

Abstract
Background
M protein mutant vesicular stomatitis virus (M51R-VSV) has oncolytic properties against many cancers. However, some cancer cells are resistant to M51R-VSV. Herein, we evaluate the molecular determinants of VSV resistance in pancreatic adenocarcinoma cells.
Methods
Cell viability and the effect of β-interferon (IFN) was analyzed using MTS assay. Gene expression was evaluated via microarray analysis. Cell infectability was measured by flow cytometry. Xenografts were established in athymic nude mice and treated with intratumoral M51R-VSV.
Results
Four of five pancreatic cancer cell lines were sensitive to M51R-VSV, while Panc03.27 cells remained resistant (81±3% viability 72-hours after single cycle infection). Comparing sensitive MiaPaCa2 to resistant Panc03.27 cells, significant differences in gene expression was found relating to IFN signaling (p=2×10-5), viral entry (p=3×10-4) and endocytosis (p=7×10-4). MiaPaCa2 cells permitted high levels of VSV infection, while Panc03.27 cells were capable of resisting VSV cell entry even at high MOIs. Extrinsic β-IFN overcame apparent defects in IFN-mediated pathways in MiaPaCa2 cells conferring VSV resistance. In contrast, β-IFN decreased cell viability in Panc3.27 cells suggesting intact anti-viral mechanisms. VSV treated xenografts exhibited reduced tumor growth relative to controls in both MiaPaCa2 (1423 ± 345% vs 164 ± 136%, p<0.001) and Panc3.27 tumors (979 ± 153% vs 50 ± 56%, p=0.002). Significant lymphocytic infiltration was seen in M51R-VSV treated Panc03.27 xenografts.
Conclusions
Inhibition of VSV endocytosis and intact IFN-mediated defenses are responsible for M51R-VSV resistance in pancreatic adenocarcinoma cells. M51R-VSV treatment appears to induce anti-tumor cellular immunity in vivo which may expand its clinical efficacy.
Keywords: Vesicular Stomatitis Virus, Pancreatic Adenocarcinoma, Interferon, Viral Endocytosis, Xenograft
Introduction
Vesicular stomatitis virus (VSV) is among several oncolytic viruses currently being developed as anticancer therapies. VSV, the prototypical member of the family Rhabdoviridae, is a negative-stranded RNA virus whose genome encodes for five proteins: nucleocapsid (N), polymerase proteins (L and P), surface glycoprotein (G) and peripheral matrix protein (M). VSV is a potently cytolytic virus that selectively replicates in cancer cells that have down-regulated their antiviral responses[1]. The selectivity of VSV for cancer cells can be enhanced by introducing mutations in the M protein, such as in the M51R variant of VSV (M51R-VSV), which contains a single arginine for methionine amino acid substitution at position 51 in the M protein[2-3]. The mutant M protein has a decreased ability to inhibit host cell antiviral mechanisms. As a result, normal cells are able to resist M51R-VSV infection[4-5] by mounting anti-viral defenses, such as IFN-mediated antiviral signaling. In contrast, many cancer cells remain susceptible to M51R-VSV infection because they possess defects in antiviral pathways[2,6]. Several reports have shown that M51R-VSV is more selective for tumor cells and causes oncolysis in a variety of cancer types; including prostate cancer[7], breast cancer[8], glioblastoma[9], colorectal cancer[5] malignant melanoma [10] and neuroendocrine tumors[11]. Several VSV variants have been developed that are similarly selective for cancers with defective antiviral responses[1] and one of these is currently in a phase one clinical trial for treatment of hepatocellular carcinoma (http://clinicaltrials.gov/show/NCT01628640).
While these preliminary reports are encouraging, VSV is not universally oncolytic in all tumor subtypes and significant variation in VSV sensitivity exists, even among cancers from the same anatomical site[6]. For example, both VSV resistant and VSV sensitive cell lines have been described in colorectal cancer[5], prostate cancer[7], breast cancer[8], malignant melanoma[10], malignant mesothelioma[12], and bladder cancer[13]. Based on available data, VSV-resistance may be as high as 36% in pancreatic adenocarcinoma and has been observed in cells from both primary and metastatic sites[14-15]. Preserved intact anti-viral mechanisms are thought to confer resistance in VSV-resistant cell lines but additional mechanisms may also contribute to resistance[5,7,14-16].
Oncolytic virus therapy is a particularly attractive strategy for treatment of cancers for which current therapies are ineffective. As such, despite advances in many other malignancies, pancreatic adenocarcinoma remains a significant therapeutic challenge. The five-year relative survival rate for patients with pancreatic cancer is 6%—the lowest among all cancers[17]. Clinical outcomes are so poor because the majority of pancreatic cancer patients present with locally advanced or metastatic disease and pancreatic adenocarcinoma is largely resistant to traditional systemic treatments. Clearly, innovative and effective therapies are critical to improving clinical outcomes in patients with pancreatic cancer.
In the work presented herein, we confirm the results of Murphy et al.[14] and Moerdyk-Schauwecker et al.[15] and extend their finding by analyzing the oncolytic effects of VSV in a panel of additional pancreatic adenocarcinoma cells with significant variability in their VSV susceptibility. Using microarray gene analysis, we explored the genetic differences between VSV-sensitive and VSV-resistant cell lines and thereby hypothesized that differences in interferon (IFN) signaling and viral endocytosis are key molecular determinants of VSV susceptibility. In support of these hypotheses, we showed that resistant cells are capable of blocking VSV infection during the early stages of viral replication and possess intact IFN responses. We also found that the integrity IFN-signaling can explain the VSV susceptibility seen in sensitive cell lines. In a murine xenograft model, we found that tumors from both sensitive and resistant cells responded to intratumoral M51R-VSV treatment. Histological examination of treated tumor suggests that adaptive cellular immunity contributes to the oncolysis of the in vitro-resistant xenografts. Collectively, these data establish oncolytic VSV is a viable therapeutic option for pancreatic adenocarcinoma. More specifically it identifies two significant molecular mechanisms of VSV resistance and provides a framework for further research into the immunologic responses to VSV.
Materials and Methods
Cells and Viruses
Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cell lines were obtained from the American Type Culture Collection (ATCC) and were grown in DMEM (Panc 1 and MiaPaCa2) or RPMI 1640 (BxPC3, Panc 03.27, and Panc 10.05) supplemented with various additives depending on the cell line according to ATCC specifications. The recombinant VSV viruses, rwt-VSV and M51R-VSV, were isolated from infectious VSV cDNA clones and virus stocks were prepared using BHK cells as previously described[18]. Cells were grown in monolayers to 70 to 90% confluence and infected in small volumes at multiplicities of infection (MOIs) as specified in each experiment.
Cell Viability Assays
Pancreatic cancer cells were plated in 96-well plates containing 3,000 cells/well. Cells were infected with rwt and M51R viruses at various MOIs. At 24, 48, and 72 hours post-infection, the quantity of live cells were measured by MTS assay (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, Wisconsin) according to manufacturer's instructions.
Microarrays
RNA was isolated from MiaPaCa3 and Panc 03.27 cell cultures using TRIzol reagent (Invitrogen, Carlsbad, California) according to manufacturer's protocol. RNA samples were then hybridized to human genome U219 array strips (Affymetrix, Inc., Santa Clara, California) which measures the expression of more than 20,000 genes. Levels of expression were measured by Robust Multi-array Average[19] normalized log2 converted probe set pixel intensity. Differentially expressed genes were identified by empirical Bayers method[20] implemented in R Biocondoctor limma package. Selection of differentially expressed genes was determined by a fold change >2, empirical Bayers p-value <0.00005 or FDR adjusted empirical Bayers p-value <0.0001. These data were analyzed through the use of Interactive Pathway Analysis (Ingenuity Systems, www.ingenuity.com) to identify specific molecular pathways associated with the differential gene expression.
