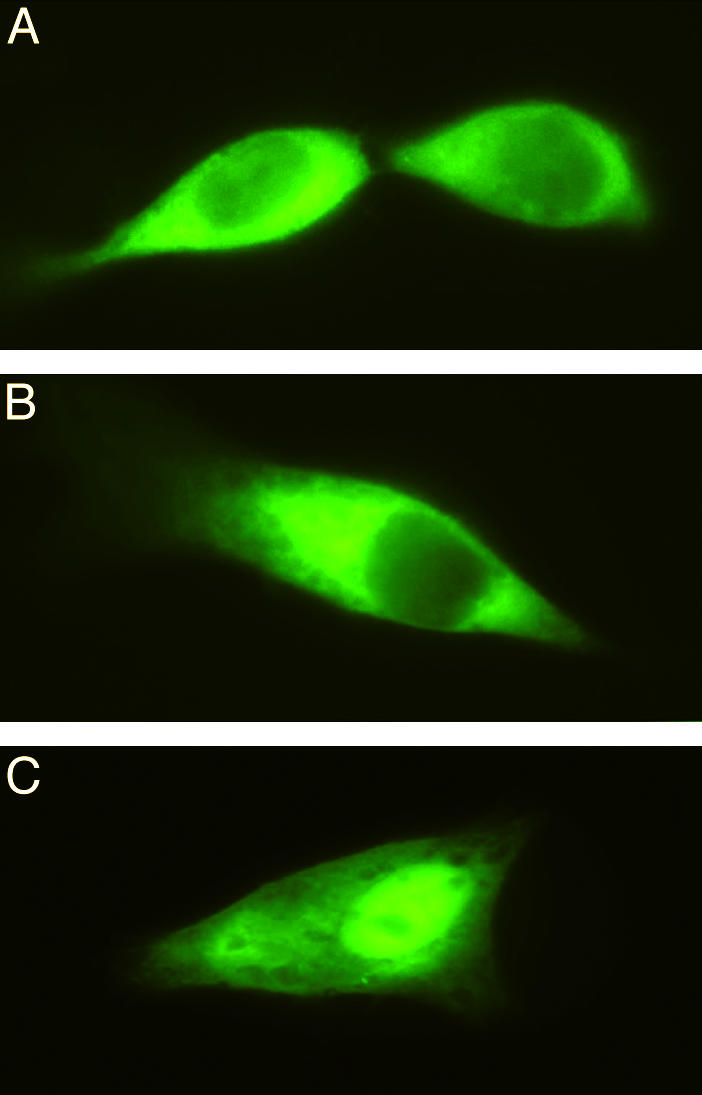
Distribution of MOCS2A, MOCS2B, and MOCS3 in HeLa cells. Fluorescence monitored after expression of MOCS3 (A), MOCS2B (B), and MOCS2A (C) C-terminal GFP fusions in transfected HeLa cells. Fluorescence was monitored with a conventional light microscope equipped with epifluorescence (Axiophot, Zeiss).
Discussion
Amino acid sequence alignments of MOCS3 showed high sequence identities to the E. coli congener MoeB. However, in addition to a 158-aa C-terminal extension displaying sequence homologies to rhodaneses, a 51-aa N-terminal extension is present in MOCS3 (Fig. 1A). To identify whether this N-terminal extension acts as a cellular localization signal, the localization of GFP fusions to MOCS3, and additionally to MOCS2A and MOCS2B, was analyzed. Because a cytosolic localization for all three proteins was identified, the role of the N-terminal amino acid extension in MOCS3 is yet to be understood. The additional nuclear localization found for MOCS2A is believed to be rather unspecific because of the small size of the MOCS2A–GFP fusion (36 kDa), which can pass through the nuclear pores without a localization signal. Because all proteins have to interact to perform the sulfur transfer reaction for the conversion of precursor Z to MPT, a congruent localization of all three proteins is expected. Moco is required for the activity of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase in humans. Although xanthine dehydrogenase was shown to be a cytosolic protein (22), sulfite oxidase, one of the major target proteins for Moco, was shown to be localized in the intermembrane space of mitochondria (23). It remains speculative whether the insertion of Moco into sulfite oxidase occurs in the cytosol or in the mitochondrial intermembrane space.
