

SUMMARY
Transforming growth factor-ß (TGF-ß) is a pleiotropic cytokine with vital homeostatic functions. Aberrant TGF-ß expression or activity is implicated in the pathogenesis of fibrosis in patients with systemic sclerosis (SSc), thus TGF-ß represents a molecular therapeutic target. Multiple strategies are available for blocking the TGF-ß pathway. A monoclonal antibody targeting TGF-ß has been evaluated in a small clinical trial for early SSc with disappointing results. Antibodies to the αvß6 integrin that prevent latent TGF-ß activation, however, show promise in preclinical studies. Small molecules inhibiting TGF-ß receptor activity are effective in animal models of fibrosis. Imatinib mesylate and related tyrosine kinase inhibitors that are currently used in cancer therapy also block TGF-ß pathways and abrogate fibrotic responses. Furthermore, some commonly used drugs have shown unanticipated anti-TGF-ß activity and, therefore, could have anti-fibrotic effects. Since TGF-ß has important physiologic functions for tissue homeostasis, blocking TGF-ß activity might lead to spontaneous immune activation, epithelial hyperplasia and impaired wound healing. Loss of immune tolerance is a potential concern in an autoimmune disease such as SSc. Novel insights from microarray-based gene expression analysis and studies of genetic polymorphisms in TGF-ß signaling could aid in identifying those patients who are most likely to respond to anti-TGF-ß treatment. Anti-TGF-ß interventions promise to have a major impact on the treatment of SSc. Significant concerns regarding efficacy, safety, questions regarding appropriate dosing and timing of therapy, identification of responders, and of biomarkers of safety and efficacy are critical challenges ahead.
INTRODUCTION
The complex pathogenesis of systemic sclerosis (SSc) is dominated by progressive fibrotic replacement of normal tissue architecture. Recent research has elucidated many of the important cellular and molecular mechanisms and mediators of pathological fibrogenesis and identified a fundamental role for transforming growth factor-ß (TGF-ß) in the process.1 TGF-ß promotes fibroblast proliferation, differentiation, migration, adhesion, and survival, induces cytokine secretion, and most importantly, upregulates the synthesis of collagen and extracellular matrix.2 In light of its key role in the pathogenesis of SSc, TGF-ß has emerged as an attractive therapeutic target. Multiple strategies for blocking the TGF-ß pathways exist (Table 1), and are currently under investigation.3 Biological therapies using antibodies for neutralizing a pathogenetic ligand have proven to be highly effective for inflammatory conditions such as rheumatoid arthritis. Small molecules that can be administered orally, however, might interrupt selected TGF-ß responses without affecting the important physiological functions of this multifunctional cytokine. Although none of these anti-TGF-ß therapies have yet reached the clinic, multiple clinical trials for various indications are ongoing (www.clinicaltrials.gov). This Review summarizes the biology of TGF-ß in the context of fibrosis and the strategies for its inhibition, and highlights recent progress toward the development of anti-TGF-ß therapies for the treatment of SSc.
Table 1.
Therapeutic strategies for blocking the TGF-ß pathway.
| Strategy | Example |
|---|---|
| Block TGF-ß production or activity | Neutralizing antibodies |
| Soluble TGF-ß receptors | |
| Anti-αvß6 integrin antibodies | |
| Nucleic acid-based (antisense, ribozyme, siRNA) | |
| Block TGF-ß receptor activation | Small molecule serine-threonine kinase inhibitors |
| Block intracellular signal transduction | Endogenous inhibitors (SMAD7); tyrosine kinase inhibitors (imatinib, dasatinib) |
| Block coactivator recruitment | Peptide aptamers (thioredoxin-A SARA) |
Abbreviation TGF- ß, transforming growth factor-ß
TGF-ß IN HEALTH AND DISEASE
TGF-ß has important homeostatic roles in the control of wound healing and tissue repair, epithelial integrity, and innate and adaptive immune responses.4 Aberrant TGF-ß regulation is associated with inherited conditions such as hereditary hemorrhagic telangiectasia and Loeys–Dietz syndrome, familial pulmonary hypertension, Camurati–Engelmann disease, Marfan syndrome and fibrodysplasia ossificans progressiva, cancers both hereditary (juvenile polyposis and Cowden syndrome) and sporadic (breast, colon, lung and pancreas), and fibrosing disorders such as post-angioplasty restenosis, pulmonary fibrosis, glomerulosclerosis and SSc.5 Reduced TGF-ß signaling resulting from decreased expression of the type I TGF-ß receptor was shown to confer a significantly increased risk of colorectal cancer first in mice and then in humans (add Zeng et al., Cancer Res 2009, 69:678-686).6 The functional duality of TGF-ß was illustrated in a mouse model of autoimmunity, where it was shown to be necessary for maintaining immune tolerance while promoting tissue fibrosis.7 These conditions demonstrate that either insufficient or excessive TGF-ß activity is harmful, and therefore therapeutic targeting of TGF-ß must consider the impact of TGF-ß blockade on physiologic as well as pathological processes.
TGF-ß AND SYSTEMIC SCLEROSIS
Excessive TGF-ß activity is a common feature of a number of fibrotic conditions of diverse etiologies, making this group of disorders potential candidates for anti-TGF-ß therapies.8 The link between aberrant TGF-ß signaling and pathological fibrosis is particularly compelling in SSc. Mice with gain-of-function mutations in the TGF-ß pathway develop progressive fibrosis in multiple organs.9,10 A subset of patients with the diffuse cutaneous SSc display a ‘TGF-ß responsive gene signature’ on gene expression profiling of the lesional skin.11,12 These patients have more severe disease (higher Rodnan skin scores and greater risk of lung involvement) than those without this pattern of gene expression.13 As patients with a TGF- ß signature might be more likely to respond to anti-TGF-ß therapies, gene expression profiling with microarray analysis could be used in the future to identify those patients who would be likely to respond to therapy.