Viral Infectability
The ability of VSV-M51R to infect pancreatic cancer cells was determined by green fluorescent protein (GFP) expression using flow cytometry analysis of infected cells. Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cells were infected with GFP labeled M51R virus (rGFP-M51R) at increasing MOIs. At specific time points, the cells were washed and fixed in 2% paraformaldehyde. GFP expression was quantified using a Becton Dickinson FASCCaliber flow cytometer.
Host Cell and Viral Protein Synthesis
To analyze the ability of rwt and M51R viruses to produce viral proteins in pancreatic cancer cells, Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cells were infected with rwt-VSV and M51R-VSV at an MOI of 5 plaque-forming unit (pfu) per cell. At various time points post-infection, cells were labeled with a 15 minute pulse of [35S]methionine (200μCi/mL) in a small volume of methionine-free medium. Cells were washed with PBS and harvested in radioimmunoprecipitation assay (RIPA) buffer. Cell extracts were normalized for protein levels by protein assay (DC Protein Assay Kit; Bio-Rad Laboratories, Hercules, California) and analyzed by 10% sodium dodocyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by phosphorescence imaging. Radioactivity of viral N protein bands and background host proteins were quantified with ImageQuant software (Molecular Dynamics, Inc., Sunnyvale, California)[18].
β-IFN Production
To determine the amount of β-IFN (IFN) produced by pancreatic cancer cells in response to VSV infection, Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cells were inoculated with rwt-VSV and M51R-VSV (MOI of 5 pfu/cell). At specified time points, 150μL of media was collected. The amount of β-IFN in each sample was quantified by enzyme-linked immunosorbent assay (VeriKine Human Interfeon-Beta ELISA Kit; PBL IFNSource, Piscataway, New Jersey) according to manufacturer's instructions.
IFN Responsiveness
Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cells were plated onto 96-well dishes and pretreated with varying amounts (0 to 25,000 IU/mL/100,000 cells) of human β-IFN (PBL IFNSource, Piscataway, New Jersey) for 8 hours. Cells were then inoculated with rwt-VSV and M51R-VSV (MOI of 5 pfu/cell). After 48 or 72 hours, the percentage of live cells was measured by MTS assay (CellTiter 96 Aqueous One Solution Cell Proliferation Assay; Promega, Madison, Wisconsin) according to manufacturer's instructions. Controls included mock-treated cells infected with virus and β-IFN treated cells not challenged with VSV.
Xenograft Treatment
MiaPaCa2 and Panc 03.27 cells were screened for animal pathogens (service provided by RADIL, Columbia Missouri) and harvested from semiconfluent cultures. After being suspended in phosphate buffer solution (PBS), 2 × 106 cells were injected subcutaneously in the right flank of athymic C57BL/6-nu/nu mice. Animals were monitored for tumor development three times a week by visual inspection and palpation of the injection site. Palpable tumors were measured with calipers and the tumor volume was calculated using the formula: volume = width2 × length/2. Once tumors reached a threshold volume of 5-10mm3, tumor-bearing mice were randomly assigned to receive intratumoral injection of 1×108 pfu of M51R-VSV or mock injection with PBS as negative controls. Tumor volume was measured three times a week. Several mice from each group were sacrificed during the first post-treatment week and the tumors were harvested for immunohistochemical analysis.
Immunohistochemistry
Harvested tumors were immersion-fixed in 10% buffered formalin and processed into paraffin blocks. Paraffin blocks were sectioned at 4μm. The tissue was stained with Hematoxylin and Eosin for histologic examination. Adjacent sections were prepared for immunohistochemical staining. Antigen retrieval was performed in Citrate buffer, pH 6.0 using a Cuisinart pressure cooker. Non-specific binding was blocked using Tris wash buffer containing 0.5% casein. For G-protein staining, sections were incubated overnight at 4ºC with rabbit polyclonal VSV-G-tag envelope glycoprotein (Fitzgerald Industries International, MA) at a 1:100 dilution. The sections were then incubated in supersensitive biotinylated goat anti-rabbit at 1:20 dilution, followed supersensitive streptavidin alkaline phosphatase at 1:20 (BioGenex, San Ramon, CA). Vector Red substrate kit I (Vector Laboratories Inc., Burlingame, CA) was used to visualize the antigen-antibody complex. Sections were counterstained in Mayer's Hematoxylin. Negative controls consisted of the omission of the primary antibody, which was substituted by non-immune rabbit serum (BioGenex, San Ramon, CA). B-cell and NK staining was performed in a similar matter using anti-CD20 (Leica Microsystems Inc, Buffalo Grove, IL) and anti-CD56 (Leica Microsystems Inc, Buffalo Grove, IL) antibodies, respectively.
Statistical analysis
A paired student t-test was used to analyze significances of differences between groups at individual time points for experiments containing two experimental groups. Repeated measures analysis of variance was used to determine significant differences between groups at different time points. Repeated measures analysis of variance was used to assess the significance of differences in tumor size relative to that seen before treatment. Specifically, we tested whether there were significant differences in the mean percent change in tumor volume between the treatment and control groups over time. Values were considered statistically significant if p values are <0.01.
Results
Variable Susceptibility of Pancreatic Adenocarcinoma Cells to VSV
The oncolytic activity of VSV in pancreatic adenocarcinoma was evaluated using a panel of five cell lines (Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05). Susceptibility to VSV was evaluated using either wild-type or M protein mutant virus (rwt-VSV and M51R-VSV, respectively). Comparing the two viruses reveals the effect of host cell responses to VSV, because rwt-VSV suppresses, while M51R-VSV induces, host cell responses[2]. At low MOIs, a small percentage of cells are initially infected and further infectivity and killing required viral replication and spread to surrounding cells. In contrast, most cells should be infected shortly after inoculation using an MOI of 10 pfu/cell and viral spread throughout the culture is not required—representing single-cycle infection.
As shown in Figure 1, three of the pancreatic cancer cell lines (Panc 1, MiaPaCa2, and BxPC3) were similarly sensitive to both rwt-VSV and M51R-VSV. In these three cell lines, cell viability decreased with time under both single-cycle and multi-cycle infection conditions. By 72 hours, cell viability was between 1 and 20% under all conditions, suggesting that these cells are susceptible to VSV and support viral replication. Panc 10.05 cells demonstrated delayed sensitivity to VSV compared to the other cells as they were moderately resistant to VSV at 48 hours, but by 72 hours cell viability was <20 % under all conditions. In contrast, Panc 03.27 remained relatively resistant to the oncolytic effects of VSV. This resistance was especially evident under multi-cycle infection conditions where cell viability was greater than 85% even after 72 hours post-infection, suggesting that Panc 03.27 cells are capable of resisting VSV replication.
Figure 1.

Pancreatic cancer cell viability after rwt-VSV or M51R-VSV infection. Cells were infection at indicated multiplicities of infection (MOIs). At 24 and 48 hours post-infection, live cells were quantified by MTS assay. Data are expressed as a percentage of mock-infected cell viability and represent the mean ± standard deviation of at least three independent experiments.
Gene Expression differs between Sensitive and Resistant Pancreatic Adenocarcinoma Cells
To begin to understand the genetic differences between VSV-sensitive and VSV-resistant pancreatic adenocarcinoma cells, the RNA expression of sensitive MiaPaCa2 and resistant Panc 03.27 cells were analyzed using microarray analysis. Genes which varied significantly between cell lines were identified and were linked to specific cell functions and cell signaling pathways. This analysis allowed us to detect global genetic differences in cell function and pathways between VSV-sensitive and VSV-resistant cell lines and potentially identify molecular determinants of VSV sensitivity.
