

Abstract
Clostridium botulinum encompasses bacteria that produce at least one of the seven serotypes of botulinum neurotoxin (BoNT/A-G). The availability of genome sequences of four closely related Type A1 or A1(B) strains, as well as the A1-specific microarray, allowed the analysis of their genomic organizations and evolutionary relationship. The four genomes share >90% core genes and >96% functional groups. Phylogenetic analysis based on COG shows closer relations of the A1(B) strain, NCTC 2916, to B1 and F1 than A1 strains. Alignment of the genomes of the three A1 strains revealed a highly similar chromosomal structure with three small gaps in the genome of ATCC 19397 and one additional gap in the genome of Hall A, suggesting ATCC 19379 as an evolutionary intermediate between Hall A and ATCC 3502. Analyses of the four gap regions indicated potential horizontal gene transfer and recombination events important for the evolution of A1 strains.
Keywords: Clostridium botulinum, genomic comparison, microarray
1. Introduction
Clostridium botulinum are Gram-positive, anaerobic bacteria that have the ability to produce spores as well as botulinum neurotoxin (BoNT), one of the deadliest toxins known to man with a lethal dose (LD50) of 1 ng per kg of body weight [Thanongsaksrikul, 2011]. There are seven serotypes of BoNTs, A, B, C1, D, E, F, and G, produced from different strains of C. botulinum, that are physiologically and phylogenetically distinct. Several C. botulinum strains are bivalent, containing combinations of bont gene clusters that are either silent or expressed at different levels [Hill, 2009]. The neurotoxin gene is located on a gene cluster together with genes for the toxin associated proteins and their regulatory components. Interestingly, the organizations and locations of these neurotoxin (bont) gene clusters varies among different toxin serotypes, as well as between strains producing the same toxin serotypes, indicating that the gene clusters have undergone horizontal gene transfers (HGTs) and recombination [Skarin, 2011].
There are a growing number of genomic sequences leading to constant revisions of the phylogenetic tree of C. botulinum and their relationship to the genus, Clostridium. Over one hundred clostridial species have been sequenced and annotated, according to the microbial genomic consortium at the Integrated Microbial Genomes and Metagenomes (IMG). Approximately twenty of those annotated genomes belong to C. botulinum, which harbors a variety of bont gene clusters located within plasmids or chromosomes [Hill, 2013; Marshall, 2007; Sebaihia, 2007]. The genomic locations of the bont gene clusters vary with toxin serotypes, subtypes, and strains. Genomic analyses of various C. botulinum strains showed multiple, but specific insertion sites for the bont gene clusters, indicating horizontal gene transfers through site-specific insertions into non-neurotoxigenic strains [Hill, 2009; Hill, 2013]. Horizontal transfers of bont gene clusters are further evident by 16S rRNA sequence similarities between the non-neurotoxigenic C. sporogenes and C. botulinum Group I, as well as others to different groups of C. botulinum [Sebaihia, 2007]. Furthermore, non-neurotoxigenic Clostridium species have shown to be able to transform into a BoNT expressing strain via lysogenic conversion [Brussow, 2004], which may foreshadow future evolutionary events.
BoNT serotypes are further grouped into subtypes (or genetic variants) based on bont sequence similarities and/or genetic clade differences among strains [Hill, 2013]. Contrasting to the large sequence differences between BoNT serotypes (~37 to 70% amino acid identities), the differences between subtypes are usually smaller [Smith, 2013]. For example, the percent amino acid identities between any pair of the five A subtypes (A1 – A5) range from ~84 to 97% [Hill, 2013]. Analysis of bont gene clusters of all serotypes and subtypes revealed two major types of bont gene arrangement, designated HA+ and orfX+ clusters [Jacobson, 2008]. The HA+ cluster is capable of producing neurotoxin complexes of 300 to 900 kDa comprising various sizes of hemagglutinins (HA), a non-hemagglutinin non-toxic protein (NTNH), and the 150 kDa BoNT [Johnson, 2001; Jacobson, 2008]. The orfX+ cluster, composed of the bont, ntnh and a number of open reading frames (orfs) of unknown functions, usually correlates to the formation of a 300 kDa neurotoxin complex of BoNT and NTNH. Among the seven BoNT serotypes, only serotype A were found to exist in both HA+ and orfX+ clusters [Hill, 2013]. Interestingly, the HA+ A clusters (including some of the A1 and A5 strains) are found to be inserted near the oppA/brnQ operon on the chromosome, while the orfX+ clusters (including some A1 and A2 strains) are inserted near the arsC operon. The orfX+ clusters of A3 and A4 are reported to be located on a plasmid [Marshall, 2007; Jacobson, 2008].
BoNT/A, particularly HA+A1, is among the most well-characterized bont gene clusters. Specifically, the HA+A1 neurotoxin gene cluster consists of the ntnh-bont operon as well as the oppositely orientated ha operon, which encodes for hemagglutinins, HA17, HA33, and HA70. In between these two operons is the gene coding for BotR, which is an alternative sigma factor of RNA polymerase and a positive regulator for genes in the bont gene cluster [Connan, 2012]. The ntnh gene, present in all serotypes and cluster types, encodes a non-toxic and non-hemagglutinin (NTNH) protein, which is part of the associated non-toxic proteins in the neurotoxin complex [Hill, 2013]. Studies have shown that, despite having different functions, the structural similarities between BoNT and NTNH may imply the genes encoding these two proteins shared a common ancestor through a gene duplication event [Doxey, 2008].
The recently available genomic sequences of several C. botulinum strains, as well as those of closely related non-neurotoxigenic clostridial strains, have opened doors to a better understanding of the evolutionary relationship among the clostridia, especially the C. botulinum strains. Additionally, sequence analysis of subtype A1 strains found that the HA+ A1 bont gene cluster of ATCC 3502 is located within positions 901,881 – 913,599 near the oppA/brnQ operon [Hill, 2009; Sebaihia, 2007; Fang, 2010]. This exact cluster and insertion site can also be found in another two assembled A1 genomes, Hall A and ATCC 19397, as well as the silent HA+ (B) cluster of NCTC 2916 [Jovita, 1998; Hill, 2013]. Interestingly, the A1 gene cluster of NCTC 2916 was found to be an orfX+ cluster located at around 48 kb upstream from the silent HA+ (B) cluster nearby the arsC operon [Hill, 2009]. Several toxin gene clusters are also found to be inserted at arsC, including A2 of strain Kyoto-F as well as F1 of strain Langeland [Hill, 2009].
While the analyses of bont gene clusters across various serotypes have supported the model of horizontal gene transfer in the evolution of the neurotoxigenic Clostridium species, analyses based on the whole genomes have been limited. A previous study using whole genome microarray analysis indicated a stable genome, showing 63% of the coding sequences of the reference strain ATCC 3502 are present in all 61 proteolytic C. botulinum and C. sporogenes strains studied [Carter, 2009]. A translocation event was also identified when aligning the genomes of three subtype A1 strains [Fang, 2010]. Our goal was to reveal the genomic connection between C. botulinum strains, particularly the closely related subtype A1 genomes, by utilizing genomic tools and genomic microarray. In this study, we focus on the analysis of A1 strains as it is one of the most well-characterized serotypes/subtypes and produces the largest neurotoxin complex. Some of the bacterial strains producing subtype A1 toxins are also known for the heat resistance of their spores, and have been used as indicators for commercial sterilization [Anderson, 2011]. The availability of three completely assembled and annotated genome sequences, ATCC 3502, ATCC 19397, and Hall A, and one partially assembled genome of A1(B), NCTC 2916, allowed us to compare these strains closely, identify the similarities and differences among them, and analyze their relationship within the genus.
2. Materials and Methods
2.1. Bacterial strains and media
The GenBank accession numbers for A1 strains, ATCC 3502 (aka: Hall A Sanger), Hall A (aka: Hyper Hall A), ATCC 19397, and NCTC 2916, are NC_009495, NC_009698, NC_009697, and ABDO02000001-49, respectively. All genomes have been completely assembled and annotated except for NCTC 2916 which is partially assembled into 49 contigs. The two Clostridium botulinum strains used in the microarray study were ATCC 3502 and Hall A. Bacteria stocks were anaerobically maintained in cooked meat medium (CMM), and revitalized at 37°C in TPGY medium (5% Trypticase peptone, 0.5% Bacto peptone, 0.4% glucose, 2% yeast extract). All procedures involving C. botulinum were performed using biosafety level 2 practices in a laboratory registered with the CDC Select Agent Program.