PRÉCIS: TGF-ß SIGNALING AXIS
TGF-ß is generally secreted from monocytes, lymphocytes and fibroblasts as a biologically inactive precursor protein. Activation of latent TGF-ß, a critical regulatory step in TGF-ß signaling, is catalyzed by serine proteases and thrombospondin, as well as cell surface integrins.14 The extracellular matrix, a major TGF-ß depot, sequesters the cytokine in a latent form. Intracellular signal transduction is initiated through sequential activation of the TGF-ß receptor complex and downstream intermediates (Figure 1). The canonical SMAD pathway is uniquely associated with TGF-ß signaling and is deregulated in SSc.15 Several non-SMAD signaling pathways that are shared by multiple cytokines and growth factors are also implicated in translating TGF-ß signal into relevant biological responses.16
Figure 1.
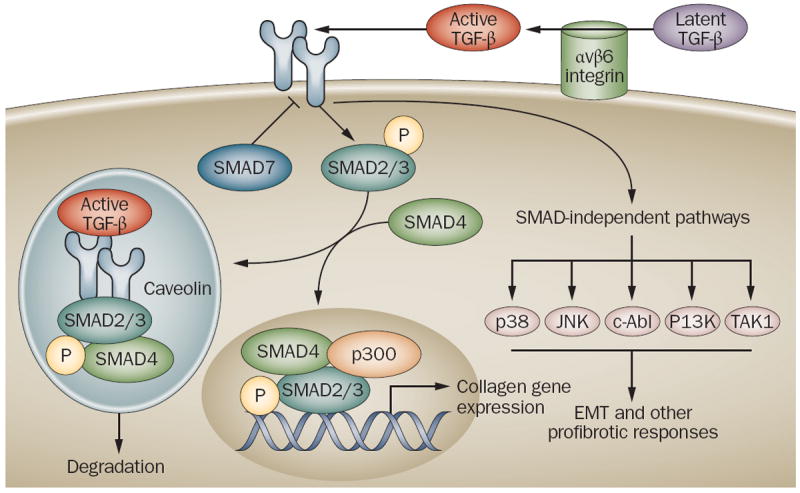
Major components of the TGF-ß signaling pathway. TGF-ß secreted from monocytes, macrophages, lymphocytes and fibroblasts is sequestered in the extracellular matrix in a biologically inactive latent form. Latent TGF-ß activation is catalyzed by αvß6 integrin on epithelial cell membranes. Active TGF-ß binds to serine/threonine kinase cell surface receptors that phosphorylate downstream SMAD2/3. Phosphorylated SMAD2/3 complexes with SMAD4 and accumulates within the nucleus, where it collaborates with other trancription factors, and recruits cofactors such as p300 to target genes, resulting in transcription. SMAD7 is a TGF-ß-inducible endogenous SMAD inhibitor that negatively regulates TGF-ß signaling. TGF-ß can also induce cellular responses such as epithelial-mesenchymal transition (EMT) via Smad-independent pathways involving the kinases Jun N-terminal kinase (JNK), p38, phosphoinositide 3 (PI3) kinase, c-Abl and TGF-ß-activated kinase TAK1. The duration of the TGF-ß signal is regulated by the uptake of the TGF-ß receptor–ligand complex into caveolin-lined endosomes that promote degradation. Excessive TGF-ß activity, or deregulated receptor trafficking or intracellular SMAD signaling result in collagen overproduction and fibrosis.
Abbreviation TGF- ß, transforming growth factor-ß
The intensity of a TGF-ß response is modulated by innate control mechanisms.17 Endogenous negative regulators of TGF-ß include SMAD7, the nuclear phosphatase PPM1A(protein phosphatase 1A [formerly 2C], magnesium-dependent, alpha isoform),18 and Man1 (also known as LEMD3), an inner nuclear membrane protein that inhibits TGF-ß signaling by sequestering SMAD2 and SMAD3 (receptor-regulated SMADs) inside the nucleus.19 Internalization of the activated TGF-ß receptor complex by caveolin-1-associated membrane lipid rafts leads to degradation of the receptor-ligand complex and cessation of TGF-ß signaling.20 Changes in caveolin-1 expression or function result in perturbed TGF-ß activity, and therefore receptor internalization represents an important regulatory mechanism. Studies in patients with SSc have revealed a marked decrease in caveolin-1 in the skin and lungs, suggesting that reduced caveolin-1 could be responsible for amplification of TGF-ß signaling contributing to progressive fibrosis in SSc. 21,22
BLOCKING THE LIGAND
Strategies to block the production and biological activity of TGF-ß include neutralizing antibodies, soluble receptors, antisense oligonucleotides and RNA interference (Figure 2). nNeutralizing antibodies to TGF-ß’ have been used in animal models to prevent organ fibrosis.23-25 In the first clinical trial of neutralizing antibodies to TGF-ß, the human monoclonal antibody, metelimumab (also known as CAT-192 OK JV), was compared with placebo in 45 patients with early SSc.26 The antibody was given by intravenous infusions at baseline and at weeks 6, 12 and 18, and patients were evaluated at 24 weeks. The trial failed to show improvements in skin scores and other disease manifestations. Adverse effects were more frequent in patients receiving CAT-192, but did not reflect enhanced autoimmunity or other spontaneous immune activation. Limitations of the study included the restricted isotype specificity of the antibody and its low binding affinity, the short treatment duration and small number of patients.
Figure 2.