Relative to MiaPaCa2 cells, 2131 genes were overexpressed and 1233 genes were underexpressed in Panc 03.27 cells. The top five pathways that displayed genetic difference between MiaPaCa2 and Panc 03.27 cells are shown in Table 1. IFN signaling and endocytotic viral entry have direct implications to the cell's potential VSV-susceptibility. As discussed above, defective IFN signaling is the leading hypothesis by which VSV is capable of infecting and destroying cancer cells while leaving normal cells relatively unharmed. The observation that IFN signaling differs significantly between VSV-sensitive and VSV-resistant pancreatic cancer cells supports the hypothesis that VSV-resistant cancer cells have intact anti-viral IFN pathways. Secondly, the genetic differences in pathways relating to viral endocytosis gives rise to a second potential mechanism of VSV-resistance. Cells that do not permit viral entry would not support the downstream steps of viral replication and protein synthesis.
Table 1.
Top five pathways displaying significant genetic differences between VSV-resistant and VSV-sensitive pancreatic cancer cells, identified by microarray gene expression and analyzed using Interactive Pathway Analysis (Ingenuity Systems, www.ingenuity.com).
| Pathway | p-value |
|---|---|
| Interferon Signaling | 2.28 × 10-5 |
| p53 Signaling | 8.75 × 10-5 |
| Virus Entry via Endocytic Pathways | 3.30 × 10-4 |
| Role of Tissue Factor in Cancer | 6.26 × 10-4 |
| Caveolar-mediated Endocytosis Signaling | 6.75 × 10-4 |
Given these results, we sought to analysis our cell lines based on VSV-susceptibility to support or challenge the hypotheses that VSV cell entry inhibition and intact IFN signaling are key molecular determinants to the oncolytic resistance in VSV-resistant pancreatic cancer cells.
VSV Infectability in Pancreatic Adenocarcinoma Cells
The kinetics of VSV cell entry and subsequent viral replication were determined by quantifying the percentage of pancreatic cancer cells infected with VSV over time. Panc 1, MiaPaCa2, BxPC3, Panc 03.27, and Panc 10.05 cells were inoculated with an M51R virus that expresses green fluorescent protein (GFP-M51R). Using varying MOIs and at specified times post-infection, the cells were harvested and analyzed by flow cytometry. Histograms from flow cytometry analysis (Figure 2) were gated on GFP expression to quantify the percentage of cells that express GFP and therefore correspond to the percentage of cells infected with GFP-M51R virus.
Figure 2.
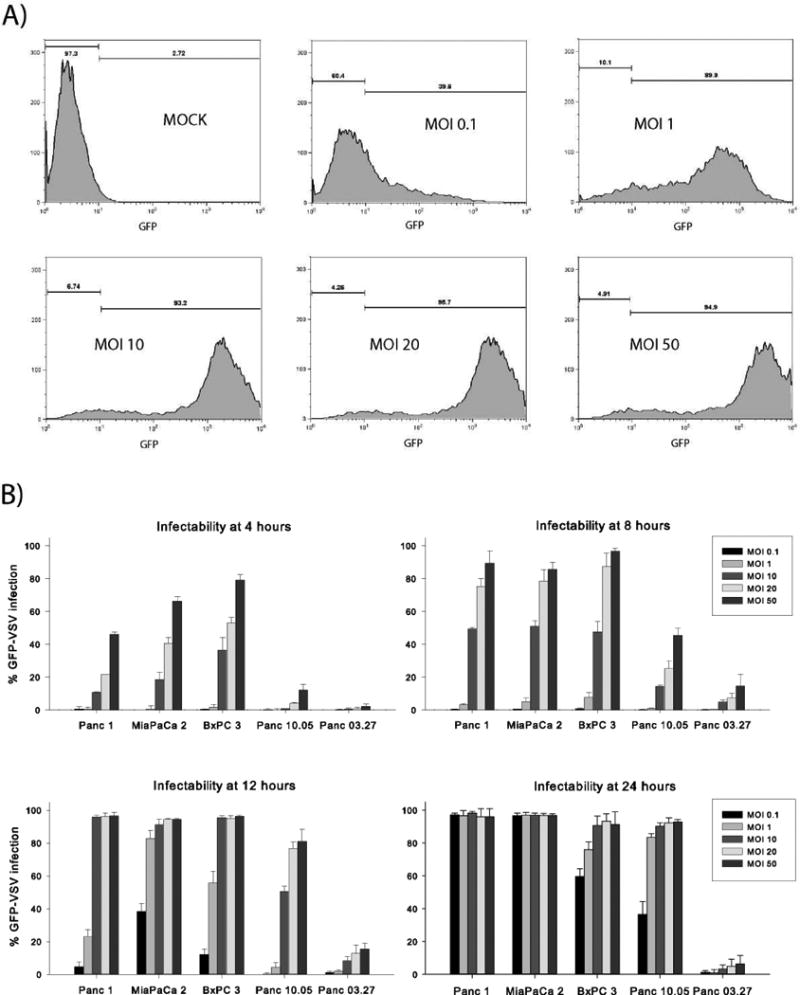
Pancreatic cancer cell infectability using GFP-labeled M51R-VSV (GFP-M51R). Cells were infected with GFP-M51R virus at increasing MOIs (0.1 to 50 pfu/cell). At indicated times post-infection, cells were analyzed for GFP expression by flow cytometry. (A) Representative histograms from MiaPaCa2 cells analyzed 12 hours after infection showing increasing proportions of cells expressing GFP at increasing MOIs. (B) The percentage of pancreatic cancer cells expressing GFP under each condition, expressed as the mean ± standard deviation from three independent experiments.
As shown in Figure 2, GFP-M51R infectability increased with increasing MOIs and over time in sensitive cell lines. By 24 hours post-infection at high MOIs (10 to 50 pfu/cell), nearly all Panc 1, MiaPaCa2, BxPC3 and Panc 10.05 cells were infected with virus. At lower MOIs (0.1 and 1 pfu/cell), BxPC3 and Panc 10.05 cells demonstrated moderate but increasing infectability, while Panc 1 and MiaPaCa2 cells were nearly completely infected, indicating that Panc 1 and MiaPaCa2 cells promote more rapid and complete viral infection and replication. Consistent with the cell viability data above, viral infectability was significantly delayed in Panc 10.05 cells compared to the other sensitive lines.
In stark contrast, Panc 03.27 cells resisted GFP-M51R infection at 24 hours even at extremely high MOIs (6.4±5.1% infectability at 50 pfu/cell). These results suggest that the VSV-resistance in Panc 03.27 cells is determined early in the replication cycle, presumably by blocking VSV entry into the cell or by inhibiting replication shortly after the virus enters the cells.
Viral and Host Cell Protein Synthesis
To further evaluate the ability of VSV to infect and replicate in pancreatic cancer cells, we examined the kinetics of viral protein expression following rwt-VSV and M51R-VSV infection. At specified time points after viral infection, the cells were methionine-starved, pulsed with [35S]-methionine, harvested and lysed. Cell lysates were subjected to protein electrophoresis and analyzed by phosphorescence imaging. The capacity of viral protein synthesis was measured by quantifying the intensity of the viral N protein (the most abundant viral protein) above background and is expressed as the percentage of intensity measured from a comparable region in mock-infected cells. Representative images and N protein quantification for each cell line is shown in Figure 3.
Figure 3.
