

Abstract
Light-induced structural changes in the bacterial reaction center were studied by a time-resolved crystallographic experiment. Crystals of protein from Blastochloris viridis (formerly Rhodopseudomonas viridis) were reconstituted with ubiquinone and analyzed by monochromatic and Laue diffraction, in the dark and 3 ms after illuminating the crystal with a pulsed laser (630 nm, 3 mJ/pulse, 7 ns duration). Refinement of monochromatic data shows that ubiquinone binds only in the “proximal” QB binding site. No significant structural difference was observed between the light and dark datasets; in particular, no quinone motion was detected. This result may be reconciled with previous studies by postulating equilibration of the “distal” and “proximal” binding sites upon extended dark adaption, and in which movement of ubiquinone is not the conformational gate for the first electron transfer between QA and QB.
Keywords: bacterial photosynthesis, secondary electron transfer, Laue diffraction, time-resolved crystallography, quinones
The photosynthetic reaction center is an integral membrane protein that converts light energy into chemical energy, by means of the separation of charge across a biological membrane. The structures of several bacterial reaction centers have been determined (1–4) and are of continuing interest due to their high degree of homology to the photosystems of higher plants (5–7). Several reviews describe the structure of the bacterial reaction center in detail (8, 9).
The terminal acceptor of electrons within the reaction center is a ubiquinone entitled QB, which binds to an active site near the cytoplasmic side of the photosynthetic membrane. While bound to the reaction center, QB accepts two electrons, which are coupled to the uptake of two protons. The transfer of one electron leads to the state P+Q–B. At room temperature and pH values ranging from 6–9, this state decays with a half-time of ≈100 ms (10, 11). Should the special pair be reduced within this time frame, the state Q–B decays with a half-time of 65 s (10). Upon transfer of a second electron, QB is released from the reaction center as dihydroquinol (12).
The mechanism of secondary electron transfer has been proposed to involve a structural change at or near the QB binding site (13–15). Recently, several crystallographic freeze-trapping experiments in reaction centers from Rhodobacter sphaeroides have suggested a significant rearrangement within the QB binding site in response to electron transfer (16–18). The ubiquinone binds in a “distal” binding site in the dark, and moves approximately 4.5 Å to a “proximal” binding site upon illumination (Fig. 1), with an accompanying 180° rotation of the aromatic ring about the isoprene tail. In these experiments, the occupancy of the QB site was increased by reconstitution with synthetic ubiquinone UQ2, which has a significantly shorter isoprenoid tail than native ubiquinone (UQ9 in Blastochloris viridis, UQ10 in R. sphaeroides).
Fig. 1.

The QB binding site and its immediate environment, showing the two positions observed for ubiquinone binding. The distal position is shown in purple, the proximal in orange. Shown are coordinates from 1AIG and 1AIJ (16). Figs. 1, 3 and 4 were prepared with pymol.
Separately, crystals of the reaction center of B. viridis were prepared in which endogenous ubiquinone was specifically depleted and reconstituted with UQ2. In this case, the refinement of a dark structure showed UQ2 to occupy only the proximal binding site (19). However, concurrent reexamination of the original structure of the B. viridis reaction center (20), which contained a minor fraction of endogenous QB, suggested partial occupancy of ubiquinone UQ9 in both the distal and proximal binding sites (21).
Two recent molecular dynamics simulations probed the position of QB in R. sphaeroides and B. viridis. In R. sphaeroides, the ubiquinone was found to occupy the distal or proximal binding sites in both neutral and semianionic states. The site occupied depended on the protonation state assigned to Glu-L212 and Asp-L213 (22). In B. viridis, spontaneous movement from the distal site to the proximal site depended on the initial orientation of the quinone in the distal site in both the PQAQB and 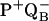 states (23). Neither simulation reproduced the 180° rotation of the aromatic ring between dark and light structures observed in the freeze-trapping experiments (16–18).
states (23). Neither simulation reproduced the 180° rotation of the aromatic ring between dark and light structures observed in the freeze-trapping experiments (16–18).
Elucidation of the structural mechanism accompanying electron transfer from QA to QB is hampered by several factors: low occupancy of or heterogeneity within the QB binding site; influence of solvent on occupancy of the proximal or distal binding site; influence of the conditions of freezing; and sensitivity to the conditions of illumination, in which the timing, intensity, and duration of illumination may lead to incomplete photoactivation or significant charge recombination between QB and the special pair.
The advent of third generation synchrotrons as sources of brilliant, pulsed, polychromatic x-ray radiation has fostered the technique of time-resolved crystallography, utilizing the Laue diffraction method (24). A laser pulse and subsequent train of synchrotron x-ray pulses represent a pump–probe experiment, in which the temporal resolution can be as short as 150 ps (25). Time-resolved crystallography with nanosecond time resolution has been successfully employed to observe the photolysis, protein relaxation, and rebinding of carbon monoxide from myoglobin (26) and the photocycle of photoactive yellow protein (27).