2.2. Comparative analysis of genomes
Some of the genomic analyses were performed using tools embedded under the Integrated Microbial Genomes (IMG) hosted by DOE Joint Genome Institute (JGI, www.jgi.doe.gov). Annotated genome sequences and statistics from GenBank can be accessed directly through IMG. The phylogenetic tree based on whole genomic information was generated using the Distance Tree in IMG, whereas dot plots were produced using the Synteny Viewer. A cladogram, analogous to a phylogenetic tree, based on Clusters of Orthologous Groups (COGs) of C. botulinum strains was obtained using the Genome Clustering tool from IMG. Artemis Comparison Tool (ACT) was used for pairwise mapping of genes. Pairwise genomic comparisons were analyzed using Vista from IMG to align toxin cluster genes. The individual and comparative circular genomic maps were created with CGView Server (www.wishart.biology.ualberta.ca/cgview/) using the FASTA files downloaded from GenBank. The Venn diagram was generated by Efficient Database framework for comparative Genome Analyses using BLAST score Ratios (EDGAR; www.edgar.cebitec.uni-bielefeld.de) to analyze the gene pools between the closely related A1 C. botulinum strains using GenBank files loaded from NCBI. The DNA sequence similarities and locations of the bont cluster genes in comparison to strain ATCC 3502 were identified using the Basic Local Alignment Search Tool (BLAST) on the NCBI website (www.ncbi.nlm.nih.gov).
2.3. Functional analysis
COG family analyses were performed by exporting the numbers of genes present in each of the 25 COG families from IMG to Microsoft Excel to tally and graph. The unique and core COG groups were calculated and displayed in Venn diagrams using Microsoft Excel after importing 3623 COG groups from the COG list tool under IMG.
2.4. Microarray analysis of genomic DNA
The genomic DNA was extracted from the bacterial culture using DNA Easy Kit (Invitrogen, Carlsbad, CA) with the lysozyme treatment at a final concentration of 6.7 mg/mL. Prior to labeling, the genomic DNA was digested with MboI (New England Biolabs, Ipswich, MA) followed by a QiaQuick PCR Purification kit (Qiagen, Valencia, CA) to remove excess restriction enzymes. The “Microbial genomic DNA aminoallyl labeling for microarrays” protocol (SOP: M009 Rev. 2; pfgrc.jcvi.org/index.php/microarray/protocols.html) was followed for labeling of the genomic DNA with aminoallyl and the subsequent incorporation of AlexaFluor Cy 3 and Cy 5 dyes (Invitrogen, Carlsbad, CA).
The C. botulinum version 2 microarray slides used for these experiments were distributed by the Pathogen Functional Genomics Resource Center under J. Craig Venter Institute (JCVI, Rockville, MD). The microarray slides were stored in a dry and dark desiccator until use. The “Hybridization of labeled DNA and cDNA probes” protocol (SOP: M008 Rev. 2.1; pfgrc.jcvi.org/index.php/microarray/protocols.html) was used to perform the hybridization experiments. After hybridization, the washed microarray slides were scanned by GenePix® 4000B microarray scanner (Molecular Devices, Sunnyvale, CA) using GenePix® Pro 6.0 to capture the images. Scanned images (.tif) of the microarray slides were analyzed using the TM4 Suite (www.tm4.org). TM4 Suite encompasses four individual software tools: SpotFinder for image processing, Microarray Data Analysis System (MIDAS) for normalization, MultiExperiment Viewer (MEV) for visualization and interpretation of microarray data, and Microarray Data Manager (MADAM) for data submission.
3. Results
3.1. Phylogenetic trees coincide with the genomic alignments of C. botulinum
Phylogenetic analyses based on the 16S rRNA gene showed diversity of bont-containing strains and the delineation following the physiological groups of C. botulinum [Hill, 2007]. As a comparison, a function based cladogram using Clusters of Orthologous Groups (COGs), was constructed using 121 clostridial genomes available through the Genome Clustering tool in Integrated Microbial Genome (IMG). C. botulinum strains fall into three different clades, which coincides with the three physiological groups of the species (I, II, and III) (Suppl. Fig. 1). The majority of C. botulinum Group I proteolytic strains such as A1 strains, A2 Kyoto-F, and F Langeland fall into the same clade as C. sporogenes ATCC 15579, and are distal from the other fermentative and cytotoxigenic clostridial species (Suppl. Fig. 1). A different clade consisting of many C. botulinum Group II non-proteolytic strains such as Eklund 17B, Alaska E43, and E1 Beluga show close relations with C. butyricum strains and C. perfringens strains. The C. botulinum Group III strains, such as C Eklund, D str. 1873, and CD strain BKT015925, clustered together on the same clade with C. novyi NT, and show a closer relation to the Group II strains than the Group I strains.
A function based phylogenetic tree was further analyzed to better separate the twenty known C. botulinum genomes (Fig. 1A). The clustering of the C. botulinum species mostly mirrors that of the neurotoxin serotypes and was similar to the cladogram shown in Supplemental Figure 1. Based on the COG functional groups, Group III branched out early followed by those strains in Group II. Three of the Group I subtype A1 strains (ATCC 3502, Hall A, and ATCC 19397) are clustered within a clade while NCTC 2916 was clustered in an out group with other Group I strains. The Group I bivalent strains, Bf and Ba, share the same large clade with the Group I strains, but are relatively more distal from the rest of the group. The recently isolated bivalent strains CFSAN001627 and CFSAN001628 produce two different toxins, A/B and A/F, respectively [Basavanna, 2013], and can be seen to cluster with Group I strains (Fig. 1A). The three available Group II non-proteolytic genomes, Eklund 17B, Alaska E43, and Beluga E, show a closer relation to Group I than Group III, which is similar to the clustering generated in Supplemental Figure 1. The relations of closely related A1 strains can be better visualized in Figure 1B, in which strain Hall A is closest to ATCC 19397 than ATCC 3502, followed by NCTC 2916.
Figure 1.

Functional and genomic relationships among C. botulinum serotypes. Phylogenetic trees based on Clusters of Orthologous Groups (COGs) were created for C. botulinum strains (A) and subtype A1 strains (B). Strains used in this study are marked by an asterisk. Analysis was performed using the genomic clustering ability with COG relationships in IMG. Synteny Dot Plots were analyzed between strains used in this study (C), closely related Group I strains (D), and between distally related Groups (E). Reference genomes lie on the x-axis, whereas, queries are found on the y-axis. Lines that travel along the upward diagonal line are the equivalent to homologous regions between the reference and query, where blue lines/dots are regions on the same strand and red lines/dots refer to regions on opposite strands. A line that has a slope of 1 signifies an undisturbed segment of conservation between the two genomes. A line with a slope −1 denotes an inverted segment of conservation. An empty space in the line indicates an insertion or deletion, whereas a break in the line favoring one axis over another shows a translocation. The Synteny Dot Plots were generated using NUCmer alignment tool from MUMmer platform in IMG.
Synteny Dot Plots provide the genomic alignment of two given genomes, revealing sequences altered during evolution as well as identifying simple sequence repeats. Several pairwise dot plots compare ATCC 3502 with closely related strains, including the three A1 (Fig. 1C), Group I but non-A1 genomes (Fig. 1D), and non-Group I genomes (Fig. 1E). When comparing among the Group I strains (Figs. 1C and D), including F Langeland (data not shown), a distinct diagonal line is observed which confirms the high degree of similarity between these strains. Only a few segment displacements and breaks are seen, which correspond to translocations and insertions, respectively.
The dot plot comparing ATCC 3502 to a closely related strain, C. sporogenes ATCC 15579, shows three major genomic alignments between these two strains (Fig. 1D). The breaks of the blue diagonal lines may have been due to translocation events and the red perpendicular line may indicate a gene inversion. Additionally, C. tetani shares a common ancestor to Group I strains (Suppl. Fig. 1), thus a dot plot comparing ATCC 3502 and C. tetani (Fig. 1D) reveals some similarities in genomic makeup as compared to other clostridial groups.
Serotypes in different groups were also compared using Synteny Dot Plots (Fig. 1E). When compared to Figures 1C and 1D, the dot plots in Figure 1E show very few similarities between strains. This confirms the different evolutionary lineages between the strains from different physiological groups of C. botulinum. This lack of genomic similarities between different physiological groups was also observed when comparing B1 Okra (Group I) and B4 Eklund 17B (Group II), even though serotype B neurotoxin is produced from both strains (Fig. 1E). These evidences indicate that the toxin has been transferred between lineages, including some more recent transfers and some rather ancient transfers.