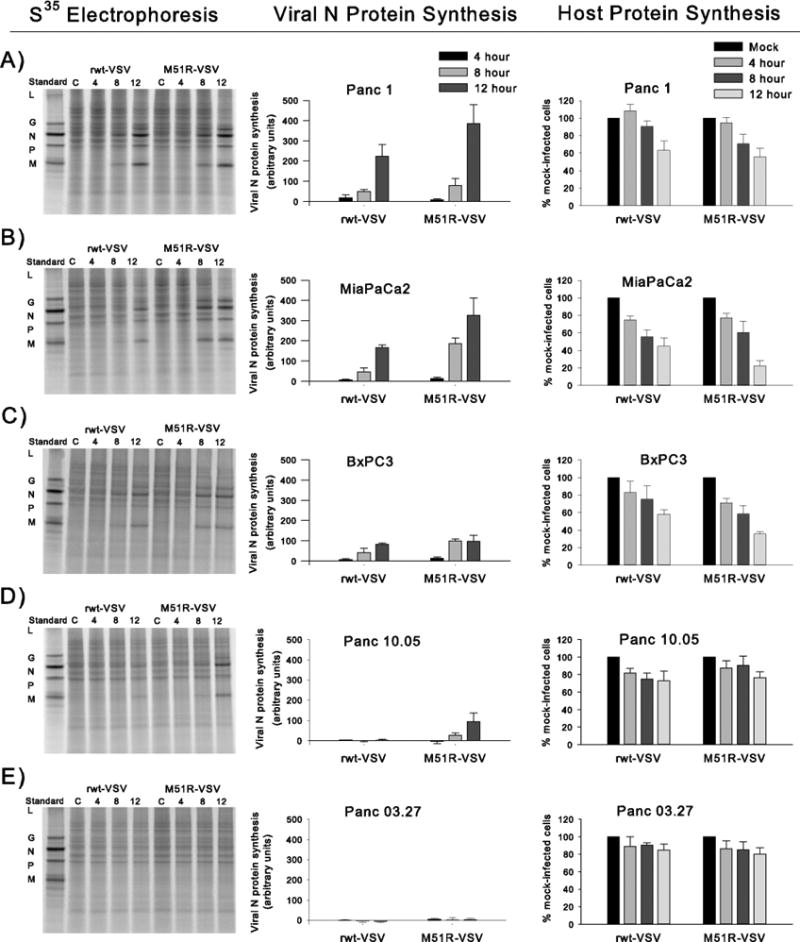
Viral and host cell protein synthesis in response to rwt-VSV and M51R-VSV infections. After viral infection at an MOI of 5 pfu/cell, cells were labeled with [35S]methionine at specified times. Proteins were analyzed by SDS-PAGE and phosphorescence imaging. In the first column representative phosphorescence images are displayed for each cell line. Standard viral proteins (L, G, N, P, and M) are shown in the first lane in each image as reference. The graphs in the middle column represent viral protein synthesis. In order to compare cell types, the radioactivity of the N protein in each lane was quantified and normalized by dividing the intensity of the N protein band by the intensity of a comparable region in mock-infected cells. In the final column, host cell protein production is quantified by measuring the signal intensity of two sections in each lane between viral protein bands and is presented as a percentage intensity from mock-infected cells. Results for each cell line are presented by row: (A) Panc 1, (B) MiaPaCa2, (C) BxPC3, (D) Panc10.05, and (E) Panc 03.27. Data in the graphs are expressed as the mean of each experimental result ± standard deviation of at least three independent experiments.
Within 8 to 12 hours there was significant viral protein synthesis in Panc 1, MiaPaCa2, and BxPC3 cells in response to both rwt and M51R viruses. In support of previous data, Panc 10.05 cells demonstrated delayed viral protein production with significant amounts of viral proteins produced only by M51R-VSV infection at 12 hours. In contrast, there was no significant viral protein synthesized by the Panc 03.27 cells in response to VSV infection. This observation indicates that Panc 03.27 cells block VSV infection prior to the establishment of significant levels of viral protein synthesis.
The ability of VSV to inhibit host cell protein synthesis was also evaluated (far right column, Figure 3). As expected, host cell protein production decreased over time in response to VSV infection in Panc 1, MiaPaCa2 and BxPC3 cells. In contrast, there was no significant decline in host cell protein synthesis in Panc 10.05 or Panc 03.27 cells within 12 hours of VSV infection.
Production of β-IFN in response to VSV Infection
As discussed above, VSV selectively targets tumor cells over normal tissue due to acquired defects in antiviral defenses, such as type I IFN responses. Some cancer cells may possess intact IFN signaling which would enable them to resist VSV oncolysis much like non-cancerous cells. To evaluate IFN signaling in pancreatic adenocarcinoma cells we measured the amount of β-IFN produced in response to VSV infection and assessed the cells response to extrinsic β-IFN.
The quantity of β-IFN produced by pancreatic cancer cells following rwt-VSV and M51R-VSV infection was measured at specified time points post-infection (Figure 4). Sensitive Panc 1 and MiaPaCa2 cells produced insignificant amounts of β-IFN in response to VSV infection, suggesting that these cells are incapable of mounting IFN-mediated anti-viral responses. Panc 10.05 produced comparatively moderate amounts of β-IFN which could explain their relative delayed sensitivity to VSV and implies that these cells retain some ability to resist VSV infection from seemingly less potent anti-viral defects. Interestingly, BxPC3 cells produced the most β- IFN despite being sensitive to VSV. Given that BxPC3 cells permit VSV infection and support VSV replication, different anti-viral signaling defects must exist to explain their sensitivity — perhaps defects downstream from β-IFN production which block the cells' response to β-IFN.
Figure 4.
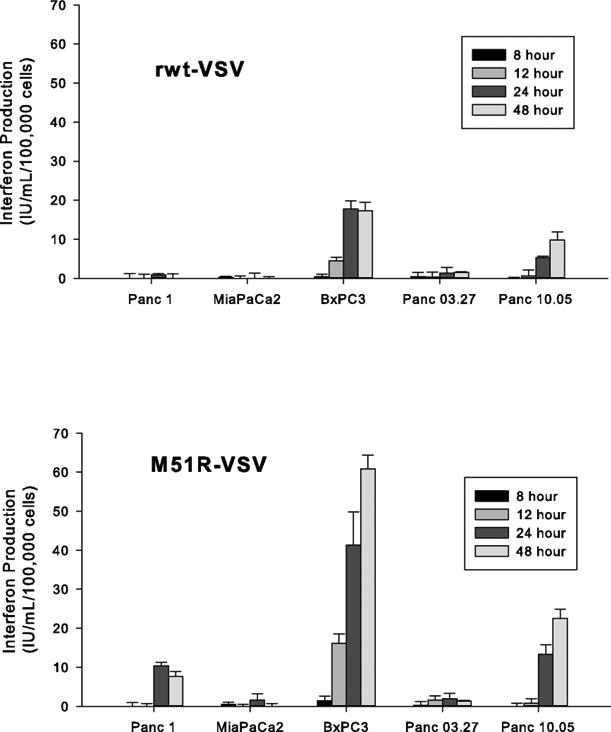
The production of β-IFN in response to rwt-VSV and M51R-VSV infection. Pancreatic cancer cells were infected at an MOI of 5 pfu/cell. At indicated times post-infection, small aliquots were removed and the amount of β-IFN (IU/mL/100,000 cells) was measured by enzyme-linked immunosorbent assay. The data are presented as the mean ± standard deviation from three independent experiments.
Panc 03.27 cells did not produce significant amounts of β-IFN, despite being VSV-resistant. This observation is not unexpected given our infectability results above. Since Panc 03.27 cells appear to block VSV infection by inhibiting early stages in virus replication, anti-viral responses would not be induced. Therefore, the resistance in Panc 03.27 does not appear to be dependent on the integrity of IFN-mediated responses.