We are motivated by the following questions. Are the quinone motion, rotation, and accompanying structural changes reported in R. sphaeroides also observed in B. viridis? If so, with what time course do they occur? To identify the position of the ubiquinone during secondary electron transfer, we performed two experiments. First, we repeated the crystallographic refinement against monochromatic x-ray data of a dark structure of the B. viridis reaction center, complexed with a naturally occurring ubiquinone (UQ10) similar to the endogenous UQ9. Second, we utilized time-resolved crystallography to conduct a pump–probe experiment that yields a dark dataset and a light dataset after a 3-ms delay, for the B. viridis reaction center complexed with the synthetic ubiquinone UQ2. Our experiments are conducted at room temperature and thus retain the possibility of observing tertiary structural changes upon electron transfer and eliminate possible artifacts due to freezing. By employing faster excitation and a shorter delay between excitation and monitoring, we minimize the possibility of spectroscopic heterogeneity and observe a homogeneous charge-separated state within the crystal.
Experimental Procedures
Reaction Center (RC) Isolation and Crystallization. Cells of B. viridis (American Type Culture Collection; ATCC 19567) were grown semianaerobically in a rich medium modified from that originally developed for Rhodopseudomonas capsulata (28). Reaction centers were purified as described (19, 29). Final buffer contained 20 mM sodium phosphate at pH 6.8, 0.1% lauryldimethylamine oxide (LDAO) (Fluka), and 10 μM EDTA. UV/Visible absorption spectroscopy was used to judge the purity (A280/A830 ≤ 2.1) and oxidation state (cytochrome c558) of the purified protein. Protein samples were exchanged into a buffer containing 50 μM UQ2 or UQ10 prior to crystallization, and the same concentration was present in all solutions prepared thereafter. Crystals were grown in the dark at 18°C, by sitting drop vapor diffusion in 24-well plates (Hampton Research, Riverside, CA) with 20 μl of sample and 1 ml of reservoir. Conditions were as originally described (30), with the presence of triethylammonium phosphate as an additional variable (31). Diffraction quality crystals grew in 1–3 weeks in 1.8 M ammonium sulfate, 0.1% LDAO, 3% 1,2,3-heptanetriol.
X-Ray Data Collection. Prior to mounting, crystals were soaked in a buffer containing 80/20 μM potassium ferri-/ferrocyanide to oxidize the high-potential hemes in the cytochrome subunit. Crystals were then transferred to an identical soak buffer without iron to remove the oxidant, which is known to interfere with secondary electron transfer (32). Crystals were mounted in 0.7- to 1.0-mm capillaries (glass no. 50, Hampton Research), with the longer dimension of the crystals (c axis) aligned along the capillary axis. Monochromatic diffraction data were collected at BioCARS beamline 14-BM-C, Advanced Photon Source, Argonne National Laboratory, at room temperature in darkness, with an x-ray wavelength of 0.9 Å. Oscillation images (180) spaced 0.5° apart were collected on an Area Detector Systems Corporation (Poway, CA) Quantum-4 charge-coupled device detector positioned 290 mm from the crystal ensure full coverage of reciprocal space.
Laue data were collected on BioCARS beamline 14-ID-B at 15°C in darkness and 3 ms after illumination by a 7-ns laser pulse from a frequency-doubled Nd:YAG pumped dye laser at 630 nm, 3 mJ/pulse. This is the minimum optical density of the sample in the visible region, which is approximately 1 per 140 μm. The pulse was delivered to the crystal by two oppositely aligned optical fibers, focused to a spot of an ≈1-mm diameter at the sample. According to calculations, the resulting fluence should achieve ≥95% photoactivation within the crystal (Supporting Methods, which is published as supporting information on the PNAS web site). The fibers were inclined at 45° to the x-ray beam, the crystal rotation axis, and the horizontal plane. The laser pulse was synchronized to the x-ray pulse by means of a millisecond x-ray shutter, such that the laser pulse arrived at the sample 3 ms before the center of a 2-ms x-ray pulse. The x-ray pulse was derived from an undulator source that produced a 1st order maximum at 1.05 Å, with significant intensity from 0.95 to 1.4 Å, delivered into a cross-sectional area of 100 × 100 μm2. Diffraction data were collected on a MAR CCD detector positioned 200 mm from the crystal, in two sets of 11 static images spaced by 9°. The second set of images were offset from the first by crystal translation along, and rotation by 4.5° about, the goniometer axis. Depending on the size of the crystal, this method resulted in exposure of 20–30% of the crystal to the x-ray beam during collection of a complete dataset whereas the entire crystal was illuminated by the laser in every light image. Dark and light data were collected at each angular setting, with ≈3 s between each light image and the subsequent dark image to allow for recovery of the dark state.