3.2. Comparison of C. botulinum A1 strains revealed a similar genomic organization
Several methods of comparison were used to better understand the differences between the four A1 containing strains of interest, Hall A, ATCC 19397, ATCC 3502, and NCTC 2916. The statistical values and more in-depth information of the four genomes are summarized in Supplemental Table 1. C. botulinum NCTC 2916 showed the largest genome size of 4,031,357 bp, which is ~ 128 kb, ~167 kb, and ~ 270 kb larger than ATCC 3502, ATCC 19397, and Hall A, respectively. Interestingly, the total number of predicted genes is highest in ATCC 3502 (3825 genes) and lowest in Hall A (3622 genes). Even though the predicted gene numbers only differ within about 200 genes, strains ATCC 3502 and NCTC 2916 have over 500 more genes coding for transmembrane proteins than Hall A and ATCC 19397. While genes missing in Gaps 1 and 3 may contribute partially to the discrepancy of these transmembrane proteins (Suppl. Table 4), the rest of the transmembrane proteins may fall under genes with unidentified function (Suppl. Table 1).
Individual circular maps of the three completely assembled A1 strains, Hall A, ATCC 19397, and ATCC 3502, were generated to show unique genomic construction (Fig. 2A). There are clear similarities amongst the three closely related strains as shown by similar gene distribution and orientation, as well as the GC% and GC skew of the chromosomes. Circular map alignments of the three assembled A1 strains using ATCC 3502 as the reference genome, shows a strong genomic correlation between these strains (Fig. 2B), which echo the synteny dot plots shown in Figure 1C. The GC skew of all three genomes follows the replication direction showing more G in the leading strand and more C in the lagging strand of replication. Interestingly, more than 80% of gene orientations follow the leading strand as shown by the dominating forward gene direction of the first half of the genome and the reverse gene direction of the second half of the genome (Fig. 2B). The skew of gene direction, also observed in other bacteria genomes such as Bacillus subtilis, may have been an evolutionary selection due to the codon usage bias [Tillier, 2007].
Figure 2.

Circular chromosome maps of Hall A, ATCC 19397, and ATCC 3502 (A). A comparative circular map of the three genomes (B) and ATCC 19397 with Hall A (C) was generated by CGView Server. The two outer blue rings represent genes on the forward and reverse strand of the indicated strains (A), or the reference strain, ATCC 3502 (B), or ATCC 19397 (C). Within the two outer circles contains tRNA (red) and rRNA (purple). Alignments of the reference genome to Hall A and ATCC 19397 are shown in red and dark green, respectively. The number of lines is proportional to the percent identity of the hit compared to the reference genome. The GC% is shown in black below and above mean. GC skew is shown in purple and green representing below and above, respectively. The inner circle displays distance of bases.
There are four gaps, designated Gaps 1 to 4 by the chromosomal positions, in the genome of Hall A when compared to ATCC 3502 (Fig. 2B). Among the four gaps, three were also observed in ATCC 19397. The third gap at around 2,280 kb position is absent in ATCC 19397. To take a closer look at the two smaller genomes, the analysis was performed using the larger ATCC 19397 genome as a reference genome and Hall A as a query sequence. As shown in Fig. 2C, the gap at the 2,280 kb position was only seen in Hall A, but not ATCC 19397. There is an additional gap at ~3,100 kb found to be missing in Hall A and ATCC 3502, but present in ATCC 19397 (Fig. 2C).
3.3. C. botulinum subtype A1 genomes share common gene functions
An analysis of the gene pool would allow a better understanding of the genes amongst the three assembled C. botulinum strains, as well as provide a comparison of genes unique or common to selected genomes. There are 3230 core genes shared among ATCC 3502, ATCC 19397, and Hall A, which is calculated to be 90.5% of ATCC 3502 gene or 95% of the smaller Hall A genome (Fig. 3). There are 263 genes (or 7.4% of total genes), 101 genes (or 2.8% of total genes), and 9 genes (or 0.26% of total genes) that are unique to ATCC 3502, ATCC 19397, and Hall A, respectively. ATCC 3502 shares additional 66 genes with ATCC 19397 and only 11 genes with Hall A, suggesting a closer relation of ATCC 3502 to ATCC 19397 than Hall A. On the other hand, Hall A is closest to ATCC 19397 as evident by the additional 148 genes shared with ATCC 19397, totaling 3378 common genes or 99.4% of its total genes. This type of analyses cannot be done on the unassembled genomes, NCTC 2916.
Figure 3.

Venn diagram comparing genes of ATCC 3502, ATCC 19397, and Hall A. The orange circle refers to the genes of ATCC 3502 whereas the green circle refers to the genes of ATCC 19397, and the blue circle refers to genes of Hall A. Numbers in parentheses under each strain name refer to the total number of genes in that particular strain. Numbers in overlapping areas are considered core genes while the number outside the overlapping area displays the number of genes unique to that specific genome. Information was gathered from Efficient Database Framework for comparative Genome Analyses using BLAST score Ratios (EDGAR) using GenBank files from NCBI and plotted in Creately.
Comparison of the genome can be done based on the distribution of COG functional groups, which allows the comparison of genomes based on functions instead of individual genes. The distribution of COG functions were analyzed on all four A1 genomes as well as representative genomes from other serotypes as a comparison (Fig. 4). The result demonstrates a similar distribution of genes within all four A1 strains of C. botulinum as well as those genomes within Group I. Group I genomes showed comparable amounts of genes among all the COG families. The strains in Group II (Eklund 17B and Alaska E43) displayed a comparable distribution of COG families, except for a higher amount of genes in carbohydrate transport and metabolism (Fig. 4). Strains BKT015925 and 1873 from Group III showed lower amounts of genes than the other clostridial groups, probably due to their smaller genomes measuring 3.2 Mbp and 2.3 Mbp, respectively (img.jgi.doe.gov/). Interestingly, Group III BKT015925 strain showed exceptionally higher amounts of genes in replication, recombination and repair as compared to the remaining strains (Fig. 4). As expected, none of the strains possessed genes towards four functional groups specific for eukaryotes: RNA processing and modification, nuclear structure, extracellular structures, and cytoskeleton. Among the genomes compared, strain Loch Maree has the largest genome size measuring 4.25 MBp, where its extra genes are mainly distributed among the common COG families.
Figure 4.

Cluster of Orthologous Group Family of selected C. botulinum strains in Groups I, II, and III. The first nine strains in the legend belong to Group I, whereas the last four strains in the legend consists of those belonging to Group II (Eklund 17B and Alaska E43) and Group III (BKT015925 and 1873). Information was generated from COG Browser on IMG and plotted by Excel.
To better visualize the functional relationship between the four A1 strains, a Venn diagram was generated using a COG list obtained from IMG (Fig. 5). The results show a core 1252 functional groups among the four strains analyzed, making up 97.1, 96.5, 96.0, and 96.3% of the functions in Hall A, ATCC 3502, ATCC 19397, and NCTC 2916, respectively. NCTC 2916 has 27 unique functional groups, whereas both ATCC 19397 and ATCC 3502 have 5, and Hall A possesses only one unique group. Three A1 strains share additional 28 functional groups together, totaling 1280 shared functional groups and making up 99.3, 98.6, and 98.2% of the functions in Hall A, ATCC 3502, and ATCC 19397, respectively. Although there are a total of 1342 functional groups identified, 2281 additional COG groups are not found in any of the four A1 strains.
Figure 5.
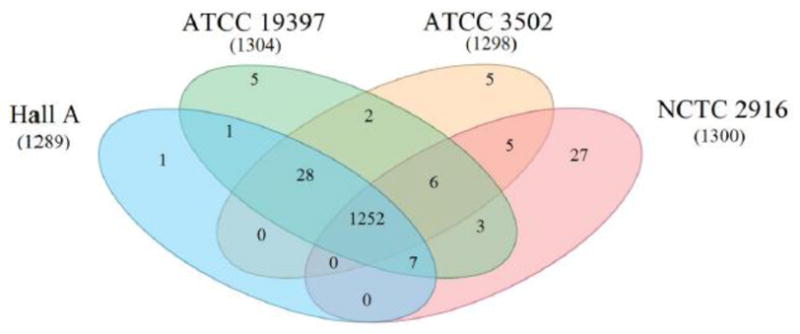
Venn diagram grouping based on COG functional relationships within C. botulinum subtype A1 strains. Numbers in parentheses under each strain name refer to the total number of COG functional groups in that particular strain. Overlapping areas signify common groups between strains. Core group number is found in the center of the figure where all four circles overlap. Functional group list was exported from IMG database and plotted in Creately.