In this report, we describe the successful purification of human MOCS3 and its C-terminal RL-domain after heterologous expression in E. coli. Both proteins displayed in vitro thiosulfate sulfurtransferase activity, using cyanide as electron acceptor. However, after heterologous expression of MOCS3 in E. coli, the majority of the MOCS3 was found in the insoluble fraction as inclusion bodies. The soluble fraction of MOCS3 exhibited thiosulfate sulfurtransferase activity, but was found to be unable to adenylate MOCS2A, showing that the N-terminal MoeB-like domain of MOCS3 was in an inactive form in the purified protein. Functional complementation studies of an E. coli moeB mutant strain showed that in vivo complementation was achieved after coexpression of MOCS3 with MOCS2A and MOCS2B, revealing that MOCS3 is expressed in an at least partly active form in E. coli and loses its activity during purification. In a defined in vitro system consisting of MOCS2A-OH, MOCS2B, Mg-ATP, and precursor Z, we replaced the MOCS3-MoeB domain by E. coli MoeB and analyzed the activity of the separately purified MOCS3-RLD in detail. The in vitro system showed that the sulfurated form of MOCS3-RLD was able to act as direct sulfur donor for the thiocarboxylation of MOCS2A, the physiological substrate of MOCS3. In general, the biological role of rhodaneses is still largely debated, and it appears unlikely that the physiological sulfur acceptor is cyanide, the substrate used in the in vitro reactions. The data presented here constitute evidence for a physiological substrate of a rhodanese-like protein in humans. Site-directed mutagenesis of all cysteine residues in MOCS3-RLD showed that C412 is the only cysteine residue in the RL-domain required for sulfurtransferase activity, and is postulated to form a persulfide during catalysis. However, it remains to be determined whether thiosulfate acts as the physiological sulfur donor for MOCS3. The possibility remains that an IscS-like l-cysteine desulfurase acts as direct sulfur donor in transferring its persulfide group further on to MOCS3. However, we could show that, at least in vitro, l-cysteine and E. coli IscS were unable to substitute for thiosulfate as a sulfur donor for MOCS3-RLD (data not shown). In addition, persulfurated MOCS3-RLD was also unable to sulfurate E. coli IscS, which eliminates the possibility that IscS can act as a mediator between the N- and C-terminal domains of MOCS3 (data not shown).
To analyze whether cysteine residues in the MOCS3-MoeB domain are involved in the sulfur transfer reaction, we mutated C239 to alanine, a conserved residue corresponding to the activesite cysteine in the E1 family of enzymes and to C187 in E. coli MoeB, which in the x-ray structure of the latter was shown to be located in a disordered loop region in close proximity to the active site (8). Functional complementation studies of an E. coli moeB mutant strain with MOCS2A, MOCS2B, and MOCS3–C239A showed that the level of nitrate reductase activity was 81% reduced compared to wild-type MOCS3. In contrast, substitution of the corresponding amino acid residue C187 in E. coli MoeB resulted only in a slightly reduced activity of the protein, consistent with the results obtained from in vitro studies (10). This shows that C239 is more important for the sulfur transfer reaction in human MOCS3 than the corresponding residue in E. coli MoeB. This observation might be explained by the fact that, in humans, a rhodanese-like protein is involved in the thiocarboxylation of MPT synthase, whereas in E. coli, an l-cysteine desulfurase performs the same reaction (9).
Summarizing the results, we are proposing a sulfur transfer mechanism for the thiocarboxylation of MOCS2A by MOCS3 (Fig. 4). The persulfide group of RLD-C412 could serve as a nucleophile at the activated MOCS2A-adenylate to form a disulfide intermediate. A disulfide intermediate for sulfur transfer was shown to exist in thiamine biosynthesis between ThiS and ThiF (24), but so far remains to be identified for Moco biosynthesis. Reductive cleavage of the disulfide bond could then occur by attack of the thiol group of C239 to form a disulfide bond with C412, and, in turn, thiocarboxylated MOCS2A is generated. The disulfide bond could be reduced by a thioredoxin system in vivo (in vitro reduction by DTT is shown).
Fig. 4.

Proposed mechanism for the formation of MOCS2A-thiocarboxylate in humans. A detailed description of the mechanism is given in the text.
For the biosynthesis of thiouridine, Mueller et al. (25) postulated a mechanism in which hydrogen sulfide is generated during the sulfur transfer from ThiI to an activated uridine species. We have observed that inorganic sulfide can substitute for sulfurated MOCS3-RLD in the thiocarboxylation of MOCS2A in vitro (data not shown) as observed earlier in an in vitro system using the E. coli proteins (10).
After identifying MOCS2A as the physiological sulfur acceptor of MOCS3-RLD, the in vivo sulfur source for MOCS3 still remains to be identified. In the future, we have to elucidate the physiological sulfur donor for Moco biosynthesis and analyze whether other proteins (e.g., cysteine desulfurases) can act as sulfur donors for the formation of the persulfide group on MOCS3-RLD.