Data Processing and Refinement. Monochromatic data were processed to 2.9 Å resolution with denzo and scalepack. Refinement was performed with cns (33), with visualization and model manipulation performed in xtalview (34). An initial model was obtained from the structure of QB-depleted RC from B. viridis, PDB code 3PRC (19). Detergent, sulfate ions, and all multiple side-chain conformations were removed. All occupancies were set to either 1 or 0, and all B-factors to 30 Å2. Following initial rigid-body refinement, several rounds of simulated annealing, minimization (maximum-likelihood function with implicit solvent mask), and model building led to a model with R (Rfree) of 0.230 (0.253). At this stage, positioning of detergent, sulfate, and QB was justified on the basis of density in the Fo – Fc map. The final stage involved restrained individual B-factor refinement followed by a final round of minimization.
Laue data was processed using laueview, which provides for indexing, integration, and scaling of Laue datasets, including harmonic deconvolution of energy overlaps (35, 36). A total of 80 images from five crystals were used to obtain a sufficiently complete dataset. Data from each crystal were integrated separately, and the resulting reflections were scaled together. During scaling, only single reflections occurring more than once were used. Single reflections observed only once were, however, included in the scaled output because these reflections could be successfully merged with reflections recovered from the harmonic deconvolution of energy overlaps. The effective resolution of the Laue datasets was 3.27 Å.
Prior to map calculation, the experimental datasets were scaled to the calculated structure factor amplitudes of the monochromatic model, omitting the ubiquinone. The phases from this omit model were used for calculation of all maps. The difference amplitudes were weighted by the factor w = 1/[1 + (ΔF/〈ΔF〉)2 + (σ/〈σ〉)2] (27). The scaling of datasets and map calculations were performed within ccp4 (37). All Laue data to 2.9Å were used for scaling, and all 39,662 reflections in common to the dark and light datasets were used for scaling and calculation of the Flight – Fdark difference electron density map.
Results
Characterization of the Sample. Oxidation of the cytochrome subunit is required for generation of the  state, which decays with a rate constant between 1 s–1 and 10 s–1 (11). Fig. 2 shows the absorption spectrum of reaction centers in solution under ambient and oxidizing conditions, and after dissolution of the crystals used in the Laue diffraction experiment. The Inset shows the expanded wavelength range around the absorption band of the bacteriopheophytin, in which the absorption of reduced hemes appears as a shoulder at 558 nm. Evidently, the conditions used in the initial crystal soak buffer were sufficient to fully oxidize the bound cytochrome subunit of the B. viridis RC, and the cytochrome subunit remained oxidized throughout the course of the Laue experiment. Furthermore, all three spectra show equivalent magnitudes of the absorption bands below 600 nm and a negligible extent of bacteriochlorophyll oxidation as judged by absorbance at 680 nm. This result demonstrates that the special pair was not oxidized by the addition of ferricyanide to the sample and that the cofactors remained intact throughout the experiment. The increased background absorption in the spectrum of dissolved crystals is due to the fact that the spectra were normalized to the absorption maxima at 600, 830, and 970 nm.
state, which decays with a rate constant between 1 s–1 and 10 s–1 (11). Fig. 2 shows the absorption spectrum of reaction centers in solution under ambient and oxidizing conditions, and after dissolution of the crystals used in the Laue diffraction experiment. The Inset shows the expanded wavelength range around the absorption band of the bacteriopheophytin, in which the absorption of reduced hemes appears as a shoulder at 558 nm. Evidently, the conditions used in the initial crystal soak buffer were sufficient to fully oxidize the bound cytochrome subunit of the B. viridis RC, and the cytochrome subunit remained oxidized throughout the course of the Laue experiment. Furthermore, all three spectra show equivalent magnitudes of the absorption bands below 600 nm and a negligible extent of bacteriochlorophyll oxidation as judged by absorbance at 680 nm. This result demonstrates that the special pair was not oxidized by the addition of ferricyanide to the sample and that the cofactors remained intact throughout the experiment. The increased background absorption in the spectrum of dissolved crystals is due to the fact that the spectra were normalized to the absorption maxima at 600, 830, and 970 nm.
Fig. 2.

Absorption spectrum of RC, 20 mM sodium phosphate, pH 6.8, 0.1% lauryldimethylamine oxide (LDAO). a, aerobic solution; b, including 80/20 μM potassium ferro-/ferricyanide; c, crystals redissolved following analysis by Laue diffraction. Absolute scale of a and b to c is 22.5:1. (Inset) Absorption spectrum from 490–580 nm, after linear baseline subtraction.