3. 4. Analyses of gaps reveal the evolutionary relation of subtype A1 strains
The four gap areas that are missing in Hall A as compared to ATCC 3502 (Fig. 2B) were investigated further using Vista Browser (pairwise genomic alignment), Artemis Comparison Tool (ACT; pairwise mapping of genes), and microarray (Fig. 6). The information of the gaps is summarized in Table 1 and a list of genes that are missing in each gap is provided in Supplemental Table 4. All but Gap 3 were also present in the closely related strain ATCC 19397, suggesting that this strain may have been an evolutionary intermediate between ATCC 3502 and Hall A. This correlation is also seen by the 16S rRNA based phylogeny (Fig. 1B). Pairwise genomic alignments of ATCC 3502 and ATCC 19397 by VistaBrowser and ACT confirmed the presence of all Gap 3 genes in ATCC 19397 (Suppl. Fig. 2). For the convenience, “gap genes” are used to indicate genes that are missing in the gap area in Hall A or ATCC 19397 when compared to ATCC 3502.
Figure 6.

Analysis of genes in the four gaps of ATCC 3502 and Hall A. (A) Graphs panel of VistaPoint showing ATCC 3502 (reference genome) at the top and Hall A at the bottom. The blue arrows above each graph show the length and orientation of each gene and chromosomal positions of the two ends of the displayed fragments are labeled. The exact boundaries of each gap are listed in Table 2. The homology is displayed as the peak height in pink for the non-coding and purple for the coding sequences. (B) Artemis Comparison Tool (ACT) showing gap areas of ATCC 3502 on the top strand and Hall A genome at the bottom. Black brackets above ATCC 3502 strand represent the gap area. (C) Scatter plots of Log2 ratios with upstream (light grey diamonds), gap (dark grey diamonds), and downstream genes (light grey diamonds). ANOVA results are shown below each plot with SEM. The statistical significance of the tested value against that of the gap genes are indicated as single asterisks for P<0.05 and double asterisks for P <0.1. Statistical significance (P<0.05) between upstream and downstream genes are noted by an “Ł”.
Table 1.
Gap information of Hall A and ATCC 19397 based on ATCC 3502 genome.
| Gat) | # of Missing Genes in Gao | Average Length of Missing Genes in Gao | Hall A | ATCC 19397 | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| ATCC 3502 Range | Gap Size | Chromosome Position | ATCC 3502 Range | Gap Size | Chromosome Position | |||
| 1 | 21 | 1203±1195.7 | 438,395 – 467,981 | 29,587 | 431 Kb | 438,500 – 467,981 | 29,482 | 431 Kb |
| 2 | 74 | 489.73±394.04 | 1,822,688 – 1,860,408 | 37.721 | 1793 Kb | 1,822,687 – 1,864,850 | 42,163 | 1793 Kb |
| 3 | 21 | 1027.43±543.04 | 2,351,399–2,379,228 | 27,830 | 2280 Kb | - | - | - |
| 4 | 84 | 606.85±555.84 | 2,466,361 – 2,523,053 | 56,693 | 2367 Kb | 2,466,361 – 2,523,053 | 56,693 | 2395 Kb |
Gap 1, occurred at around 431 kb position on the chromosomes of Hall A and ATCC 19397, consists of 21 missing genes and 29.5 kb in length spanning 438,395 – 467,981 on the chromosome of ATCC 3502 (Table 1). Missing genes in this gap are mostly membrane associated, including several ABC transporters and cell surface proteins (Suppl. Table 4). The pairwise genomic alignment showed limited matches to Hall A within the gap area, whereas the sequences surrounding the gap exhibited high conservation as colored by the purple (coding sequences) and pink (non-coding sequences) areas (Fig. 6A). To determine if these missing genes in Gap 1 were relocated elsewhere on the genome of Hall A, the DNA fragment from ATCC 3502 was further aligned to the genome of Hall A using ACT, which shows lack of alignment to anywhere on the genome of Hall A (Fig. 6B). Interestingly, the immediate downstream of Gap 1 exhibited hot spots of recombination as shown in the ACT alignment between ATCC 3502 and Hall A genomes (Fig. 6B).
Gap 2, mapping to 1,822,688 – 1,860,408 (~37.7 kb in length) on ATCC 3502, was absent at the 1,793 kb position on the chromosomes of Hall A and ATCC 19379. Unlike the distinct transition between 0 and 100% identities bordering the Gap 1 area, the pairwise alignment shows two taller peaks of homologies toward the downstream area of Gap 2 (Fig. 6A), indicating several gene recombination events may have occurred when the gap was formed by either an insertion into ATCC 3502 or a deleted in Hall A. The ATCC 3502 sequences mapped to the Gap 2 area consists of 74 genes with an average gene size of < 500 bp (Table 1 and Suppl. Table 4). Among these short open reading frames, 64 are directly phage related (Suppl. Table 4), implying the involvement of bacteriophage-mediated gene transfer events. However, 20 genes at the 3′ end of Gap 2 showed recombination events (Fig. 6B). Gaps 3 and 4 are only 87 kb apart on the chromosome of ATCC 3502. Gap 3, ranging from 2,351,399 – 2,379,228 on ATCC 3502, is flanked by high levels of conserved regions (Fig. 6A). The gap is ~27.8 kb in length and consists of a total of 21 genes present in ATCC 3502 (Table 1). These genes encode for an array of functions, including membrane proteins such as ferredoxin and gluconate permease, as well as several hydrogenases/dehydrogenases involved in energy transportation within the cells (Suppl. Table 4). The last gap, Gap 4, mapped to the fragment at 2,466,361 – 2,523,053 (~ 56.6 kb in length) on ATCC 3502 with several areas of high conservation between the aligned sequences, as seen by the coding (purple) peaks within the gap area (Fig. 6A). As seen in Gap 2, the ATCC 3502 sequences mapped to the Gap 4 area also consists of many short open reading frames of unknown or phage related functions (Suppl. Table 4), suggesting a role of bacteriophage(s) in rearrangements of this region.
The presence/absence of genes in the gaps was further verified by the 2-color genomic microarray analysis using the ATCC 3502-based oligo microarray slides made by the J. Craig Venter Institute. The dye intensities of Cy5-labeled ATCC 3502 gDNA and Cy3-labeled Hall A gDNA, as well as the Log2 ratios of Cy5/Cy3 signals were listed for individual gap genes (Suppl. Table 4) and the Log2 ratios of genes within and flanking the gaps were plotted (Fig. 6C). Genes with Log2 ratios close to zero are considered to be similar between ATCC 3502 and Hall A, while a higher Log2 ratio may imply the absence of gene in Hall A. Among the four gap areas, only Gap 3 behaved as expected where close to zero Log2 ratios were observed in genes upstream and downstream of the gap and significantly higher Log2 ratios (p<0.05) were found in Gap 3 genes (Fig. 6C). As expected, there is no significant difference in the Cy5 (ATCC 3502) intensities between the gap and flanking genes. However, Cy3 (Hall A) shows a significantly lower average intensity for the genes upstream and downstream the gap with P values lower than 0.05 and 0,1, respectively. (Fig. 6C). Gap 1 analysis also demonstrated a somewhat similar pattern as seen in Gap 4 except that the Cy5 (ATCC 3502) dye intensities of the gap genes was unexpectedly low (Fig. 6C). Gaps 2 and 4 are missing 74 and 84 short open reading frames, respectively, which may have contributed to their background-leveled dye intensities (<1000 RFU) for both Cy5 (ATCC 3502) and Cy3 (Hall A) channels. Therefore, the Log2 ratios could not be used to validate the presence and absence of genes in these two gaps. Nevertheless, none of the missing gap genes in Hall A showed positive hybridization results. In summary, the four gap areas contribute to a total of 151.8 kb and 200 genes missing in Hall A, which encompasses the majority of the genomic discrepancy between Hall A and ATCC 3502 (Suppl. Table 1). Missing genes in Gaps 1 and 3 encode for mostly membrane associated proteins, while missing genes in Gaps 2 and 4 are short with phage-related or unknown functions (Suppl. Table 4). The 2-color microarray used in this study could successfully identify the presence and absence of Gap 3 genes in ATCC 3502 and Hall A, respectively. However, it failed to detect the presence and absence of genes in Gaps 1, 2, and 4 in both strain (Fig. 6C).