Responsiveness of Pancreatic Adenocarcinoma Cells to Extrinsic β-IFN prior to VSV Treatment
The ability of VSV-permissive pancreatic cancer cells to mount IFN-mediated anti-viral mechanisms was further evaluated by pre-treating cells with increasing concentrations of β-IFN prior to VSV infection. As shown in Figure 5, high levels of β-IFN protected Panc 1, MiaPaCa2, BxPC3, and Panc 10.05 cells from the oncolytic effects of VSV. The ability of high-dose β-IFN to confer VSV-resistance suggests that defects in IFN-mediated anti-viral mechanisms exist in these cells. IFN-signaling is not completely defective however, but requires high levels of β-IFN.
Figure 5.
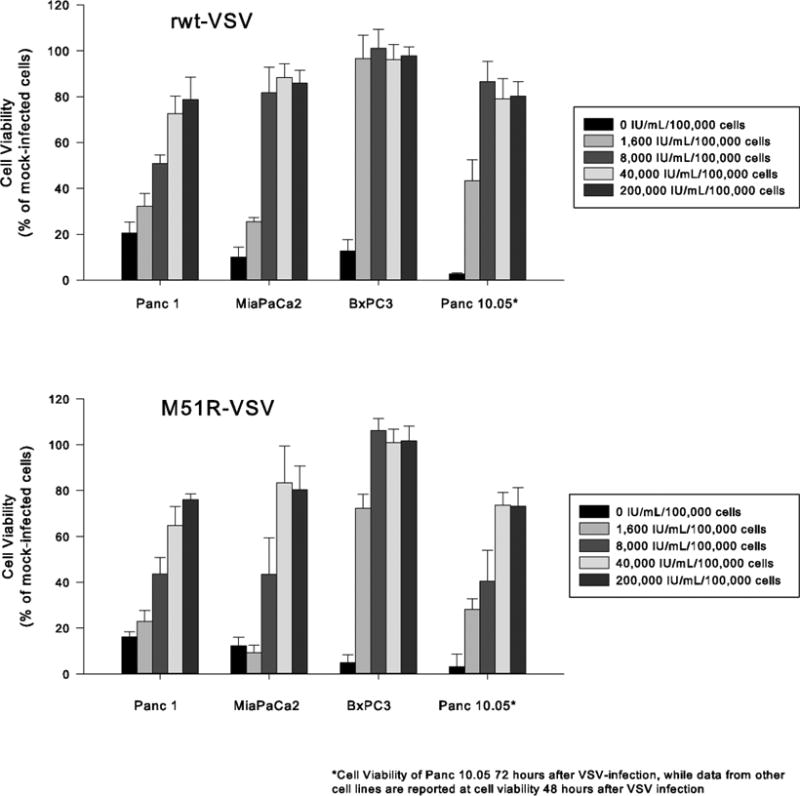
The responsiveness of Panc1, MiaPaCa2, BxPC3, and Panc10.05 pancreatic cancer cells to β-IFN. Cells were incubated with varying concentrations of β-IFN (0 to 40,000 IU/mL/100,000 cells) for 8 hours and then challenged with rwt-VSV or M51R-VSV (MOI of 5 pfu/cell). Cell viability was measured by MTS assay 48 hours (Panc1, MiaPaCa2 and BxPC3) or 72 hours (Panc10.05) after VSV infection. Data are expressed as the percentage of β-IFN treated, mock-infected cells and presented as the mean ± standard deviation of three independent experiments.
In contrast to the VSV-sensitive pancreatic cancer cells, pre-treatment with even low-dose β-IFN had a detrimental effect on virus infection in Panc 03.27 cells (see Figure 6). Given their inherent resistance to VSV replication, it was not surprising that β-IFN did not enhance Panc 03.27 cells' resistance to the oncolytic effects of VSV. Instead, normal levels of β-IFN appear to inhibit cell proliferation and/or induce apoptosis suggesting that IFN-mediated pathways remain intact in Panc 03.27 cells.
Figure 6.
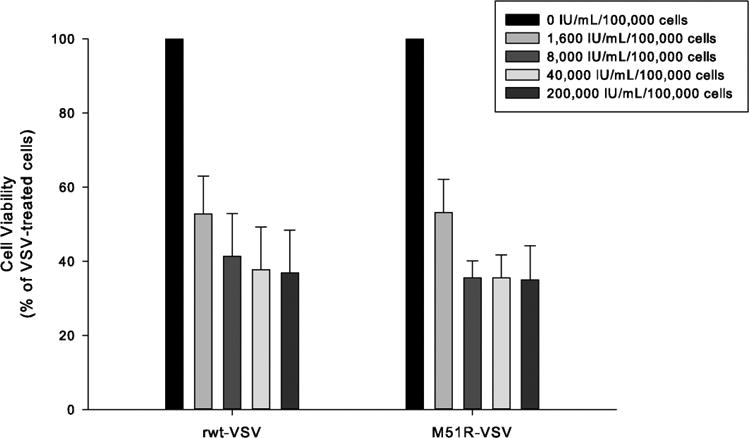
Cytotoxic effect of β-IFN in Panc 03.27 cells the setting of VSV infection. Cells were incubated with varying concentrations of β-IFN (0 to 40,000 IU/mL/100,000 cells) and challenged with rwt-VSV or M51R-VSV (MOI of 5 pfu/cell). Cell viability was measured by MTS assay 48 hours after VSV treatment. Data are expressed as the percentage of VSV-treated cells and presented as the mean ± standard deviation of three independent experiments.
In vivo Effects of M51R-VSV in a Murine Xenograft Model
As an extension of our in vitro analysis, we tested the in vivo oncolytic effects of M51R-VSV in a murine xenograft model. VSV-sensitive MiaPaCa2 or VSV-resistant Panc 03.27 cells were injected subcutaneously into the flanks of athymic nude mice. After approximately two weeks, the resultant xenografts were treated with a single intratumoral injection of M51R-VSV (1×108 pfu) or mock injection as controls. Tumor volume was measured three times a week as an indicator of treatment effect and tumor growth over time in each group is shown in Figure 7. Mock-treated xenografts from both cell lines grew exponentially, although the Panc 03.27 xenografts grew more slowly. Consistent with our in vitro observations, MiaPaCa2 tumors responded to M51R-VSV treatment as the xenografts did not grow appreciably after treatment. In fact, 40% of the tumors completely resolved by the end of the experiment and no macroscopic evidence of tumor was seen at necropsy. By post-treatment day 4, the percent change of tumor growth in the M51R-VSV treated xenografts was significantly lower compared to mock treated tumors (-11 ± 41% versus 76 ± 26%, p<0.0001) and this difference remained significant throughout the remainder of the study period. By post-treatment day 30, the percentage of tumor growth in the M51R-VSV treated xenografts was 164 ± 136% compared to 2138 ± 572% in the mock-treated group (p=0.003).
Figure 7.

Intratumoral M51R-VSV treatment of pancreatic cancer xenografts derived from VSV-sensitive MiaPaCa2 and VSV-resistant Panc 03.27 cells. Subcutaneous xenografts were established in the right flank of athymic nude mice. Once palpable tumors formed, the mice were randomly assigned to a single intratumoral injection of 1×108 pfu M51R-VSV (n=10) or culture medium as negative controls (n=9). Tumor volume was measured with calipers. Tumor growth is presented as the percentage of tumor size on day 0 post-infection and is expressed as the mean ± standard error of the mean. P-values shown represent the difference in percent change in tumor growth between M51R-VSV treated and mock-treated xenografts at post-treatment day 30.