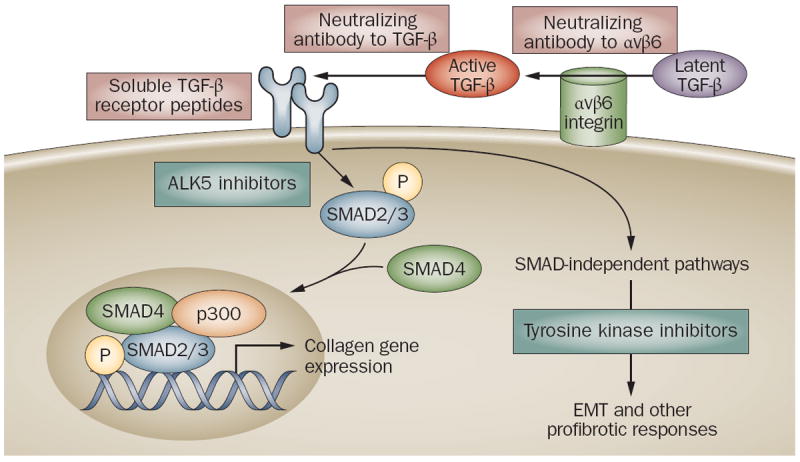
Strategies for blocking TGF-ß pathways. Biological therapies targeting TGF-ß are shown in grey boxes; small molecule therapies are shown in yellow boxes. Neutralizing antibodies to αvß6 integrin inhibit latent TGF-ß activation only at sites of injury where αvß6 integrin is expressed. Neutralizing antibodies to TGF-ß sequester the active ligand, and soluble receptor peptides prevent its binding to cell surface receptors. Small molecule kinase inhibitors of type 1 TGF-ß receptor block SMAD2/3 activation and abrogate SMAD-dependent profibrotic responses such as collagen synthesis and myofibroblast transformation. Tyrosine kinase inhibitors such as imatinib mesylate block SMAD-independent intracellular TGF-ß signaling.
Abbreviation TGF- ß, transforming growth factor-ß
A monoclonal neutralizing antibody to TGF-ß2 (lerdelimumab, CAT-152) has been tested for the prevention of scarring following glaucoma surgery.27 A pan-specific monoclonal antibody (GC1008) targeting all three TGF-ß isoforms is currently in a phase I trial for idiopathic pulmonary fibrosis.28 Disrupting TGF-ß receptor activation by sequestrating the ligand has been shown to prevent fibrosis in animal models. For instance, soluble type III TGF-ß receptor (TGFBR3) prevented diabetic glomerulosclerosis,29 and a topically administered small peptide fragment of TGFBR3 prevented bleomycin-induced SSc.30
BLOCKING TGF-ß SIGNALING[JV]
Targeting integrin function
A novel strategy for therapeutic disruption of TGF-ß involves blocking the activation of matrix-bound latent TGF-ß by targeting the αvß6 integrin, which is expressed on epithelial cells. This membrane integrin catalyzes the activation of latent TGF-ß in the local microenvironment.31 Mice with targeted deletion of αvß6 integrin developed spontaneous lung inflammation, but were protected from bleomycin-induced fibrosis.32 Recent studies demonstrated that antibodies to αvß6 integrin blocked latent TGF-ß activation and prevented the development of lung fibrosis induced by intratracheal bleomycin or radiation.33,34 A theoretical advantage of targeting αvß6 integrin is that such an approach might not interfere with homeostatic TGF-ß, since TGF-ß activation is blocked only at sites of injury where αvß6 integrin is induced.35,36
Targeting TGF-ß receptor activity
Small molecules that bind to the ATP binding domain of the serine/threonine kinase TGFBR1 prevent ligand-induced SMAD2/3 phosphorylation and consequent fibrotic responses in vitro,37,38 ameliorate experimental fibrosis in the kidneys, liver, blood vessels, and lungs, and prevent diabetic nephropathy in (db/db).39-43 Whether orally available TGFBR1 antagonists will ultimately be investigated in clinical trials for the treatment of SSc remains uncertain. The cross-reactivity characteristically associated with kinase inhibitors raises concern that TGFBR1 inhibition could disrupt multiple TGF-ß pathways, in addition to off-target effects unrelated to TGF-ß signaling, resulting in undesirable effects on immune regulation, cancer surveillance and wound healing.