3.5. The bont A1 gene clusters and the insertion sites are highly conserved
Studies on the sequence of the bont gene clusters have shown the involvement of horizontal gene transfer in the acquisition of the neurotoxin [Hill, 2013]. To further investigate the insertion site of the toxin clusters, sequences of the bont gene clusters as well as the flanking regions were analyzed. Alignments of genes ±10 kb flanking the bont gene cluster showed over 99% identities of ATCC 3502 with Hall A (Fig. 7A) and ATCC 19397 (Fig. 7B). The BLAST alignment demonstrated a single transition mutation from an A in the intergenic region between a hypothetical protein (CBO0790) and the dihydrodipicolinate reductase (CBO0791) at chromosome position 893,493, about 8.4 kb upstream of the bont gene cluster in ATCC 3502, to a G in Hall A and ATCC 19397 at 865,987 and 865,820, respectively.
Figure 7.

Analysis of botulinum neurotoxin gene clusters of ATCC 3502, Hall A, ATCC 19397, and NCTC 2916. (A–C) VistaSynteny alignments showing neurotoxin gene cluster (CBO0801–CBO0806) (orange rectangle) using ATCC 3502 as the reference genome against Hall A (A), ATCC 19397 (B), and NCTC 2916 (C). VistaSynteny of ATCC 3502 between Hall A and ATCC 19397 include genes ±10 kb flanking neurotoxin gene cluster. VistaSynteny and ACT of ATCC 3502 and NCTC 2916 include genes +10 kb downstream and −48 kb upstream from the neurotoxin gene cluster on ATCC 3502. The bont A1 gene cluster, HA+ (B) cluster, and orfX+ A1 cluster are indicated by rectangular boxes colored orange, red, and blue, respectively. Parallel lines within the VistaSynteny of ATCC 3502 and NCTC 2916 represent ~24 kb of homology between the two genomes. The dark grey and green bars in the VistaSynteny graphs represent the homologous fragments of the reference genome (top) and the query genome (bottom), respectively. The homologous fragments are connected using grey lines. Genome structures were obtained from GenBank via IMG.
Two bont gene clusters, orfX+ A1 cluster and HA+ (B) cluster, have been found in the A1(B) strain, NCTC 2916 [Jovita, 1998; Franciosa, 2004]. We investigated the clusters closely within the partially assembled genome of NCTC 2916. The two bont gene clusters are located closely at approximately 41 kb apart in the same orientation on Contig 1 of 1.43 Mb in length (Fig. 7C). Contrary to the 100% identities found in the toxin clusters of the three A1 genomes studied, the two toxin clusters of NCTC 2916 share a lowered sequence identity to the HA+ A1 cluster ranging from 88 to 100%, with the two ntnh genes being the least similar (Fig. 7C and Suppl. Table 3). The ntnh in the HA+ A1 cluster of ATCC 3502 shares a higher sequence similarity to the NCTC 2916 HA+ (B) cluster (94% identity) than the orfX+ A1 cluster (88% identity). The upstream 10 kb region of HA+ (B) cluster of NCTC 2916 showed 94–98% sequence identities to other A1 strains; while ~96% identity to A1 strains resumes at approximately 1.8 kb downstream from the HA+ (B) cluster (BLAST; data not shown). This 1.8 kb downstream from the HA+ cluster encodes for three putative transposase(s) in ATCC 3502 and one transposase in NCTC 2916, which may play a role in the transfer of the HA+ clusters. BLAST analysis further demonstrated that the downstream area from the HA+ (B) cluster of NCTC 2916 is more closely related to H04402 065 (A5), 657 (Ba4), and Loch Maree (A3) strains, except for the first 600 bp which showed a 95% sequence identity to the immediate downstream regions of the toxin clusters of the pCL plasmids of Okra (B1) and Eklund 17B. Thus, indicating a potential involvement of plasmids in the recombination and evolution of HA+ (B) cluster (data not shown).
To analyze the insertion site of orfX+ A1 cluster in NCTC 2916, the 48 kb upstream of the HA+ A1 cluster of ATCC 3502 was extended to align with the region spanning the two toxin clusters in NCTC 2916. As shown in Figure 7C, the majority of this region in ATCC 3502 mapped to the corresponding region in NCTC 2916, except a 6 kb fragment (~861 to 867 kb position on ATCC 3502). Instead, NCTC 2916 harbors a 17 kb nonhomologous sequence containing the orfX+ A1 cluster, as well as the upstream 4 kb (~808 to 812 kb position on the chromosome) and downstream 3 kb (~822 to 825 kb) fragments (Fig. 7C). A closer look at the annotated genes around the nonhomologous regions in ATCC 3502 shows the presence of the ars operon and its related genes within the upstream region of the 6 kb fragment, whereas the downstream area of the this 6 kb fragment consists of a couple ATP binding proteins as well as a phosphomethylpyrimidine kinase (data not shown). The nonhomologous regions flanking the orfX+ A1 cluster in NCTC 2916 encode for a few hypothetical proteins in the region upstream from the A1 cluster and LycA in the downstream area. There is no strong evidence of mobile elements around this region except for a few phage related genes found in the upstream region of the 6 kb fragment in ATCC 3502.
4. Discussion
Clostridium botulinum and other neurotoxin-producing clostridial species can be categorized by the botulinum neurotoxin serotypes (A to G) produced, as well as the four physiological groups based on gene sequences and biochemical reactions such as their carbohydrate utilization and proteolytic activities. The neurotoxins A - G are classified by serology and sub-classified into “subtypes” or “subserotype” by DNA sequence analysis (e.g. A1, A2, A3 etc.). The disease, botulism, is inflicted by the toxicity from a functional bont gene cluster found in C. botulinum strains as well as a few other Clostridium species such as C. baratii and C. butyricum, suggesting horizontal gene transfer (HGT) occurred within the four groups of C. botulinum strains as well as within the genus [Skarin, 2011]. Analysis of HGT events was hampered due to lack of genetic tools and difficulty in manipulating the bacteria due to its toxicity and anaerobic nature. With the availability of sequenced genomes and strain-specific microarrays, it has enabled a closer view into the species at the genomic level.
Our genomic alignments using Synteny dot plots clearly demonstrated the close relatedness of genomes of Group I strains, as well as C. sporogenes, regardless of BoNT serotypes (Fig. 1D). This supports the observation of a genomic microarray study showing approximately 63% of the CDSs of ATCC 3502 to be similar among 61 proteolytic C. botulinum strains (Group I) and C. sporogenes surveyed [Carter, 2009]. A study of 42 BoNT/A2 strains and four BoNT/A3 strains by MLST and PFGE methods also revealed limited genetic diversity among these Group I strains [Luquez, 2012]. A similarly close relatedness was also observed within the Group II C. botulinum strains by genomic indexing using the microarray based on a Group II strain, Eklund 17B [Stringer, 2013]. Stringer et al. (2013) further showed that, among Group II strains, serotypes B and F strains clustered closely together and were more distal to serotype E strains. Like those comparisons within Group I strains, our Synteny plots comparing the genomes within Group II or Group III strains also demonstrated close relatedness as shown by a visible diagonal line (data not shown). On the contrary, when comparing the genomes across different groups using Synteny dot plots, no apparent similiarity was observed as expected (Fig. 1E). The Group III genomes (BKT015925 and 1873) analyzed in our study did not show any apparent alignment with Group I and II genomes (Fig. 1E). They also had fewer genes in each of the COG families than Group I and II genomes, which may be due to their smaller genome size (Fig. 4). All analyzed Group I genomes show very similar distributions of genes among the COG families, and the distributions vary only slightly to Group II genomes (Fig. 4). These analyses based on COG families further confirmed the close relatedness within Group I strains and their closer relations to the Group II than the Group III strains, as shown in the function-based phylogenic tree (Fig. 1A and Suppl. Fig. 1). Our function-based whole genome phylogeny, as well as the COG functional group analyses (Fig. 4), yielded phylogenetic relationships similar to those observed using 16S rRNA [Hill, 2007; Hill, 2009], but different from the dendrograms generated based on bont or ntnh genes [Zhang, 2003; Hill, 2007], implying horizontal transfer of bont gene clusters between bacteria.