Surprisingly, the M51R-VSV treated Panc 03.27 xenografts also responded to in vivo treatment, despite being resistant to VSV in vitro. By post-treatment day 14, the change in tumor growth between M51R-VSV treated xenografts and mock-treated xenografts was significantly different (-18 ± 16% versus 135 ± 34% respectively, p=0.0006). Furthermore by the end of the experiment, the percentage of tumor growth in the M51R-VSV treated tumors was 50 ± 52% while it was 979 ± 153% in the mock-treated tumors (p<0.0001). Although none of the Panc 03.27 xenografts resolved completely, 50% of them decreased in size after M51R-VSV treatment.
Xenografts were harvested a various times post-treatment and stained with hematoxylin and eosin (H&E). To evaluate in vivo VSV replication and spread, immunohistochemical analysis was also performed by staining tumors with antibodies against VSV surface glycoprotein (G protein). Shown in Figure 8, mock-treated MiaPaCa2 xenografts exhibited uniform cells with well-defined borders and nuclei. No significant necrosis or G-protein staining was seen in the mock-treated tumors. In contrast, MiaPaCa2 tumors treated with M51R-VSV showed areas of necrosis throughout the tumor, characterized by loss of nuclear staining, increased cytoplasmic eosinophilia and loss of cellular detail and borders. Extensive cytoplasmic G-protein staining correlated to the areas of patchy necrosis seen on H&E staining.
Figure 8.

Histological and immunohistochemical analysis of MiaPaCa2 xenografts. Mock-infected tumors with injected with culture medium. M51R-VSV treated tumors received a single intratumoral injection (1×108 pfu). Tumors were harvested at post-infection day three. Representative sections stained with hematoxylin and eosin (H&E) and immunohistochemical staining for VSV surface glycoprotein (G-protein) are shown from mock-treated and M51R-VSV treated xenografts as indicated.
Representative histological and immunohistochemical staining of Panc 03.27 xenografts are shown in Figure 9. Panc 03.27 tumors developed more glandular tissue, consistent with a well-differentiated adenocarcinoma. Mock-treated tumors were again characterized by uniform cells with clear borders and nuclei with no areas of necrosis or VSV G-protein staining. Xenografts harvested three days post-treatment did not shown significant effects of VSV infection as there was no significant necrosis and any immunohistochemical staining seen was non-specific nuclear staining. However, tumors harvested seven days after treatment displayed patchy areas of VSV G-protein staining which correlated to small areas of cellular necrosis on H&E. More significant, however, was the marked lymphocytic infiltration seen in the VSV-treated tumors seven days after treatment. To further characterize the lymphocytic infiltration seen in VSV-treated Panc 03.27 xenografts, tumors were stained for B-cells and NK cells. As represented in Figure 10, both B-cell and NK cell were visualized in the areas of lymphocytic infiltration. These findings suggest that the treatment response from M51R-VSV seen in the Panc 03.27 xenografts was immune-mediated rather than solely from viral oncolysis.
Figure 9.
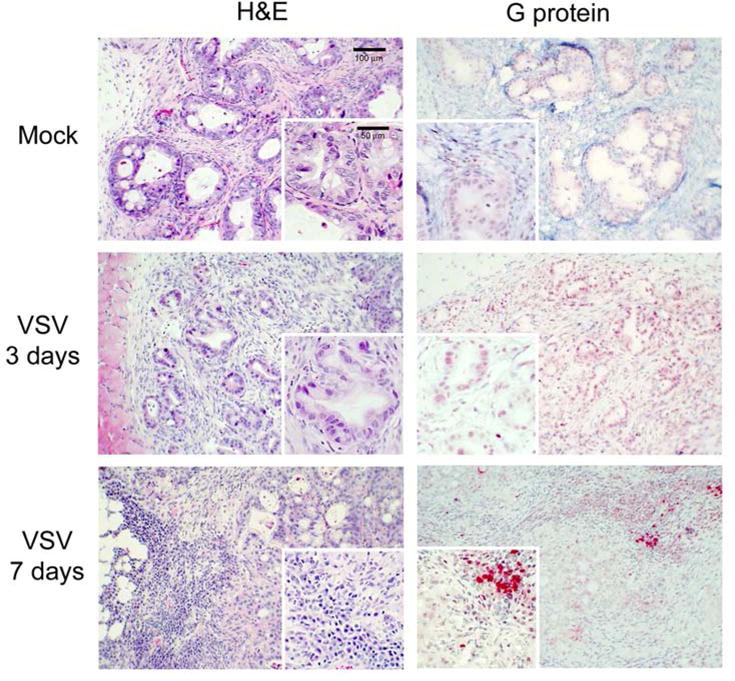
Histological and immunohistochemical analysis of Panc 03.27 xenografts. Mock-infected tumors with injected with culture medium. M51R-VSV treated tumors received a single intratumoral injection (1×108 pfu). Tumors were harvested three or seven days after treatment as indicated. Representative sections stained with hematoxylin and eosin (H&E) and immunohistochemical staining for VSV surface glycoprotein (G-protein) are shown from mock-treated and M51R-VSV.
Figure 10.

H&E staining (A) and immunohistochemical staining of B-cells (B) and NK cells (C) seven days following intratumoral M51R-VSV treatment.
Discussion
The results presented here are consistent with those of Murphy, et al.[14], who found significant heterogeneity in VSV-susceptibility among 13 pancreatic cancer cells and described similar results in terms of IFN responsiveness[14]. Given the aggressiveness of pancreatic adenocarcinoma and its insensitivity to traditional chemotherapy, it is encouraging that the majority of cell lines tested to date are sensitive to VSV. Our results support previous findings that VSV-sensitive cancer cells possess acquired defects in IFN-mediated anti-viral pathways which explain their susceptibility to VSV[6, 10, 14-15, 21-25].
However, there is considerable variability in IFN responses among pancreatic cancer cells following VSV infection which does not always correlate to VSV-sensitivity[14-15] and suggests that different defects along the IFN pathway exist in different sensitive cell lines. Our results indicate that defects exist not only in anti-viral signaling that stimulate β-IFN production but also in the cells' downstream response to IFN. Several specific defects in IFN-mediated pathways have been described in a variety of malignant cell lines and can be directly or indirectly implicated to confer VSV sensitivity. For example, Zhang, et al.[26] recently reported that high-grade bladder cancer cells expressing low levels of type I IFN receptors were susceptible to VSV whereas normal levels of type I IFN receptors correlated to VSV resistance in normal bladder cells and low-grade bladder cancer cells. In addition, by knocking down type I IFN receptors with siRNA, VSV-resistant bladder cancer cells became susceptible to the oncolytic effects of VSV. Marozin, et al.[27] described splicing variants in IFN regulatory factor-3 in hepatocellular carcinoma cells leading to inhibition of β-IFN signaling and enhanced viral replication in response to M51R-VSV infection. Additionally, normal IFNα-induced expression of MxA, an anti-viral protein known to inhibit VSV replication, was found to be deficient in several cancer cell lines as a result of ERK signaling activation[28]. Finally, other investigators have described defects in STAT1 and STAT3 expression[29-30] which correlate to IFN responsiveness.