Intense synchrotron radiation is capable of photoreduction of metal centers such as those in cytochrome P450cam (38), and photoreduction of QB within the crystal may occur during crystallographic studies of the reaction center. This possibility may be discounted in our experiment for the following reasons. The high-potential hemes in the cytochrome subunit are more easily reduced than QB by reductants in solution; hence, photoreduction of the ubiquinone by radicals in solution would be preceded by photoreduction of cyt c-558, which is not observed (Fig. 2). Even if the ubiquinone were directly photoreduced within the reaction center, the electron would slowly recombine with oxidized c-558 leading to reduction of the cytochrome subunit, which again is not observed.
Charge-Neutral (Dark) Structure of the RC. Table 1 provides the parameters and statistics for the monochromatic and Laue x-ray data collection, processing, and refinement. The final structure obtained from refinement of monochromatic data, with R (Rfree) of 0.201 (0.228), varies little from the starting model. Several detergent molecules and sulfate ions could not be well refined as illustrated by uneven density or high B-factors, presumably due to the lower resolution of the dataset. Water molecules were omitted from the model.
Table 1. X-ray data collection and refinement statistics.
| PQB (Bragg dark) | PQB (Laue dark) |
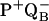 (Laue light) (Laue light) |
||
|---|---|---|---|---|
| Data collection* | ||||
| Wavelength, ņ | 0.9 | 0.9-1.4 | ||
| Data range, Å | 42-2.9 | 42-2.9 | ||
| Observations | 445,487 | 373,404 | 366,170 | |
| Unique reflections | 62,939 | 52,845 | 49,977 | |
| Redundancy‡ | 7.1 | 7.1 | 6.5 | |
| Rsym (%) | 8.7 (44.7) | 8.1 (12.1) | 9.8 (12.8) | |
| Resolution, Å | 2.9 | 3.27 | ||
| Completeness§ (%) | 99.5 (95) | 88.2 (62.3) | 83.1 (50.8) | |
| I/σ¶ | 23.0 (3.8) | 16.0 (1.9) | 12.4 (1.5) | |
| Refinement∥ | ||||
| No. of reflections used | 61,245 | 45,590 | 41,648 | |
| No. of atoms in model | 10,136 | 10,113 | 10,113 | |
| Rcryst (%) | 20.1 | 25.2 | 27.1 | |
| Rfree** (%) | 22.8 | 27.1 | 29.6 | |
| rmsd bond length, Å | 0.008 | 0.009 | 0.010 | |
| rmsd bond angles, ° | 1.5 | 1.6 | 1.6 | |
| RQAQ†† | 0.55 | — | — | |
| RQBQ†† | 0.35 | — | — | |
Crystal dimensions (μm): monochromatic, 500 × 500 × 800; Laue, 150 × 300 × 600; 250 × 350 × 800; 250 × 350 × 700; 100 × 250 × 600; 150 × 150 × 300. Space group: P43212. Unit cell dimensions: 223.5 × 223.5 × 112.5 Å.
Wavelength for Laue data collection corresponds to that used during scaling.
Laue data redundancy is calculated from single reflections used in scaling whereas completeness is calculated following harmonic deconvolution.
Completeness for the Laue datasets reported by for the restricted resolution range 42-3.27 (3.44-3.27): for the resolution range 42-2.9 (3.0-2.9) Å the completeness is 71.6% (19.5%) for the dark dataset and 65.3% (13.1%) for the light dataset.
I/σ for the Laue datasets estimated from F and σ(F), using the relations I = F2, σ(I)/I = 2σ(F)/F. At the resolution limit (3.44-3.27 Å), the values of I/σ are 4.2 and 3.2 for the dark and light datasets, respectively.
Monochromatic refinement statistics are for the final model including QB. Laue refinement statistics are from a single round of simulated annealing and minimization starting from the refined monochromatic model, omitting QB.
Rfree calculated from 10% of dataset formonochromatic refinement, from 5% of dataset for Laue refinement.
Relative Quinone Quality factor, see ref. 21.
Density for ubiquinone was clearly identified in initial omit maps as a shovel-shaped contour at 1σ (Fig. 3A). This density could not be adequately represented by an alternative model of a detergent molecule and several tightly bound water molecules. Following refinement, the quinone head-group, with the exception of the methoxy atoms O2 and CM2, is contained within the 1σ contour of electron density. The quinone is clearly bound in the proximal binding site, as seen in the previous structure of the RC-UQ2 complex (19).
Fig. 3.