It was proposed that the acquisition of the bont gene clusters in clostridia may have occurred more recently than the divergence of the neurotoxigenic clostridia due to the relatively high degree of similarity among the toxin clusters [Jacobson, 2008]. The HGTs were further evident by the presence of mobile elements flanking the bont gene clusters, as well as the localization of A3, A4, or B1 toxin clusters on a plasmid [Marshall, 2007; Smith, 2007] and C and D toxin clusters on bacteriophages [Johnson, 2001]. It has been suggested that insertion sequence (IS) elements may have contributed to the HGT of the bont gene clusters among strains and species of Group III C. botulinum. However, the tendency to lose unneccesary genetic materials in Group III genomes has made it difficult to trace [Skarin, 2011]. IS elements have been found near the bont gene clusters in all subtypes of serotype A strains, indicating the likelihood of its involvment in HGTs of the toxin clusters [Smith, 2007]. Our sequence analysis showing the presence of three transposases immediately downstream from the bont gene clusters further supports HGT and recombination via mobile elements. Interestingly, although there are only a limited number of transposases in the A1 genomes, many IS elements were identified throughout the genome of C. botulinum ATCC 3502 when analyzed by ISFinder Database (data not shown). The IS elements have been found to be prevalent in large genomes such as firmicutes and the abundance of the IS elements has been shown to correlate to HGT regions [Touchon, 2007]. Therefore, it is likely that C. botulinum genomes are active in HGTs which may have contributed to the mobility of the toxin cluster as well as many functions not yet studied.
Within the toxin gene clusters, the bont gene may have been acquired and evolved distinctly from the ntnh and ha genes, based on sequence analyses of representative subtype A1-A4 bont gene clusters [Arndt, 2006; Jacobson, 2008]. It was proposed that BoNT may have evolved from polyproteins harboring viral proteases [DasGupta, 2006]. The present study analyzing the positions and neighboring sequences of the bont gene clusters on ATCC 3502 revealed similar chromosomal positions of the bont gene locus in Hall A (Fig. 7A) and ATCC 19397 (Fig. 7B). Sequence comparison showed 100% identities of all genes of these A1 bont gene clusters (Suppl. Table 3). These findings varied slightly to a previous study comparing the toxin clusters between Hall A and ATCC 3502, which showed 99–100% amino acid identities of bont, ha17 and ha 33 genes, but 93% and 97% identity for ntnh and ha70 genes, respectively [Zhang, 2003]. These variations may have been errors in the earlier sequencing results or differences in the bacterial culture used in individual laboratories. Analysis of the toxin clusters and their ±10 kb flanking regions revealed a 99% nucleotide sequence identity between Hall A and ATCC 19397 (data not shown), and a G to A transitional substitution in ATCC 3502 at about 8.4 kb upstream of the bont gene cluster. The bont gene clusters in these three A1 strains belong to the HA+ cluster that was found near the oppA/brnQ operon on the chromosome [Hill, 2009; Hill, 2013]. The identical insertion sites provide strong evidence that these three A1 strains were closely related and may have diverged after the acquisition of the bont gene cluster. In addition, the identical A to G substitution in the upstream region of the toxin clusters in both Hall A and ATCC 19397 suggests that the two strains are more closely related as compared to ATCC 3502. A similar relationship was also observed in our COG-based phylogram, showing that Hall A strain is closer to ATCC 19397 than to ATCC 3502 (Fig. 1B).
The availability of the partially assembled genome of NCTC 2916 provides an opportunity to reveal the evolutionary relation between the A1 and A1(B) strains. As reported previously, NCTC 2916 harbors a silent bont/B in its HA+ cluster and a bont/A1 in its orfX+ cluster [Jovita, 1998; Hill, 2013]. The silent bont/B has the full coding region but with a stop codon in amino acid position 128 [Jovita, 1998]. The two toxin gene clusters are located 41 kb apart in the same orientation on Contig 1 (Fig. 7C). DNA sequence identities of the toxin clusters between ATCC 3502 and NCTC 2916 vary from 88% to 100%, with the two ntnh genes being the least similar (Fig. 7C and Suppl. Table 3). Between the two copies of ntnh in NCTC 2916, the one in the HA+ (B) cluster exhibits a higher sequence identity (94%) to the ntnh in ATCC 3502, as compared to only 88% identity for the ntnh in the orfX+ A1 cluster (Suppl. Table 3). This suggests that the composition of the HA+ (B) cluster is more similar to the HA+ A1 clusters in the A1 strains, with exception to the silent bont/B. It was hypothesized that the silent bont/B could have been swapped with the bont/A1, or vice versa, through a potential recombination site within ntnh [East, 1996; Smith, 2007; Jacobson, 2008]. Further alignments of the orfX+ A1 and HA+ (B) clusters of NCTC 2916 showed a chimeric gene organization, where the 3′ end of orfX+ A1 cluster and the 5′ end of HA+ (B) cluster matched to the HA+ A1 cluster. Consequently, this recombination site was proposed to reside in the central 1413 bp overlapping region between the two distant copies of ntnh in NCTC 2916 [Dineen, 2003; Jovita, 1998; Jacobson, 2008]. Like the HA+ A1 cluster, the HA+ (B) cluster is located near the oppA/brnQ operon on the chromosome of NCTC 2916 [Hill, 2013]. Analyses of the short sequences immediately flanking the HA+ (B) cluster of NCTC 2916 indicated a high degree of similarity to the HA+ A1 cluster in the upstream, but not the downstream, region [Dineen, 2003]. To better understand the genomic environment of the insertion sites of the toxin gene clusters, we compared the regions spanning the two clusters of NCTC 2916 on Contig 1 to the A1 genomes, as well as the database in GenBank. The upstream 10 kb region of HA+ (B) cluster of NCTC 2916 shares >95% sequence identity to other A1 strains; while ~96% identity to A1 strains resumes at approximately 1.8 kb downstream region (Fig. 7C and BLAST data not shown). Both the HA+ (B) and HA+ A1 clusters harbor their unique transposase(s) within this 1.8 kb downstream area, providing strong evidence that the transposases were part of the bont gene cluster unit of transfer. The BLAST analysis further showed that the downstream 10 kb region of HA+ (B) cluster is more closely related to A3-A5 strains than the A1 strains, suggesting a closer genetic background between NCTC 2916 and these subtypes. Interestingly, the immediate 600 bp downstream of HA+ (B) cluster showed a 95% sequence identity to the same downstream region of the HA+ B clusters on the pCL plasmids of Okra B1 (Group I) and Eklund 17B (Group II), suggesting a potential origin as well as the involvement of plasmids in the recombination and evolution of the HA+ (B) cluster. It is likely that the HA+ (B) cluster of NCTC 2916 was acquired through homologous recombination between HA+ clusters on a chromosome and a plasmid. Our results further support the hypothesis that the neurotoxin clusters may be evolved from several recombination events based on the study of the HA+ and orfX+ clusters in the A1-A4 strains [Jacobson, 2008].
Although most comparative studies have mainly emphasized the bont gene clusters and their surrounding regions, it is also important to analyze the genomic background of the host bacteria that house the bont gene cluster(s). The genome sizes among the A1 or A1(B) strains range from 3.76 to 4.03 Mb, with Hall A being the smallest and NCTC 2916 being the largest (Suppl. Table 1). These sizes are comparable to most of the genomes sequenced in the genus of Clostridium, but are relatively small when compared to another spore forming bacterial genus, Bacillus, which has its typical genome sizes ranging from 4 to 6.5 kb (http://img.jgi.doe.gov/). Alignments of the genomes of ATCC 3502 to Hall A and/or ATCC 19397 show highly similar genomic arrangements (Figs. 1C and 2B). The function-based phylogenetic analysis of subtype A1 strains displays ATCC 3502, ATCC 19397, and Hall A in sister branches while NCTC 2916 is in an outgroup (Fig. 1B), demonstrating a higher similarity between the former three. The observation was further verified by the Venn diagrams based on COG functional groups (Fig. 5). The four A1 or A1(B) strains share 1252 core COG groups, or 96.5%, of those found in ATCC 3502; while the three A1 strains shared additional 28 functional groups totaling 1280 shared functional groups or 98.2% of those found in ATCC 3502 (Fig. 5). In addition, the Venn diagram showing the gene pool analyses among the A1 genomes revealed over 90% of the genes falling into the core gene pool (Fig. 3). Among the three A1 strains, Hall A and ATCC 19397 share the most number of common genes (99.4%, Fig. 3) and functional groups (99.4%, Fig. 5), which echoes the phylogenetic relationship shown in Figure 1B. Our gene pool analysis also provided evidence that ATCC 3502 genome is closer to ATCC 19397 than Hall A (Fig. 3) as represented by the number of shared genes. A recent study analyzing the A1 genomes identified four major genomic blocks where two smaller blocks were translocated at ~500 kb chromosome position in Hall A when compared to ATCC 3502 and ATCC 19397 [Fang, 2010], which also suggests that Hall A is farther distal from ATCC 3502 than ATCC 19397.