Targeting intracellular signal transduction: the SMAD pathway
Strategies to disrupt intracellular SMAD signaling (Figure 2) include endogenous SMAD inhibitors, SMAD sequestration or targeting degradation.17,44 Gene transfer of inhibitory SMAD7 ameliorated pulmonary, renal and peritoneal fibrosis and vitreous retinopathy in animal models.45-48 Hepatocyte growth factor, also known as scatter factor, is a naturally occurring anti-fibrotic cytokine that works in part by inducing inhibitory SMAD7.49 A synthetic analog of hepatocyte growth factor, BB3, is undergoing preclinical evaluation for the treatment of hepatic fibrosis.50 Some of the anti-fibrotic activities of interferon-γ might also be attributed to endogenous SMAD7.51 Paclitaxel (Taxol®, Bristol-Myers-Squibb, Princeton, NJ, USA), a cancer drug that works by stabilizing the microtubules, attenuated constitutive SMAD2 activation in skin grafts of SSc patients xenotransplanted onto severe combined immunodeficient mice.52 An intriguing novel anti-TGF-ß strategy uses peptide aptamers to selectively block intracellular signal transduction. Introduction of thioredoxin-A SARA aptamers into mammalian cells blocked epithelial-to-mesenchymal transformation and related TGF-ß responses without generally inhibiting SMAD-dependent signaling.53
Targeting non-SMAD intracellular signal transduction
The panoply of non-SMAD signal transduction pathways downstream of TGF-ß (Figure 2) provides multiple opportunities for therapies targeting TGF-ß. An activating mutation of the protein tyrosine kinase c-Abelson (c-Abl), a member of the Src family, underlies the pathogenesis of chronic myelogenous leukemia (CML). In recent studies, c-Abl, was shown to be activated by TGF-ß in fibroblasts, and to mediate some of the profibrotic effects independent of SMAD signaling.54,55 Moreover, c-Abl was found to be constitutively phosphorylated in the lesional skin in patients with SSc.56,57
Imatinib mesylate (Gleevec®, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA), a selective protein tyrosine kinase inhibitor active against oncogenic Bcr-Abl, as well as the platelet-derived growth factor receptor and c-kit, is now a widely used and highly effective oral therapy for CML.58 Imatinib was shown to block the induction of c-Abl activity and fibrotic gene responses elicited by TGF-ß, and normalized collagen overproduction in explanted SSc fibroblasts.54,59, 60 The anti-TGF-ß effects of imatinib are associated with blockade of the activation of SMAD1 and early growth response early growth response factor-1.56,61 Although imatinib prevented fibrosis in the lung, kidney and skin of mice,59,62,63 it failed to arrest the progression of established lung fibrosis in animal models.62,64 It has been suggested, therefore, that the anti-fibrotic effects of imatinib might be more prophylactic than therapeutic.59,61
Anecdotal reports suggest that imatinib might be effective in ameliorating skin fibrosis in some patients with SSc, and in fibrosing conditions such as chronic graft-versus-host disease, nephrogenic fibrosing syndrome and localized forms of SSc.65-70 An intriguing study in mice demonstrated that bleomycin-induced lung injury was associated with a marked increase in serum and tissue levels of the acute phase reactant α1-acid glycoprotein (AGP), a protein known to neutralize imatinib.71 Furthermore, levels of AGP were shown to be elevated in patients with pulmonary fibrosis,71 and in the serum and bronchoalveolar lavage fluid of patients with SSc.72,73 These observations suggest that AGP mediates drug resistance, and its induction by tissue injury could account for the failure of imatinib to ameliorate fibrosis.
The therapeutic efficacy of imatinib as an anti-fibrotic agent is currently under evaluation in clinical trials. Preliminary results from a placebo-controlled multicenter clinical trial of imatinib in idiopathic pulmonary fibrosis do not indicate a significant treatment advantage (C Daniels, personal communication). The lack of detectable clinical response to imatinib in this trial could have been a result of either the incomplete blockade of TGF-ß signaling achieved with the drug doses employed, or the accumulation of AGP, which neutralizes the effects of imatinib, as discussed above. Observations in patients with CML and other forms of malignancies have shown that imatinib is generally well tolerated, although mild side effects are common.74 Of potential importance for SSc, imatinib was shown to ameliorate pulmonary arterial hypertension, a frequent complication of SSc, in mouse models of pulmonary hypertension, individual case reports, a small phase II clinical trial.75-78 Dasatanib, a novel tyrosine kinase that is active against multiple members of the Src family of kinases, as well as c-Abl and the PDGF receptor, is in early-stage clinical trials for SSc.79
In light of their relative tolerability, ease of administration, favorable pharmacokinetics, and anti-fibrotic activity predicted from in vitro experiments and animal studies, protein tyrosine kinase inhibitors are promising candidates for the treatment of various forms of fibrosis and SSc.65 Clinical experience with their use in fibrotic conditions, however, is limited and their safety profile in this setting remains unknown. Currently, therefore, we do not recommend that patients with SSc be treated with imatinib and other protein tyrosine kinase inhibitors off-label, but would encourage such patients to enroll in randomized clinical trials.
TGF-ß-induced stimulation of collagen synthesis involves chromatin remodeling, which is mediated through the recruitment of histone acetyltransferases such as p300. Accumulation of p300 on a specific gene is associated with locus-specific hyperacetylation of histone H4, resulting in enhanced gene transcription (AK Ghosh et al., personal communication). The histone deacetylase inhibitor trichostatin A, which is used for the treatment of prostate cancer, blocked in vitro the stimulatory effects of TGF-ß on collagen gene expression in cultured normal skin fibroblasts, and normalized the activated phenotype of SSc fibroblasts.81-83 These findings suggest that pharmacological modulation of histone activity could be a novel strategy in the treatment of fibrosis.
CONCERNS ABOUT INTERFERING WITH TGF-ß BIOLOGY
Given the exceptionally broad range of biological activities ascribed to TGF-ß and its fundamental physiological roles, non-selective TGF-ß blockade could have undesired consequences. Complete abrogation of TGF-ß signaling could lead to loss of immune tolerance with uncontrolled activation of T and B cells and inhibition of regulatory T cell (CD4+ CD25+) function, resulting in inflammation and spontaneous autoimmunity. Indeed, upregulated immunity induced by TGF-ß blockade could be desirable in cancer therapy.67 Interestingly, spontaneous autoimmunity has not been observed in preclinical studies with anti-TGF-ß antibodies or soluble receptors.68,69 Even in lupus-prone NZBxNZW mice, anti-TGF-ß antibody did not exacerbate autoimmunity.6 It is thought that since neutralizing antibodies, soluble receptors and natural antagonists achieve only partial TGF-ß deficiency, they interfere with excess TGF-ß activity without altering homeostatic TGF-ß signaling or abrogating pathological TGF-ß responses such as fibrosis, while preserving homeostatic functions. Long-term observation of TGF-ß blockade in clinical trials will be required to validate this concept.