Electron density for the QB binding pocket. 2Fo – Fc maps show density from a simulated-annealing omit map omitting QB (green) and density for the refined model (cyan). (A) Monochromatic dataset, 2.9-Å resolution, contour level 0.5σ.(B) Dark Laue dataset, 3.3-Å resolution, contoured at 0.3σ.
The ubiquinone in the QB site was modeled as a UQ2 molecule even though the crystal contains UQ10, with an extended isoprenyl tail. A detergent molecule occupies the QB site in the absence of quinone (19), and the disordered tail of one of the four bacteriochlorophyll molecules also intrudes into this region. Hence, the observed density is presumably a composite of these three different molecules, which we are unable to adequately model at the present resolution. Based upon a comparison of the average B-factor for the UQ2 model with the B-factors of surrounding residues (Ile-L189, His-L190, Glu-L212, Asn-L213, Phe-L216, Tyr-L222, Ser-L223, and Ile-L224), we estimate the QB site occupancy to be 0.6 ± 0.1.
Processing of Laue Datasets. The time-resolved experiment was performed on five crystals of the RC center complexed with UQ2. For each crystal, the initial dark images showed good diffraction to 2.9-Å resolution. The effect of the laser pulse was evident in the streaking and splitting of reflections in the diffraction pattern. This commonly observed phenomenon is believed to be physical rather than chemical or structural in origin. Nonuniform absorption of light due to the appreciable optical density of the crystal leads to a small, transient temperature gradient across it that dissipates in the millisecond time range (39). This effect was fully reversible; the subsequent dark pattern regained excellent reflection profiles. However, significant overall degradation of the diffraction pattern was apparent over the course of two data collection passes, most likely arising from secondary radiation damage.
In all cases, the first set of 11 images was integrated to 2.9-Å resolution, and the second set was integrated to 3.1–3.5 Å. Data were of similar quality and redundancy for the different crystals, and the parameters of the scaling such as the wavelength normalization curve were very similar. Hence, the integrated images from all crystals were scaled simultaneously. Within the redundant subset of data from a single crystal, harmonic deconvolution of energy overlaps recovered up to 10% of the total dataset, but, when all five datasets were scaled together, the increase in completeness after harmonic deconvolution was insignificant, indicating that the majority of deconvoluted reflections were successfully merged with otherwise nonredundant single reflections.
The presence of ubiquinone in the Laue crystals was confirmed for the dark dataset by calculation of a 2Fo – Fc map omitting QB from the model (Fig. 3B).
Calculation of Difference Fourier Maps. Three Flight – Fdark maps were calculated. The first (Fig. 4A) used the experimental structure factor amplitudes and errors. The second (Fig. 4B) was a “control” map in which the difference amplitudes were simply the difference in the experimental errors. The third and fourth difference maps were prepared from calculated light and dark structure factors to which the experimental errors were added. The calculated structure factors were obtained from models with two different occupancies of ubiquinone in the distal or proximal binding sites. A “worst case” map (Fig. 4C) assumed 10% occupancy of both distal and proximal binding positions in the dark and 20% occupancy of the proximal binding site after illumination.
Fig. 4.

(Flight – Fdark)·w maps of the QB-Fe-QA region. (A) Experimental structure factors. (B) Fdark = σ(Fdark), Flight = σ(Flight). (C) Simulated map: dark, 10% distal and proximal; light, 20% proximal. Positive density is shown in blue, negative density in red. Maps contoured at ±3σ.
The experimental difference map (Fig. 4A) is devoid of any significant features. That is, the positive and negative peaks are distributed essentially uniformly throughout the asymmetric unit at all contour levels. The same is true for the control map although there is a greater proportion of outlying features in this map, presumably reflecting some correlation of errors between the light and dark datasets. Both simulated difference maps show a large signal, in particular large positive and negative lobes about a nodal surface containing the C5—C6 bond of the quinone ring. The qualitative distinction between the simulated and experimental maps is revealed by the ratio of ΔFmax to ΔFrms: 9.4 and 12.4 for the experimental maps, 47 for the simulated map.
Discussion
We report here a time-resolved crystallographic structural study of the photosynthetic RC, the largest protein to be studied in this manner, and an integral membrane protein. Our monochromatic structure of the RC-UQ10 complex in B. viridis exhibits proximal binding of ubiquinone. This result is in agreement with published models 1PRC (20) and 2PRC (19). Because our monochromatic model is not significantly overdetermined, we are unable to perform the grouped occupancy refinement used in reporting partial occupancy of the distal site in the original dataset (21).