ATCC 3502, the largest genome (3.9 Mbp) among the three A1 strains studied, has its extra gene contents clustered in four segments on the genome, designated Gaps 1 – 4 (Fig. 2B). Our analyses focused primarily on the four gaps based on the sequence alignment between ATCC 3502 and Hall A. Strain ATCC 19379, being a slightly larger genome than Hall A, was also referenced in some analyses. All of the four gap areas absent in Hall A were also absent in ATCC 19397, except for Gap 3 (Figs. 2B and C). Genes missing from the four gap areas are mostly absent from Hall A with only a few fragments at the boundaries aligned elsewhere on the genome, as shown by the cross lines on the ACT plots (Fig. 6B). In addition, the areas surrounding the gaps are highly homologous between ATCC 3502 and Hall A (Fig. 6B), indicating the two strains, as well as ATCC 19379 (Suppl. Fig. 2), share the same genomic background. The truncation and/or insertion of these genes in the gap areas may involve bacteriophages, as suggested by the presence of genes for integrases/resolvase and phage related proteins within the ATCC 3502 sequences corresponding to areas of Gaps 2 and 4 (Suppl. Table 4). Gaps 2 and 4 lack the most numbers of ORFs, at 74 and 84 ORFs, respectively (Table 1). These small ORFs, with an average length of ≤600 bp, encode for either phage or unknown proteins (Suppl. Table 4), which further suggests that these ORFs may have been left from previous recombination events. Gap 3, which is positioned at 27.8 kb for Hall A, is not observed when aligned ATCC 3502 to ATCC 19397. Additionally, genes missing from Gap 3 include those coding for proteins in energy transportation.
Strain Hall A is known for its high yield of BoNT, and therefore has been used in the production of toxin for research and therapeutic applications [Malizio, 2000]. It has been reported that Hall A could produce a significantly higher amount of BoNT during the later stage of growth under a specific toxin producing medium [Bradshaw, 2004; Rao, 2007]. It is not clear whether the lack of energy-related genes from the Gap 3 area in Hall A has contributed to its unique hyper toxinogenic phenotype. Positioned at 29.6 kb, Gap 1 in Hall A lacks genes coding for several membrane proteins (Suppl. Table 4). The missing genes of Gap 1 and Gap 3 that encode for many membrane associated proteins may account partially for the decreased transmembrane proteins found in Hall A and ATCC 19397 (Suppl. Table 1).
To further verify the missing genes by an experimental method, gDNA from ATCC 3502 and Hall A were co-hybridized to the ATCC 3502-based microarray from JCVI. Among the four gap areas in Hall A, only the missing genes of Gap 3 were successfully validated by the microarray experiments showing high Log2 (Cy5/Cy3) ratios, indicating the lack of genes in the gap but not the surrounding areas in Hall A (Fig. 6C). Analysis of the data within and around the gap regions show that our microarray experiments can effectively detect the presence of genes surrounding all four gaps for both strains, as well as Gap 3 genes in ATCC 3502 (Fig. 6C). Surprisingly, missing genes in Gaps 1, 2, and 4 exhibited only background levels of hybridization signals for both ATCC 3502 and Hall A (Fig. 6C and Suppl. Table 4). The lack of signals for our positive control genome, ATCC 3502, is unlikely to be a problem of our microarray hybridization experiments, since the surrounding genes in these gap areas showed the expected positive signals for both ATCC 3502 (Cy5) and Hall A (Cy3). Genomic microarray is known to be prone to hybridization bias due to many factors, such as the dye labeling efficiency, design of the oligos on the microarray, hybridization strength between the oligos and the labeled gDNA, as well as the sizes of the labeled gDNA [Nowak, 2007]. Our microarray data have gone through rigorous normalization to avoid dye labeling bias and variations among repeats within the slides. In addition, two independent studies were performed. The fact that negative hybridization results were observed in the ATCC 3502 genes corresponding to the Gaps 1, 2, and 4 areas, but not the surrounding genes strongly suggests that these gap genes either have weak hybridization strengths, possibly due to their short ORF sizes, or these gap genes are also missing in the ATCC 3502 strain used in this study. Further studies are required to verify if Gaps 1, 2, and 4 genes are present in the ATCC 3502 strain used in our study, and whether it is different from the ATCC 3502 strain used in the sequencing project [Sebaihia, 2007].
The present study reported an in-depth analyses of the genomes of four A1 strains, including one A1(B) strain, with foci on the comparison of the genomic background and the extended region surrounding the bont gene clusters. Our results showed that the four strains share over 90% core genes and over 96% of the functional groups (Figs. 3 and 5). The A1(B) strain, NCTC 2916, exhibited a relatively distal relation to the rest of the A1 strains and may have been more closely related to A3-5 strains based on the BLAST analysis of an extended region downstream of the HA+ (B) cluster (data not shown). The three A1 strains, ATCC 3502, ATCC 19397, and Hall A, exhibited high degrees of homology throughout the genomes, except for a few gaps (Fig. 6) and one translocation [Fang, 2010]. Based on the near 100% homology of the the bont gene clusters and their surrounding regions, it is convincing that the bont gene cluster has been present prior to the divergence of the three A1 strains. Our sequence analyses of the gaps and the extended regions surrounding the bont gene clusters of the three A1 strains provided additional insights to support the phylogenetic relations shown in Figure 1B. ATCC 3502, the larger genome of the three A1 strains, may have diverged earlier from the other two A1 strains as evident by the lack of gaps and the A to G transitional substitution in the upstream region of the toxin cluster. Hall A could have been branched from ATCC 19397 later through a translocation event [Fang, 2010] and the loss of the Gap 3 region. In addition, our BLAST analysis of the immediate downstream region of the HA+ (B) cluster in NCTC 2916 suggested a possible recombination between the HA+ clusters located on the plasmid and chromosome.
Supplementary Material
Highlights.
Three Clostridium botulinum A1 genomes were compared.
The genomes of A1 strains show high degrees of sequence identities.
Four gaps found in Hall A and three in ATCC 19397 when compared to ATCC 3502.
ATCC 19379 may be an evolutionary intermediate between Hall A and ATCC 3502.
Acknowledgments
The authors would like to thank Dr. Eric A. Johnson of University of Wisconsin-Madison for providing the bacterial strain, Drs. Ren-Jang Lin and Carlotta Glackin of Beckman Research Institute of the City of Hope for their assistance in the microarray study, the Pathogen Functional Genomics Resource Center of J. Craig Venter Institute for the microarray slides, and DOE Joint Genomic Institute for the workshop on genomic analysis tools. This project is partially funded by NIH grant 1SC3GM086303.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Virginia Ng, Email: vng@csupomona.edu.
Wei-Jen Lin, Email: weijenlin@csupomona.edu.