PERSPECTIVES ON ANTI-TGF-ß THERAPIES FOR SYSTEMIC SCLEROSIS
Perturbed TGF-ß expression and function is a fundamental abnormality underlying the pathogenesis of distinct fibrosing disorders, and TGF-ß is the molecular target of choice for anti-fibrotic therapy. Multiple platforms for blocking TGF-ß exist in the pipeline. The pleiotropic biological activities of TGF-ß regulate both physiologic and pathological processes and can be beneficial or detrimental. Selectivity of TGF-ß inhibition could be achieved spatially by, for instance, blocking TGF-ß activation only at sites where integrin αvß6 is expressed; or by selective inhibition of deleterious effects by blocking target gene-specific coactivator interactions using aptamers. Both biological therapies and orally available small molecules to block TGF-ß are currently undergoing investigation.
In additions to statins and kinase inhibitors, a number of other drugs that are currently in clinical use show anti-TGF-ß activity (Table 2). The angiotensin II type 1 receptor blocker losartan antagonizes TGF-ß signaling through inhibition of the renin–angiotensin axis. In a mouse model of mutant fibrillin-1 that phenocopies Marfan syndrome, losartan abrogated the activation of TGF-ß signaling and restored normal tissue architecture.87,88 The anti-cancer drug paclitaxel attenuated SMAD activation in SSc tissue grafts in a human skin-severe combined immunodeficiency mouse transplantation model in vivo,52 but has been linked to SSc-like reactions in some cancer patients, and remains to be evaluated in patients with SSc.89,90 Insulin-sensitizing drugs such as rosiglitazone and pioglitazone, which are widely used in the treatment of type 2 diabetes, activate the peroxisome proliferator-activated receptor-γ. These drugs inhibit TGF-ß-induced fibrotic responses and SMAD-mediated transcription, in part by targeting early growth response protein 1 and blocking the recruitment of the p300 histone acetyltransferase coactivator.91,92 Furthermore, treatment of mice with peroxisome proliferator-activated receptor-gamma-γ ligands, such as rosiglitazone, prevents or attenuates lung fibrosis and bleomycin-induced SSc.93-95
Table 2.
Currently used drugs with anti-TGF-ß activity
| Drug | Clinical indication | Putative anti-TGF-ß mechanism |
|---|---|---|
| Losartan | hypertension | Blocks angiotensin II induction of TGF-ß; reduced levels of TGF-ß |
| AT1 receptor blocker | ||
| HMG CoA reductase inhibitors (statins) | Hypercholesterolemia | Blocks TGF-ß stimulation of collagen synthesis |
| Endothelin-1 blockers (bosentan, ambrisentan) | Pulmonary hypertension | Blocks TGF-ß signaling |
| Imatinib mesylate; related kinase inhibitors (dasatnib, nilotinib) | chronic myelogenous leukemia, GIST | Blocks SMAD-independent TGF-ß signaling, PDGF receptor activation, |
| PPAR-γ ligands (rosiglitazone, pioglitazone) | Type 2 diabetes | Disrupts intracellular TGF-ß-SMAD signal transduction |
| Tranilast (approved in Japan) | Allergy, keloid, hypertrophic scars | Inhibits TGF-ß secretion, activity |
| Paclitaxel Cancer | Cancer | Microtubule stabilization; blocks SMAD activation and SMAD-dependent responses |
Abbreviations GIST, gastrointestinal stromal tumors; PDGF, Platelet-derived growth factor; PPAR-γ, peroxisome proliferator-activated receptor-γ; TGF- ß, transforming growth factor-ß
The endothelin-1 receptor antagonist bosentan, which is used to treat idiopathic and SSc-associated pulmonary hypertension, blocks TGF-ß-mediated fibrotic responses in vitro.96 A randomized clinical trial of bosentan, however, failed to show significant benefit in patients with SSc.97 Tranilast, a synthetic tryptophan metabolite long used in Japan for the treatment of allergic condition, keloids and hypertrophic scars, was found to inhibit collagen production by cultured fibroblasts and prevent fibrosis in animal models.98 The putative mechanism of action involves reduced expression of the TGF-ß receptors and blockade of SMAD2 activation.99 The established safety profile of tranilast, together with its combined anti-fibrotic and anti-inflammatory activities, make this an appealing drug for further clinical development in SSc.
CONCLUSION
The pleiotropic roles of TGF-ß in tissue homeostasis suggest that blocking TGF-ß activity might be associated with significant toxicity. To date, however, preclinical studies have not found this to be the case, possibly because only partial abrogation of TGF-ß activity can be achieved. The modest toxicities associated with anti-TGF-ß interventions provide confidence for pursuing clinical development of such therapies for SSc. The selection of appropriate patients who would best respond, such as those with the ‘TGF-ß signature’, elevated levels of circulating TGF-ß or genetically determined elevations in basal levels of endogenous TGF-ß signaling, is a critical consideration, as is the optimal dose and route of administration. In addition, the intervention is likely to be most effective when initiated in early-stage disease when fibrosis is TGF-ß-driven and potentially reversible. Carefully designed long-term clinical trials that incorporate biomarkers of biological response and clinical efficacy will be required for evaluating novel therapies targeting the TGF-ß pathway in SSc. These studies will necessitate optimized study design, and intensive collaboration among investigators from multiple centers.
KEY POINTS.
Systemic sclerosis (SSc) is a highly heterogeneous fibrotic condition that to date has no effective disease-modifying therapy. Arresting the progression of the disease and reversing organ damage will require the use of effective anti-fibrotic therapies.
Fibrosis is associated with fibroblast activation mediated by transforming growth factor-ß (TGF-ß); therefore, blocking TGF-ß pathways is a rational approach to anti-fibrotic therapy. Gene expression analysis shows evidence of a TGF-ß-driven response in a subset of patients with diffuse cutaneous SSc.