The experimental difference Fourier map (Fig. 4A) comparing the light and dark state contains no significant features. The time-resolved experiment is very sensitive to changes in electron density by virtue of the light and dark datasets being collected on the same crystals at very nearly the same time. We would readily observe motion of ≈4.5 Å postulated by wholesale displacement of ubiquinone from the distal to the proximal position, with as little as 10% change in occupancy (Fig. 4C).
Assuming the QB site to be 60% occupied on the basis of the monochromatic model, and that our method of illumination was sufficient to at least 20% photoactivation within the crystal, our result does not support a large-scale motion of QB between the PQB and P+Q–B states at pH 6.8 in B. viridis. This finding differs from observations made at pH 8.0–8.5 in the R. sphaeroides RC (16–18). However, it is in agreement with a previous attempt to measure light-induced structural changes in B. viridis using monochromatic x-ray diffraction (40) and with recent investigations of secondary electron transfer in R. sphaeroides at pH 7 by Fourier transform IR spectroscopy (41–43).
Our experimental result, however, does not contradict the existence of an energetic minimum for QB in the distal site, nor does it contradict the original conclusion that movement from the distal to the proximal position is a prerequisite for electron transfer from QA to QB (16). Our result is easily explained if the distal position is not a stable binding position on the timescale of generation 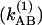 and decay
and decay  of the
of the 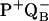 state. That is, the motion of neutral ubiquinone from the distal position to the proximal position is not reversible on the timescale of our pump–probe experiment, which is approximately 6 s from one laser pulse to the next (with an intervening dark image collected). Even if a certain amount of RCs in the crystal occupied the distal binding site, they would move to the proximal position upon the first laser flash, and remain thereafter in the proximal position. However, several other effects may also explain the discrepancy between our result obtained from RC of B. viridis and those obtained from RC of R. sphaeroides, such as sequence differences between the two species (for example residues L209 and L213) and the differing pH and ionic strength of the mother liquor in the crystallographic studies.
state. That is, the motion of neutral ubiquinone from the distal position to the proximal position is not reversible on the timescale of our pump–probe experiment, which is approximately 6 s from one laser pulse to the next (with an intervening dark image collected). Even if a certain amount of RCs in the crystal occupied the distal binding site, they would move to the proximal position upon the first laser flash, and remain thereafter in the proximal position. However, several other effects may also explain the discrepancy between our result obtained from RC of B. viridis and those obtained from RC of R. sphaeroides, such as sequence differences between the two species (for example residues L209 and L213) and the differing pH and ionic strength of the mother liquor in the crystallographic studies.
We propose a model for the first electron transfer to QB that accounts for the observation of a single binding site for ubiquinone in Fourier transform IR difference studies (41, 42), the dependence of ubiquinone position on the protonation or position of ionizable residues within the QB pocket, as suggested by simulations (22, 23, 44–46), and the observed distal binding of ubiquinone in the dark (16–18, 21). The distal binding site is a metastable binding site for neutral ubiquinone, but it does not correspond to a conformational gate in the reaction. Rather, it is a site that becomes populated during longer periods of dark adaption, such as those that occur during x-ray data collection of the charge-neutral state. Equilibration between the distal and proximal position occurs through dissociation to the solvent, or perhaps by means of an intermediate binding position, such as that observed in simulations in B. viridis (23) in which the quinone headgroup is free to rotate with respect to the isoprenyl tail.
Upon illumination, an electron arrives at QA. The proximal position is the only active position for electron transfer, as originally suggested (16). The actual conformational gate involves the response of ionizable residues near the QB pocket to the charge at QA, or to the movement of the electron to QB, and may be coupled to proton uptake as concluded in a recent Fourier transform IR study (43). Once QB becomes a semiquinone, it is tightly bound in the proximal position, in agreement with the light structures from freeze-trapping studies (16–18), where it is stabilized by additional electrostatic or hydrogenbonding interactions due to the proton uptake by surrounding residues. Meanwhile, ubiquinone is still free to exchange with the solvent or intermediate site, and so the equilibrium shifts to complete proximal binding, as described by the pull-transition suggested previously (44). This equilibration is slower than the time delay in our experiment (3 ms), but shorter than the timescale of freezing in freeze-trapping studies (≥150 ms). After charge recombination, the time for reequilibration of the distal position is slower than the time allowed for relaxation in our time-resolved crystallographic experiment (3–6 s), and also slower than the acquisition time in previous Fourier transform IR studies (23 s) (41).
Future time-resolved crystallographic studies may test this hypothesis by varying the time-delay between the laser pulse and acquisition of the light dataset, and by extending our investigation to R. sphaeroides. Crystallographic studies should also be conducted at varying pH as suggested (46), and at the same pH when comparing results from different species. Further improvement in the time-resolved technique should focus on obtaining better resolution and determining the conditions for optimal photoactivation in the crystal, and independent estimation of quinone content in the crystal. Time-resolved crystallography can thus be used to gain fresh insights into the mechanism of quinone oxidation and reduction in bioenergetic proteins.