Our microarray gene expression analysis identified three potential molecular pathways which could explain differences in VSV-susceptibility: 1) IFN signaling, 2) viral entry via endocytosis, and 3) caveolar-mediated endocytosis. Intact IFN signaling appears to play a significant role in VSV-resistance as described above. Our data suggests that inhibition of VSV cell entry via endocytosis is an alternative mechanism of VSV-resistance in some cell lines. VSV was incapable of significantly infecting Panc 03.27 cells even at extremely high concentrations and virtually no viral protein synthesis occurred relative to the other cell lines following VSV inoculation. Furthermore, VSV did not invoke significant β-IFN production as expected. Collectively, these observations imply that VSV replication is thwarted in Panc 03.27 cells during the early stages of infection. Similarly, Murphy, et al.[14] reported that VSV mRNA levels were relatively reduced in resistant pancreatic cancer cell lines following VSV infection also indicating early inhibition of VSV replication. Furthermore, our lab has previously reported on PC3 prostate cancer cells whose VSV resistance occurs shortly after cell entry and includes delays in virus penetration[31].
Molecular variations in any of the replication steps from VSV cell attachment to viral RNA transcription could explain the VSV-resistance seen in Panc 03.27 cells. Previously it was thought that nonspecific electrostatic and hydrophobic interactions promoted VSV attachment to the cell membrane[32]. However, recently the LDL receptor was identified as the main receptor for VSV cell entry[33]. Once bound to its receptor, VSV penetration proceeds via clathrin-dependent endocytosis. As the endosomal pH is lowered, VSV G-protein undergoes a conformational change which is capable of fusing the viral envelope with the endosomal membrane thus releasing the internal virion components into the cytoplasm. Following endosomal release, viral replication proceeds immediately via a cell independent VSV-associated RNA polymerase made up of L and P proteins of VSV[32].
It is unlikely that inhibition of VSV replication occurs at the receptor level. The LDL receptor is ubiquitously expressed in all human cells[34]. Furthermore, cells deficient in LDL receptors remain susceptible to VSV, albeit less effective, via other LDL receptor family members which possess the same class A cysteine-rich repeats at their ligand-binding site[33]. Taken together, the process of VSV endocytosis is a likely site of resistance in this pancreatic cancer cell line. As many as 80 different kinases have been identified in the regulation of VSV endocytosis and silencing many of these kinases has been shown to effectively block VSV infection[35]. Once specific targets are identified, strategies to inhibit or circumvent these anti-viral defenses could be developed using synchronous systemic therapy or genetically engineered VSV designed to deliver specific viral vectors. Interestingly, our microarray data found differences in the caveolae-dependent endocytosis pathway between the sensitive and resistant cell lines, while it is well described that VSV penetration occurs via clathrin-dependent endocytosis. The fact that there is overlap between the caveolae and clarthrin-dependent pathways[35] may explain the results of our microarray analysis.
Despite our findings of VSV resistance at the early stages of infection, low levels of cell infectability were seen in Panc03.27 cells and by 72 hours cell viability began to decrease under single-cycle infection conditions suggesting that high levels of VSV are capable of overcoming VSV-inhibition. We found that exposure to low levels of extrinsic β-IFN significantly decreased cell viability. The fact that cells responded to low levels of β-IFN suggests that IFN-mediated pathways are intact in these cells and are designed to inhibit cell growth or trigger apoptosis as a mechanisms to limit viral replication and spread. IFN-induced apoptosis has been shown in many malignant cell lines, including melanoma[36-39], breast cancer[40], colorectal cancer[41], neuroendocrine tumors[42], hepatocellular carcinoma[43], as well as, pancreatic cancer[44].
Despite the mechanisms of VSV resistance in vitro, Panc 03.27 xenografts responded to intratumoral treatment of M51R-VSV. One explanation for this observation is that β-IFN from normal cells could induce apoptosis in the Panc 03.27 cells based on our in vitro IFN responsiveness results; however, IFNs are species specific[45] and therefore IFNs produced by normal murine cells would not be expected to stimulate anti-viral responses in human pancreatic cancer cells. Moreover, oncolysis from the direct effects of VSV cannot completely explain the response of in vitro-resistant xenografts because there was no necrosis or VSV G-protein expression in in vitro-resistant xenografts three days after M51R-VSV treatment. However, we observed significant lymphocytic infiltration in these tumors seven days after treatment. This observation was unexpected since the tumors were produced in athymic nude mice, yet both NK cells and B cells were seen in the tumors treated with VSV. While further studies are necessary to evaluate the immune effects of VSV in immunocompetent hosts, these observations suggest that VSV-mediated oncolysis may be able to stimulate adaptive immunity by generating tumor antigens. Stimulated NK cells and other antigen-presenting cells may then prime T and B lymphocytes against the remaining viable tumor cells. Diaz, et al. showed a significant lymphocytic response in immunocompetent mice using syngeneic B16 melanoma tumors treated with intratumoral VSV[46]. They found that NK cells and cytotoxic T cells are important contributors to the in vivo oncoloytic effects of VSV and that adoptive T cell transfer can be synergic with the direct oncolytic VSV effects. The ability of VSV to stimulate adaptive immunity has the potential to expand the clinical efficacy of VSV into malignancies which are inherently more resistant to direct VSV-oncolysis.
The final notable observation from our results is the minimal differences we found in the oncolytic activity of M51R-VSV compared to rwt-VSV in these pancreatic cancer cell lines. Previous work has shown similar oncolysis between rwt-VSV and M51R-VSV in some, but not all cell lines[5,7,10]. There are two major advantages of M51R-VSV over rwt-VSV. First, M51R-VSV allows normal cells, to mount anti-viral defenses against M51R-VSV infection, while many malignant cells, which possess defective anti-viral responses, remain sensitive to VSV killing. This enhanced selectivity for malignant cells is a distinguishing characteristic of M51R-VSV as an anti-cancer therapy. Secondly, while wild-type VSV is thought to cause a non-specific flu-like illness in humans, it causes encephalitis in mice resulting in hind limb paralysis and eventually death[47-49]. In multiple studies, M51R-VSV has been administered, both locally and systemically at high doses without associated morbidity or mortality in immunodeficient and immunocompetent mice[5, 7-11, 50] while retaining its oncolytic activity. These studies establish the pre-clinical safety of M51R-VSV in preparation for human trials. The fact that M51R-VSV is equally oncolytic in pancreatic adenocarcinoma cells compared to rwt-VSV—yet is safer and has enhanced selectivity for cancer cells—makes it an exceptionally attractive candidate for future clinical applications.
In conclusion, M51R-VSV is a viable option for the future treatment of pancreatic adenocarcinoma. The integrity of IFN-mediated anti-viral mechanisms explains VSV-sensitivity; although there is evidence that different defects exist at various stages of IFN signaling which inhibit the cells' ability to resist VSV oncolysis. While VSV-resistant pancreatic cancer cells appear to possess intact IFN-mediated pathways, these cells also block the early stages of viral replication likely by inhibiting viral endocytosis. Finally, xenografts from in-vitro resistant cells respond to local M51R-VSV treatment, presumably by inducing anti-tumor adaptive immunity. Future studies will evaluate the in-vivo immunologic effects of VSV using syngeneic models and will define specific IFN-mediated signals and endocytosis mediators which can be exploited using viral vectors or other strategies to expand the therapeutic potential of M51R-VSV.
Acknowledgments
This work was supported by Grant no. K08-CA131482 from the National Cancer Institute (JS), 63527 from the Robert Wood Johnson Foundation Harold Amos Faculty Development Award (JS), R01-AI32983 from the National Institute of Allergy and Infectious Diseases (DL), and the Bradshaw Surgical Resident Research Endowment (AB).