Targeting the TGF-ß pathway using biological therapies with antibodies or soluble receptors to TGF-ß has been shown to prevent fibrosis in animal models, but has not yet been shown to be effective in patients with SSc
Small molecule inhibitors of tyrosine kinases, which are increasingly employed in cancer therapy and generally well tolerated, block c-Abl-mediated intracellular TGF-ß signaling and prevent TGF-ß-driven fibrotic responses in vitro and in vivo. Clinical trials are under way to evaluate the efficacy and safety of imatinib and dasatinib in patients with SSc.
As TGF-ß plays fundamental roles in regulating both homeostatic and pathological processes, blocking the TGF-ß pathway could be associated with adverse effects such as loss of immune tolerance and spontaneous autoimmunity, epithelial hyperplasia, and defective tissue repair.
The key challenges for the further clinical development of anti-TGF-ß therapies in systemic sclerosis include the a priori identification of responder patient subsets, delineation of the optimal timing of therapy (early disease) and dosing (continuous versus intermittent), as well as the development of biomarkers of biological response and clinical efficacy, robust study design, and network of collaborating sites.
Biographies
John Varga is an academic rheumatologist with long-standing interest in fibrosis and systemic sclerosis. His research focuses on the identification of molecular targets of therapy, and their validation in clinical trials. Recent studies from his laboratory have demonstrated the fundamental role of transforming growth factor-beta in fibrosis, and have uncovered some of the underlying signaling mechanisms.
Boris Pasche identified and characterized TGFBR1*6A, a common hypomorphic TGFBR1 variant. He was the first to propose that a combined assessment of naturally-occurring variants of the TGF-β pathway may affect disease susceptibility and outcome. He was the first to demonstrate that constitutively decreased TGFBR1 expression confers an increased risk of colorectal cancer.
Contributor Information
John Varga, Department of Medicine, Northwestern University, Feinberg School of Medicine, Chicago, IL.
Boris Pasche, Medicine and Chief of the Division of Hematology/Oncology and UAB Comprehensive Cancer Center, University of Alabama at Birmingham, AL, USA.
References
- 1.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blobe GC, et al. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 3.Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–1091. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- 4.Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through SMADs. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 5.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Valle L, et al. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–1365. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxena V, et al. Dual roles of immunoregulatory cytokine TGF-beta in the pathogenesis of autoimmunity-mediated organ damage. J Immunol. 2008;180:1903–1912. doi: 10.4049/jimmunol.180.3.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonnylal S, et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007;56:334–344. doi: 10.1002/art.22328. [DOI] [PubMed] [Google Scholar]
- 10.Wu M, Varga J. In perspective: murine models of scleroderma. Curr Rheumatol Rep. 2008;10:173–182. doi: 10.1007/s11926-008-0030-9. [DOI] [PubMed] [Google Scholar]
- 11.Whitfield ML, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100:12319–12324. doi: 10.1073/pnas.1635114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milano A, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sargent J, et al. The TGF-ß responsive gene signature identifies systemic sclerosis patients with more severe disease. Arthritis Rheum. unpublished data et al. (in revision) [Google Scholar]
- 14.Annes JP, et al. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 15.Varga J. Scleroderma and SMADs: dysfunctional SMAD family dynamics culminating in fibrosis. Arthritis Rheum. 2002;46:1703–1713. doi: 10.1002/art.10413. [DOI] [PubMed] [Google Scholar]
- 16.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 17.Derynck R, Zhang YE. SMAD-dependent and SMAD-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 18.Lin X, et al. PPM1A functions as a SMAD phosphatase to terminate TGFbeta signaling. Cell. 2006;125:915–928. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin F, et al. MAN1, an integral protein of the inner nuclear membrane, binds SMAD2 and SMAD3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet. 2005;14:437–445. doi: 10.1093/hmg/ddi040. [DOI] [PubMed] [Google Scholar]
- 20.Di Guglielmo GM, et al. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 21.Galdo FD, et al. Decreased expression of caveolin 1 in patients with systemic sclerosis: Crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008;58:2854–2865. doi: 10.1002/art.23791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tourkina E, et al. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2008;294:L843–L861. doi: 10.1152/ajplung.00295.2007. [DOI] [PubMed] [Google Scholar]
- 23.Ziyadeh FN, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCormick LL, et al. Anti-TGF-beta treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999;163:5693–5699. [PubMed] [Google Scholar]
- 25.Fukasawa H, et al. Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting SMAD/TGF-beta signaling. Kidney Int. 2004;65:63–74. doi: 10.1111/j.1523-1755.2004.00393.x. [DOI] [PubMed] [Google Scholar]
- 26.Denton CP, et al. Cat-192 Study Group; Scleroderma Clinical Trials Consortium. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–333. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- 27.Mead AL, et al. Evaluation of anti-TGF-beta2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest Ophthalmol Vis Sci. 2003;44:3394–3401. doi: 10.1167/iovs.02-0978. [DOI] [PubMed] [Google Scholar]
- 28. [20 January 2009];ClinicalTrials.gov. http://clinicaltrials.gov/ct/show/NCT00125385.