Supplementary Material
Acknowledgments
We thank Zhong Ren for assistance in Laue data processing. Jason Key, Sudarshan Rajagopal, and Spencer Anderson are thanked for many fruitful discussions. This work was supported by Department of Energy Grant DE-FG02-96ER14675 (to J.R.N.), and by National Institutes of Health Grant GM36452 (to K.M.). BioCARS is supported by National Institutes of Health Grant RR07707 (to K.M.). R.H.G.B. was supported by the Brian Ledley Scholarship from the Gowrie Trust Fund of Australia, and Fellowship 1001774 from the Burroughs–Wellcome Interfaces Program. We acknowledge the Burroughs Wellcome Fund Interfaces Cross–Disciplinary Training Program at the University of Chicago for partial support of the Chemistry Department computer cluster.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: RC, reaction center.
Data deposition: The monochromatic model coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1R2C).
References
- 1.Deisenhofer, J., Epp, O., Miki, K., Huber, R. & Michel, H. (1984) J. Mol. Biol. 180, 385–398. [DOI] [PubMed] [Google Scholar]
- 2.Allen, J. P., Feher, G., Yeates, T. O., Rees, D. C., Deisenhofer, J., Michel, H. & Huber, R. (1986) Proc. Natl. Acad. Sci. USA 83, 8589–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang, C.-H., Tiede, D., Tang, J., Smith, U., Norris, J. & Schiffer, M. (1986) FEBS Lett. 205, 82–86. [DOI] [PubMed] [Google Scholar]
- 4.Nogi, T., Fathir, I., Kobayashi, M., Nozawa, T. & Miki, K. (2000) Proc. Natl. Acad. Sci. USA 97, 13561–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zouni, A., Witt, H.-T., Kern, J., Fromme, P., Krauss, N., Saenger, W. & Orth, P. (2001) Nature 409, 739–743. [DOI] [PubMed] [Google Scholar]
- 6.Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W. & Krauss, N. (2001) Nature 401, 909–917. [DOI] [PubMed] [Google Scholar]
- 7.Kamiya, N. & Shen, J.-R. (2003) Proc. Natl. Acad. Sci. USA 100, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michel, H. & Deisenhofer, J. (1988) Biochemistry 27, 1–7. [Google Scholar]
- 9.Lancaster, C. R. D., Ermler, U. & Michel, H. (1995) in Anoxygenic Photosynthetic Bacteria, Advances in Photosynthesis, eds. Blankenship, R. E., Madigan, M. T. & Bauer, C. E. (Kluwer, Dordrecht, The Netherlands), Vol. 2, pp. 503–526. [Google Scholar]
- 10.Shopes, R. J. & Wraight, C. A. (1985) Biochim. Biophys. Acta 806, 348–356. [DOI] [PubMed] [Google Scholar]
- 11.Gao, J.-L., Shopes, R. J. & Wraight, C. A. (1991) Biochim. Biophys. Acta 1056, 259–272. [Google Scholar]
- 12.McPherson, P. H., Okamura, M. Y. & Feher, G. (1990) Biochim. Biophys. Acta 1016, 289–292. [DOI] [PubMed] [Google Scholar]
- 13.Kleinfeld, D., Okamura, M. Y. & Feher, G. (1984) Biochemistry 23, 5780–5786. [DOI] [PubMed] [Google Scholar]
- 14.Baciou, L. & Sebban, P. (1995) Photochem. Photobiol. 62, 271–278. [Google Scholar]
- 15.Tiede, D. M., Utschig, L., Hanson, D. K. & Gallo, D. M. (1998) Photosynth. Res. 55, 267–273. [Google Scholar]
- 16.Stowell, M. H. B., McPhillips, T. M., Rees, D. C., Soltis, S. M., Abresch, E. & Feher, G. (1997) Science 276, 812–816. [DOI] [PubMed] [Google Scholar]
- 17.Axelrod, H. L., Abresch, E. C., Paddock, M. L., Okamura, M. Y. & Feher, G. (2000) Proc. Natl. Acad. Sci. USA 97, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritzsch, G., Koepke, J., Kuglstatter, A. & Baciou, L. (2002) Acta Crystallogr. D 58, 1660–1663. [DOI] [PubMed] [Google Scholar]
- 19.Lancaster, C. R. D. & Michel, H. (1997) Structure 5, 1339–1359. [DOI] [PubMed] [Google Scholar]