References
- Anderson NM, Larkin JW, Cole MB, Skinner GE, Whiting RC, Gorris LGM, Rodriguez A, Buchanan R, Stewart CM, Hanlin JH, Keener L, Hall PA. Food safety objective approach for controlling Clostridium botulinum growth and toxin production in commercially sterile foods. Journal of Food Protection. 2011;74:1956–1989. doi: 10.4315/0362-028X.JFP-11-082. [DOI] [PubMed] [Google Scholar]
- Arndt JW, Jacobson MJ, Abola EE, Forsyth CM, Tepp WH, Marks JD, Johnson EA, Stevens RC. A structural perspective of the sequence variability within botulinum neurotoxin subtypes A1-A4. Journal of Molecular Biology. 2006;362:733–742. doi: 10.1016/j.jmb.2006.07.040. [DOI] [PubMed] [Google Scholar]
- Basavanna U, Gonzalez-Escalona N, Timme R, Datta S, Schoen B, Brown EW, Zink D, Sharma SK. Draft genome sequence of a Clostridium botulinum isolate from water used for cooling at a plant producing low-acid canned foods. Genome Announcements. 2013;1:e00200–12. doi: 10.1128/genomeA.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw M, Dineen SS, Maks ND, Johnson EA. Regulation of neurotoxin complex expression in Clostridium botulinum strains 62A, Hall A-hyper, and NCTC 2916. Anaerobe. 2004;10:321–333. doi: 10.1016/j.anaerobe.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiology and Molecular Biology Reviews. 2004;68:560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AT, Paul CJ, Mason DR, Twine SM, Alston MJ, Logan SM, Austin JW, Peck MW. Independent evolution of neurotoxin and flagellar genetic loci in proteolytic. Clostridium botulinum BMC Genomics. 2009;10 doi: 10.1186/1471-2164-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connan C, Brueggemann H, Mazuet C, Raffestin S, Cayet N, Popoff MR. Two-component systems are involved in the regulation of botulinum neurotoxin synthesis in Clostridium botulinum type A strain Hall. PLoS One. 2012;7:e41848. doi: 10.1371/journal.pone.0041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta BR. Botulinum neurotoxins: perspective on their existence and as polyproteins harboring viral proteases. The Journal of General and Applied Microbiology. 2006;52:1–8. doi: 10.2323/jgam.52.1. [DOI] [PubMed] [Google Scholar]
- Dineen SS, Bradshaw M, Johnson EA. Neurotoxin gene clusters in Clostridium botulinum type A strains: sequence comparison and evolutionary implications. Current Microbiology. 2003;46:345–352. doi: 10.1007/s00284-002-3851-1. [DOI] [PubMed] [Google Scholar]
- Doxey AC, Lynch MDJ, Muller KM, Meiering EM, McConkey BJ. Insights into the evolutionary origins of clostridial neurotoxins from analysis of the Clostridium botulinum strain A neurotoxin gene cluster. BMC Evolutionary Biology. 2008;8 doi: 10.1186/1471-2148-8-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East AK, Bhandari M, Stacey JM, Campbell KD, Collins MD. Organization and phylogenetic interrelationships of genes encoding components of the botulinum toxin complex in proteolytic Clostridium botulinum types A, B, and F: evidence of chimeric sequences in the gene encoding the nontoxic nonhemagglutinin component. International Journal of Systematic Bacteriology. 1996;46:1105–1112. doi: 10.1099/00207713-46-4-1105. [DOI] [PubMed] [Google Scholar]
- Fang P-K, Raphael BH, Maslanka SE, Cai S, Singh BR. Analysis of genomic differences among Clostridium botulinum type A1 strains. BMC Genomics. 2010;11 doi: 10.1186/1471-2164-11-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciosa G, Floridi F, Maugliani A, Aureli P. Differentiation of the gene clusters encoding botulinum neurotoxin type A complexes in Clostridium botulinum type A, Ab, and A(B) strains. Applied and Environmental Microbiology. 2004;70:7192–7199. doi: 10.1128/AEM.70.12.7192-7199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KK, Smith TJ. Genetic diversity within Clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Current Topics in Microbiology and Immunology. 2013;364:1–20. doi: 10.1007/978-3-642-33570-9_1. [DOI] [PubMed] [Google Scholar]
- Hill KK, Xie G, Foley BT, Smith TJ, Munk AC, Bruce D, Smith LA, Brettin TS, Detter JC. Recombination and insertion events involving the botulinum neurotoxin complex genes in Clostridium botulinum types A, B, E and F and Clostridium butyricum type E strains. BMC Biology. 2009;7 doi: 10.1186/1741-7007-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KK, Smith TJ, Helma CH, Ticknor LO, Foley BT, Svensson RT, Brown JL, Johnson EA, Smith LA, Okinaka RT, Jackson PJ, Marks JD. Genetic diversity among botulinum neurotoxin-producing clostridial strains. Journal of Bacteriology. 2007;189:818–832. doi: 10.1128/JB.01180-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MJ, Lin G, Raphael B, Andreadis J, Johnson EA. Analysis of neurotoxin cluster genes in Clostridium botulinum strains producing botulinum neurotoxin serotype A subtypes. Applied and Environmental Microbiology. 2008;74:2778–2786. doi: 10.1128/AEM.02828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EA, Bradshaw M. Clostridium botulinum and its neurotoxins: a metabolic and cellular perspective. Toxicon. 2001;39:1703–1722. doi: 10.1016/s0041-0101(01)00157-x. [DOI] [PubMed] [Google Scholar]
- Jovita MR, Collins MD, East AK. Gene organization and sequence determination of the two botulinum neurotoxin gene clusters in Clostridium botulinum type A(B) strain NCTC 2916. Current Microbiology. 1998;36:226–231. doi: 10.1007/s002849900299. [DOI] [PubMed] [Google Scholar]
- Luquez C, Raphael BH, Joseph LA, Meno SR, Fernandez RA, Maslanka SE. Genetic diversity among Clostridium botulinum strains harboring bont/A2 and bont/A3 genes. Applied and Environmental Microbiology. 2012;78:8712–8718. doi: 10.1128/AEM.02428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malizio CJ, Goodnough MC, Johnson EA. Purification of Clostridium botulinum type A neurotoxin. Methods in Molecular Biology. 2000;145:27–39. doi: 10.1385/1-59259-052-7:27. [DOI] [PubMed] [Google Scholar]
- Marshall KM, Bradshaw M, Pellett S, Johnson EA. Plasmid encoded neurotoxin genes in Clostridium botulinum serotypes A subtypes. Biochemical and Biophysical Research Communications. 2007;361:49–54. doi: 10.1016/j.bbrc.2007.06.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak NJ, Miecznikowski J, Moore SR, Gaile D, Bobadilla D, Smith DD, Kernstine K, Forman SJ, Mhawech-Fauceglia P, Reid M, Stoler D, Loree T, Rigual N, Sullivan M, Weiss LM, Hicks D, Slovak ML. Challenges in array comparative genomic hybridization for the analysis of cancer samples. Genetics in Medicine. 2007;9:585–595. doi: 10.1097/gim.0b013e3181461c4a. [DOI] [PubMed] [Google Scholar]
- Rao S, Starr RL, Morris MG, Lin WJ. Variations in expression and release of botulinum neurotoxin in Clostridium botulinum type A strains. Foodborne Pathogens and Disease. 2007;4:201–207. doi: 10.1089/fpd.2006.0076. [DOI] [PubMed] [Google Scholar]
- Sebaihia M, Peck MW, Minton NP, Thomson NR, Holden MTG, Mitchell WJ, Carter AT, Bentley SD, Mason DR, Crossman L, Paul CJ, Ivens A, Wells-Bennik MHJ, Davis IJ, Cerdeno-Tarraga AM, Churcher C, Quail MA, Chillingworth T, Feltwell T, Fraser A, Goodhead I, Hance Z, Jagels K, Larke N, Maddison M, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Sanders M, Simmonds M, White B, Whithead S, Parkhill J. Genome sequence of a proteolytic (Group I) Clostridium botulinum strain Hall A and comparative analysis of the clostridial genomes. Genome Research. 2007;17:1082–1092. doi: 10.1101/gr.6282807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarin H, Segerman B. Horizontal gene transfer of toxin genes in Clostridium botulinum: involvement of mobile elements and plasmids. Mobile Genetic Elements. 2011;1:213–215. doi: 10.4161/mge.1.3.17617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Hill KK, Foley BT, Detter JC, Munk AC, Bruce DC, Doggett NA, Smith LA, Marks JD, Xie G, Brettin TS. Analysis of the neurotoxin complex genes in Clostridium botulinum A1-A4 and B1 strains: BoNT/A3, /Ba4 and /B1 clusters are located within plasmids. PLoS One. 2007;2:e1271. doi: 10.1371/journal.pone.0001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer SC, Carter AT, Webb MD, Wachnicka E, Crossman LC, Sebaihia M, Peck MW. Genomic and physiological variability within group II (non-proteolytic) Clostridium botulinum. BMC Genomics. 2013;14 doi: 10.1186/1471-2164-14-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanongsaksrikul J, Chaicumpa W. Botulinum neurotoxins and botulism: a novel therapeutic approach. Toxins. 2011;3:469–488. doi: 10.3390/toxins3050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillier ERM, Collins RA. The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J MOl Evol. 2000;50:249–257. doi: 10.1007/s002399910029. [DOI] [PubMed] [Google Scholar]
- Touchon M, Rocha EPC. Causes of insertion sequences abundance in prokaryotic genomes. Mol Biol Evol. 2007;24(4):969–981. doi: 10.1093/molbev/msm014. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lin WJ, Li S, Aoki KR. Complete DNA sequences of the botulinum neurotoxin complex of Clostridium botulinum type A-Hall (Allergan) strain. Gene. 2003;315:21–32. doi: 10.1016/s0378-1119(03)00792-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.