Acknowledgments
We thank Prof. B. M. Jockusch (Technical University Braunschweig) for help with cell localization and Dr. M. Nimtz (Gesellschaft für Biotechnologische Forschung Braunschweig) for mass spectrometry. This research was funded by National Institutes of Health Grant GM44283 (to K.V.R.) and Deutsche Forschungsgemeinschaft Grant Le1171/2-2 (to S.L.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MPT, molybdopterin; Moco, molybdenum cofactor; RLD, rhodanese-like domain.
References
- 1.Marquet, A. (2001) Curr. Opin. Chem. Biol. 5, 541–549. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan, K. V., Johnson, J. L. & Hainline, B. E. (1982) Fed. Proc. 41, 2608–2612. [PubMed] [Google Scholar]
- 3.Mendel, R. R. & Schwarz, G. (2002) Met. Ions Biol. Syst. 39, 317–368. [PubMed] [Google Scholar]
- 4.Johnson, J. L. & Duran, M. (2001) in The Metabolic and Molecular Bases of Inherited Disease, eds. Scriver, C. R., Beaudet, A. L., Sly, W. S., Valle, D., Childs, B. & Vogelstein, B. (McGraw-Hill, New York), 8th Ed., pp. 3163–3177.
- 5.Reiss, J. (2000) Hum. Genet. 106, 157–163. [DOI] [PubMed] [Google Scholar]
- 6.Stallmeyer, B., Drugeon, G., Reiss, J., Haenni, A. L. & Mendel, R. R. (1999) Am. J. Hum. Genet. 64, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leimkühler, S., Freuer, A., Santamaria Araujo, J. A., Rajagopalan, K. V. & Mendel, R. R. (2003) J. Biol. Chem. 278, 26127–26134. [DOI] [PubMed] [Google Scholar]
- 8.Lake, M. W., Wuebbens, M. M., Rajagopalan, K. V. & Schindelin, H. (2001) Nature 414, 325–329. [DOI] [PubMed] [Google Scholar]
- 9.Leimkühler, S. & Rajagopalan, K. V. (2001) J. Biol. Chem. 276, 22024–22031. [DOI] [PubMed] [Google Scholar]
- 10.Leimkühler, S., Wuebbens, M. M. & Rajagopalan, K. V. (2001) J. Biol. Chem. 276, 34695–34701. [DOI] [PubMed] [Google Scholar]
- 11.Wuebbens, M. M. & Rajagopalan, K. V. (2003) J. Biol. Chem. 278, 14523–14532. [DOI] [PubMed] [Google Scholar]
- 12.Bordo, D. & Bork, P. (2002) EMBO Rep. 3, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leimkühler, S. & Rajagopalan, K. V. (2001) J. Biol. Chem. 276, 1837–1844. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, M. E. & Rajagopalan, K. V. (1987) J. Bacteriol. 169, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones, R. W. & Garland, P. B. (1977) Biochem. J. 164, 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Temple, C. A. & Rajagopalan, K. V. (2000) Arch. Biochem. Biophys. 383, 281–287. [DOI] [PubMed] [Google Scholar]
- 17.Westley, J. (1973) Adv. Enzymol. 39, 327–368. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, J. L., Hainline, B. E., Rajagopalan, K. V. & Arison, B. H. (1984) J. Biol. Chem. 259, 5414–5422. [PubMed] [Google Scholar]
- 19.Appleyard, M. V. C. L., Sloan, J., Kana'n, G. J. M., Heck, I. S., Kinghorn, J. R. & Unkles, S. E. (1998) J. Biol. Chem. 273, 14869–14876. [DOI] [PubMed] [Google Scholar]
- 20.Nagahara, N., Okazaki, T. & Nishino, T. (1995) J. Biol. Chem. 270, 16230–16235. [DOI] [PubMed] [Google Scholar]
- 21.Schlesinger, P. & Westley, J. (1974) J. Biol. Chem. 249, 780–788. [PubMed] [Google Scholar]
- 22.Jarash, E.-D., Grund, C., Bruder, G., Heid, H. W., Keenan, T. W. & Franke, W. W. (1981) Cell 25, 67–82. [DOI] [PubMed] [Google Scholar]
- 23.Cohen, H. J., Betcher-Lange, S., Kessler, D. L. & Rajagopalan, K. V. (1972) J. Biol. Chem. 247, 7759–7766. [PubMed] [Google Scholar]
- 24.Xi, J., Ge, Y., Kinsland, C., McLafferty, F. & Begley, T. P. (2001) Proc. Natl. Acad. Sci. USA 98, 8513–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller, E. G., Palenchar, P. M. & Buck, C. J. (2001) J. Biol. Chem. 276, 33588–33595. [DOI] [PubMed] [Google Scholar]
- 26.Westley, J. (1981) Methods Enzymol. 77, 285–291. [DOI] [PubMed] [Google Scholar]