- 29.Juárez P, et al. Soluble betaglycan reduces renal damage progression in db/db mice. Am J Physiol Renal Physiol. 2007;292:F321–F329. doi: 10.1152/ajprenal.00264.2006. [DOI] [PubMed] [Google Scholar]
- 30.Santiago B, et al. Topical application of a peptide inhibitor of transforming growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J Invest Dermatol. 2005;125:450–455. doi: 10.1111/j.0022-202X.2005.23859.x. [DOI] [PubMed] [Google Scholar]
- 31.Munger JS, et al. The integrin alphavbeta6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 32.Kaminski N, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A. 2000;97:1778–1783. doi: 10.1073/pnas.97.4.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horan GS, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 34.Puthawala K, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177:82–90. doi: 10.1164/rccm.200706-806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munger JS, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 36.Horan GS, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177:56–65. doi: 10.1164/rccm.200706-805OC. [DOI] [PubMed] [Google Scholar]
- 37.Mori Y, et al. Selective inhibition of activin receptor-like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum. 2004;50:4008–4021. doi: 10.1002/art.20658. [DOI] [PubMed] [Google Scholar]
- 38.Ishida W, et al. Intracellular TGF-beta receptor blockade abrogates SMAD-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol. 2006;126:1733–1744. doi: 10.1038/sj.jid.5700303. [DOI] [PubMed] [Google Scholar]
- 39.Moon JA, et al. IN-1130, a novel transforming growth factor-beta type I receptor kinase (TGFBR1) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 70:1234–1243. doi: 10.1038/sj.ki.5001775. [DOI] [PubMed] [Google Scholar]
- 40.de Gouville AC, et al. Inhibition of TGF-beta signaling by an TGFBR1 inhibitor protects rats from dimethylnitrosamine-induced liver fibrosis. Br J Pharmacol. 145:166–177. doi: 10.1038/sj.bjp.0706172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu K, et al. SM16, an orally active TGF-beta type I receptor inhibitor prevents myofibroblast induction and vascular fibrosis in the rat carotid injury model. Arterioscler Thromb Vasc Biol. 2008;28:665–671. doi: 10.1161/ATVBAHA.107.158030. [DOI] [PubMed] [Google Scholar]
- 42.Bonniaud P, et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active TGFBR1 kinase inhibitor. Am J Respir Crit Care Med. 171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 43.Petersen M, et al. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008;73:705–715. doi: 10.1038/sj.ki.5002717. [DOI] [PubMed] [Google Scholar]
- 44.Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/SMAD signal transduction. Curr Opin Cell Biol. 2007;19:176–184. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 45.Nakao A, et al. Transient gene transfer and expression of SMAD7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nie J, et al. SMAD7 gene transfer inhibits peritoneal fibrosis. Kidney Int. 2007;72:1336–1344. doi: 10.1038/sj.ki.5002533. [DOI] [PubMed] [Google Scholar]
- 47.Saika S, et al. Effect of SMAD7 gene overexpression on transforming growth factor beta-induced retinal pigment fibrosis in a proliferative vitreoretinopathy mouse model. Arch Ophthalmol. 2007;125:647–654. doi: 10.1001/archopht.125.5.647. [DOI] [PubMed] [Google Scholar]
- 48.Lan HY. SMAD7 as a therapeutic agent for chronic kidney diseases. Front Biosci. 2008;13:4984–4992. doi: 10.2741/3057. [DOI] [PubMed] [Google Scholar]
- 49.Shukla MN, et al. Hepatocyte Growth Factor Inhibits Epithelial to Myofibroblast Transition in Lung Cells Via SMAD7. Am J Respir Cell Mol Biol. 2008 Nov 6; doi: 10.1165/rcmb.2008-0217OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angion Biomedica Corp. [20 January 2009]; http://angion.com/products.asp.
- 51.Weng H, et al. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor SMADs. J Hepatol. 2007;46:295–303. doi: 10.1016/j.jhep.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Liu X, et al. Paclitaxel modulates TGFbeta signaling in scleroderma skin grafts in immunodeficient mice. PLoS Med. 2005;2:e354. doi: 10.1371/journal.pmed.0020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao BM, Hoffmann FM. Inhibition of transforming growth factor-beta1-induced signaling and epithelial-to-mesenchymal transition by the SMAD-binding peptide aptamer Trx-SARA. Mol Biol Cell. 2006;17:3819–3831. doi: 10.1091/mbc.E05-10-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daniels CE, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishida W, et al. Novel role of c-Abl tyrosine kinase in profibrotic TGF-Beta responses: selective modulation by the anticancer drug imatinib methylate (gleevec) Arthitis Rheum. 2006;54:S776. [Google Scholar]
- 56.Bhattacharyya S, et al. c-Abl mediates profibrotic responses in fibroblasts via Egr-1: inhibition by imatinib. Oncogene. 2009 in press. [Google Scholar]
- 57.Chung L, et al. Molecular Framework for Response to Imatinib Mesylate in Systemic Sclerosis. Arthritis Rheum. 2009 doi: 10.1002/art.24221. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 59.Distler JH, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007;56:311–322. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- 60.Soria A, et al. The effect of imatinib (Glivec) on scleroderma and normal dermal fibroblasts: a preclinical study. Dermatology. 2008;216:109–117. doi: 10.1159/000111507. [DOI] [PubMed] [Google Scholar]
- 61.Pannu J, et al. SMAD1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58:2528–2537. doi: 10.1002/art.23698. [DOI] [PubMed] [Google Scholar]
- 62.Aono Y, et al. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005;171:1279–1285. doi: 10.1164/rccm.200404-531OC. [DOI] [PubMed] [Google Scholar]
- 63.Wang S, et al. Imatinib mesylate blocks a non-SMAD TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- 64.Vittal R, et al. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther. 2007;321:35–44. doi: 10.1124/jpet.106.113407. [DOI] [PubMed] [Google Scholar]