- 20.Deisenhofer, J., Epp, O., Sinning, I. & Michel, H. (1995) J. Mol. Biol. 246, 429–457. [DOI] [PubMed] [Google Scholar]
- 21.Lancaster, C. R. D. (1999) Biochem. Soc. Trans. 27, 591–596. [DOI] [PubMed] [Google Scholar]
- 22.Grafton, A. K. & Wheeler, R. A. (1999) Biochemistry 103, 5380–5387. [Google Scholar]
- 23.Zachariae, U. & Lancaster, C. R. D. (2001) Biochim. Biophys. Acta 1503, 280–290. [DOI] [PubMed] [Google Scholar]
- 24.Moffat, K. (1998) Acta Crystallogr. A 54, 833–841. [DOI] [PubMed] [Google Scholar]
- 25.Schotte, F., Lim, M., Jackson, T. A., Smirnov, A. V., Soman, J., Olson, J. S., Phillips, G. N., Jr., Wulff, M. & Anfinrud, P. A. (2003) Science 300, 1944–1947. [DOI] [PubMed] [Google Scholar]
- 26.Šrajer, V., Ren, Z., Teng, T.-Y., Schmidt, M., Ursby, T., Bourgeois, D., Pradervand, C., Schildkamp, W., Wulff, M. & Moffat, K. (2001) Biochemistry 40, 13802–13815. [DOI] [PubMed] [Google Scholar]
- 27.Ren, Z., Perman, B., Šrajer, V., Teng, T.-Y., Pradervand, C., Bourgeois, D., Schotte, F., Ursby, T., Kort, R., Wulff, M. & Moffat, K. (2001) Biochemistry 40, 13788–13801. [DOI] [PubMed] [Google Scholar]
- 28.Weaver, P. F., Wall, J. D. & Gest, H. (1975) Archiv. Microbiol. 105, 207–216. [DOI] [PubMed] [Google Scholar]
- 29.Fritzsch, G. (1998) Methods Enzymol. 297, 57–77. [Google Scholar]
- 30.Michel, H. (1982) J. Mol. Biol. 158, 567–572. [DOI] [PubMed] [Google Scholar]
- 31.Lancaster, C. R. D. (1996) Ph.D. thesis (Johann Wolfgang Goethe Universität, Frankfurt am Main).
- 32.Wraight, C. A., Stein, R. R., Shopes, R. J. & McComb, J. C. (1984) in Advances in Photosynthesis Research, ed. Sybesma, C. (Nijhoff/Junk, Dordrecht, The Netherlands), Vol. 1, pp. 629–636. [Google Scholar]
- 33.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et. al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 34.McRee, D. E. (1999) J. Struct. Biol. 125, 156–165. [DOI] [PubMed] [Google Scholar]
- 35.Ren, Z. & Moffat, K. (1995) J. Appl. Crystallogr. 28, 461–481. [Google Scholar]
- 36.Ren, Z. & Moffat, K. (1995) J. Appl. Crystallogr. 28, 482–493. [Google Scholar]
- 37.Bailey, S. (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 38.Schlichting, I., Berendzen, J., Chu, K., Stock, A. M., Maves, S. A., Benson, D. E., Sweet, R. E., Ringe, D., Petsko, G. A. & Sligar, S. G. (2000) Science 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- 39.Moffat, K., Chen, Y., Ng, K. M., McRee, D. & Getzoff, E. D. (1992) Philos. Trans. R. Soc. London A 340, pp. 175–190. [Google Scholar]
- 40.Buchanan, S., Michel, H. & Gerwert, K. (1990) in Reaction Centers of Photosynthetic Bacteria, Springer Series in Biophysics, ed. Michel-Beyerle, M. E. (Springer-Verlag, Berlin), Vol. 6, pp. 75–85. [Google Scholar]
- 41.Breton, J., Boullais, C., Mioskowski, C., Sebban, P., Baciou, L. & Elaine, N. (2002) Biochemistry 41, 12921–12927. [DOI] [PubMed] [Google Scholar]
- 42.Nabedryk, E., Breton, J., Sebban, P. & Baciou, L. (2003) Biochemistry 42, 5819–5827. [DOI] [PubMed] [Google Scholar]
- 43.Remy, A. & Gerwert, K. (2003) Nat. Struct. Biol. 10, 637–644. [DOI] [PubMed] [Google Scholar]
- 44.Rabenstein, B., Ullmann, G. M. & Knapp, E.-W. (2000) Biochemistry 39, 10487–10496. [DOI] [PubMed] [Google Scholar]
- 45.Walden, S. E. & Wheeler, R. A. (2002) J. Phys. Chem. B 106, 3001–3006. [Google Scholar]
- 46.Taly, A., Sebban, P., Smith, J. C. & Ullmann, G. M. (2003) Biophys. J. 84, 2090–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.