- 65.van Daele PL, et al. Is imatinib mesylate a promising drug in systemic sclerosis? Arthritis Rheum. 2008;58:2549–2552. doi: 10.1002/art.23648. [DOI] [PubMed] [Google Scholar]
- 66.Sabnani I, et al. A novel therapeutic approach to the treatment of scleroderma-associated pulmonary complications: safety and efficacy of combination therapy with imatinib and cyclophosphamide. Rheumatology (Oxford) 2009;48:49–52. doi: 10.1093/rheumatology/ken369. [DOI] [PubMed] [Google Scholar]
- 67.Sfikakis PP, et al. Imatinib for the treatment of refractory, diffuse systemic sclerosis. Rheumatology (Oxford) 2008;47:735–737. doi: 10.1093/rheumatology/ken104. [DOI] [PubMed] [Google Scholar]
- 68.Magro L, et al. Efficacy of imatinib mesylate in the treatment of refractory sclerodermatous chronic GVHD. Bone Marrow Transplant. 2008;42:757–760. doi: 10.1038/bmt.2008.252. [DOI] [PubMed] [Google Scholar]
- 69.Moreno-Romero JA, et al. Imatinib as a potential treatment for sclerodermatous chronic graft-vs-host disease. Arch Dermatol. 2008;144:1106–1109. doi: 10.1001/archderm.144.9.1106. [DOI] [PubMed] [Google Scholar]
- 70.Kay J, High WA. Imatinib mesylate treatment of nephrogenic systemic fibrosis. Arthritis Rheum. 2008;58:2543–2548. doi: 10.1002/art.23696. [DOI] [PubMed] [Google Scholar]
- 71.Azuma M, et al. Role of alpha1-acid glycoprotein in therapeutic antifibrotic effects of imatinib with macrolides in mice. Am J Respir Crit Care Med. 2007;176:1243–1250. doi: 10.1164/rccm.200702-178OC. [DOI] [PubMed] [Google Scholar]
- 72.Kucharz EJ, et al. Acute-phase proteins in patients with systemic sclerosis. Clin Rheumatol. 2000;2:165–166. doi: 10.1007/s100670050039. [DOI] [PubMed] [Google Scholar]
- 73.Fietta A, et al. Analysis of bronchoalveolar lavage fluid proteome from systemic sclerosis patients with or without functional, clinical and radiological signs of lung fibrosis. Arthritis Res Ther. 2006;8:R160. doi: 10.1186/ar2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Druker BJ, et al. IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 75.Schermuly RT, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perros F, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 77.Patterson KC, et al. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann Intern Med. 145:152–153. doi: 10.7326/0003-4819-145-2-200607180-00020. [DOI] [PubMed] [Google Scholar]
- 78.Ghofrani HA, et al. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 79.Clinical Trial Registration Number: NCT00764309
- 80.Rosenbloom J, Jiménez SA. Molecular ablation of transforming growth factor beta signaling pathways by tyrosine kinase inhibition: the coming of a promising new era in the treatment of tissue fibrosis. Arthritis Rheum. 2008;58:2219–2224. doi: 10.1002/art.23634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghosh AK, et al. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 354:420–426. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 82.Wang Y, et al. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54:2271–2279. doi: 10.1002/art.21948. [DOI] [PubMed] [Google Scholar]
- 83.Huber LC, et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007;56:2755–2764. doi: 10.1002/art.22759. [DOI] [PubMed] [Google Scholar]
- 84.Pennison M, Pasche B. Targeting transforming growth factor-beta signaling. Curr Opin Oncol. 2007;19:579–585. doi: 10.1097/CCO.0b013e3282f0ad0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruzek MC, et al. Minimal effects on immune parameters following chronic anti-TGF-beta monoclonal antibody administration to normal mice. Immunopharmacol Immunotoxicol. 2003;25:235–257. doi: 10.1081/iph-120020473. [DOI] [PubMed] [Google Scholar]
- 86.Yang YA, et al. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohn RD, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Angelis R, et al. Diffuse scleroderma occurring after the use of paclitaxel for ovarian cancer. Clin Rheumatol. 2003;22:49–52. doi: 10.1007/s10067-002-0635-8. [DOI] [PubMed] [Google Scholar]
- 90.Kupfer I, et al. Scleroderma-like cutaneous lesions induced by paclitaxel: a case study. J Am Acad Dermatol. 2003;48:279–281. doi: 10.1067/mjd.2003.30. [DOI] [PubMed] [Google Scholar]
- 91.Ghosh AK, et al. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305–1318. doi: 10.1002/art.20104. [DOI] [PubMed] [Google Scholar]
- 92.Sime PJ. The antifibrogenic potential of PPARgamma ligands in pulmonary fibrosis. J Investig Med. 2008;56:534–538. doi: 10.2310/JIM.0b013e31816464e9. [DOI] [PubMed] [Google Scholar]
- 93.Milam JE, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L891–L901. doi: 10.1152/ajplung.00333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Genovese T, et al. Effect of rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur Respir J. 2005;25:225–234. doi: 10.1183/09031936.05.00049704. [DOI] [PubMed] [Google Scholar]
- 95.Wu M, et al. Rosiglitazone modulates fibrotic responses in mice via peroxisome proliferators activated receptor gamma: implications for systemic sclerosis. Am J Pathol. 2009 in press. [Google Scholar]
- 96.Shi-wen X, et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56:4189–4194. doi: 10.1002/art.23134. [DOI] [PubMed] [Google Scholar]
- 97.Silver RM. Endothelin and scleroderma lung disease. Rheumatology (Oxford) 2008;47(Suppl 5):v25–v26. doi: 10.1093/rheumatology/ken283. [DOI] [PubMed] [Google Scholar]
- 98.Yamada H, et al. Tranilast inhibits collagen synthesis in normal, scleroderma and keloid fibroblasts at a late passage culture but not at an early passage culture. J Dermatol Sci. 1995;9:45–47. doi: 10.1016/0923-1811(94)00355-i. [DOI] [PubMed] [Google Scholar]
- 99.Platten M, et al. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) inhibits transforming growth factor-beta release and reduces migration and invasiveness of human malignant glioma cells. Int J Cancer. 2001;93:53–61. doi: 10.1002/ijc.1289. [DOI] [PubMed] [Google Scholar]