We thank Hermina Borgerink (Wake Forest School of Medicine, Department of Comparative Medicine) for staining of tissue sections and Lou Craddock (Wake Forest School of Medicine, Department of Biochemistry) for her help with our microarray analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–27. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed M, McKenzie MO, Puckett S, et al. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J Virol. 2003;77:4646–57. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed M, Lyles DS. Identification of a consensus mutation in M protein of vesicular stomatitis virus from persistently infected cells that affects inhibition of host-directed gene expression. Virology. 1997;237:378–88. doi: 10.1006/viro.1997.8808. [DOI] [PubMed] [Google Scholar]
- 4.Ebert O, Shinozaki K, Huang TG, et al. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Res. 2003;63:3605–11. [PubMed] [Google Scholar]
- 5.Stewart JH, Ahmed M, Northrup SA, et al. Vesicular stomatitis virus as a treatment for colorectal cancer. Cancer Gene Ther. 2011;18:837–49. doi: 10.1038/cgt.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stojdl DF, Lichty BD, tenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–75. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed M, Cramer SD, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 8.Ahmed M, Puckett S, Lyles DS. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther. 2010;17:883–92. doi: 10.1038/cgt.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cary ZD, Willingham MC, Lyles DS. Oncolytic Vesicular Stomatitis Virus Induces Apoptosis in U87 Glioblastoma Cells by a Type II Death Receptor Mechanism and Induces Cell Death and Tumor Clearance In Vivo. J Virol. 2011;85:5708–17. doi: 10.1128/JVI.02393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackham AU, Northrup SA, Willingham M, et al. Variation in susceptibility of human malignant melanomas to oncolytic vesicular stomatitis virus. Surgery. 2013;153:333–43. doi: 10.1016/j.surg.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randle RW, Northrup SA, Sirintrapun SJ, Lyles DS, Stewart JH. Oncolytic Vesicular Stomatitis Virus as a Treatment for Neuroendocrine Tumors. Surgery. 2013 doi: 10.1016/j.surg.2013.04.050. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saloura V, Wang LC, Fridlender ZG, et al. Evaluation of an Attenuated Vesicular Stomatitis Virus Vector Expressing Interferon-beta for Use in Malignant Pleural Mesothelioma: Heterogeneity in Interferon Responsiveness Defines Potential Efficacy. Hum Gene Ther. 2010;21:51–64. doi: 10.1089/hum.2009.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadaschik BA, Zhang K, So AI, et al. Oncolytic vesicular stomatitis viruses are potent agents for intravesical treatment of high-risk bladder cancer. Cancer Res. 2008;68:4506–10. doi: 10.1158/0008-5472.CAN-08-0238. [DOI] [PubMed] [Google Scholar]
- 14.Murphy AM, Besmer DM, Moerdyk-Schauwecker M, et al. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J Virol. 2012;86:3013–87. doi: 10.1128/JVI.05640-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moerdyk-Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili V. Resistance of pancreatic cancer to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology. 2013;436:221–34. doi: 10.1016/j.virol.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carey BL, Ahmed M, Puckett S, et al. Early Steps of the Virus Replication Cycle Are Inhibited in Prostate Cancer Cells Resistant to Oncolytic Vesicular Stomatitis Virus. J Virol. 2008;82:12104–15. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jemal A, Siegel R, Xu JQ, Ward E. Cancer Statistics, 2010. CACancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 18.Kopecky SA, Willingham MC, Lyles DS. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J Virol. 2001;75:12169–81. doi: 10.1128/JVI.75.24.12169-12181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 20.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 21.Kirn DH, Wang YH, Le Boeuf F, et al. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:2001–12. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elankumaran S, Chavan V, Qiao D, et al. Type I Interferon-Sensitive Recombinant Newcastle Disease Virus for Oncolytic Virotherapy. J Virol. 2010;84:3835–44. doi: 10.1128/JVI.01553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9:1163–76. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- 24.Balachandran S, Roberts PC, Brown LE, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–41. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 25.Balachandran S, Barber GN. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life. 2000;50:135–8. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
- 26.Zhang KX, Matsui Y, Hadaschik BA, et al. Down-regulation of type I interferon receptor sensitizes bladder cancer cells to vesicular stomatitis virus-induced cell death. Int J Cancer. 2010;127:830–8. doi: 10.1002/ijc.25088. [DOI] [PubMed] [Google Scholar]
- 27.Marozin S, Altomonte J, Stadler F, et al. Inhibition of the IFN-beta Response in Hepatocellular Carcinoma by Alternative Spliced Isoform of IFN Regulatory Factor-3. Mol Ther. 2008;16:1789–97. doi: 10.1038/mt.2008.201. [DOI] [PubMed] [Google Scholar]
- 28.Noser JA, Mael AA, Sakuma R, et al. The RAS/Raf1/MEK/ERK signaling pathway facilitates VSV-mediated oncolysis: Implication for the defective interferon response in cancer cells. Mol Ther. 2007;15:1531–6. doi: 10.1038/sj.mt.6300193. [DOI] [PubMed] [Google Scholar]
- 29.Wong LH, Krauer KG, Hatzinisiriou I, et al. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3 gamma. J Biol Chem. 1997;272:28779–85. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 30.Sun WH, Pabon C, Alsayed Y, et al. Interferon-alpha resistance in a cutaneous T-cell lymphoma cell line is associated with lack of STAT1 expression. Blood. 1998;91:570–6. [PubMed] [Google Scholar]
- 31.Carey BL, Ahmed M, Puckett S, et al. Early Steps of the Virus Replication Cycle Are Inhibited in Prostate Cancer Cells Resistant to Oncolytic Vesicular Stomatitis Virus. J Virol. 2008;82:12104–15. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyles D. Rupprecht CE: Rhabdoviridae. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Lippincott Williams and Wilkins; 2007. pp. 1364–402. [Google Scholar]
- 33.Finkelshtein D, Werman A, Novick D, et al. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110:7306–11. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willnow TE. The low-density lipoprotein receptor gene family: multiple roles in lipid metabolism. J Mol Med. 1999;77:306–15. doi: 10.1007/s001090050356. [DOI] [PubMed] [Google Scholar]
- 35.Pelkmans L, Fava E, Grabner H, et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 36.Nagatani T, Okazawa H, Kambara T, et al. Effect of natural interferon-beta on the growth of melanoma cell lines. Melanoma Res. 1998;8:295–9. doi: 10.1097/00008390-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Kamiya T, Okabayashi T, Yokota S, et al. Increased Caspase-2 Activity is Associated with Induction of Apoptosis in IFN-beta Sensitive Melanoma Cell Lines. J Interferon Cytokine Res. 2010;30:349–57. doi: 10.1089/jir.2009.0015. [DOI] [PubMed] [Google Scholar]
- 38.Chawla-Sarkar M, Leaman DW, Borden EC. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha 2: Correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin Cancer Res. 2001;7:1821–31. [PubMed] [Google Scholar]
- 39.Kubo H, Ashida A, Matsumoto K, et al. Interferon-beta therapy for malignant melanoma: the dose is crucial for inhibition of proliferation and induction of apoptosis of melanoma cells. Arch Dermatol Res. 2008;300:297–301. doi: 10.1007/s00403-008-0841-6. [DOI] [PubMed] [Google Scholar]
- 40.Coradini D, Biffi A, Pirronello E, Di Fronzo G. The Effect of Alpha-Interferon, Beta-Interferon and Gamma-Interferon on the Growth of Breast-Cancer Cell-Lines. Anticancer Res. 1994;14:1779–84. [PubMed] [Google Scholar]
- 41.Choi EA, Lei HQ, Maron DJ, et al. Stat1-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand and the cell-surface death signaling pathway by interferon beta in human cancer cells. Cancer Res. 2003;63:5299–307. [PubMed] [Google Scholar]
- 42.Vitale G, de Herder WW, van Koetsveld PM, et al. IFN-beta is a highly potent inhibitor of gastroenteropancreatic neuroendocrine tumor cell growth in vitro. Cancer Res. 2006;66:554–62. doi: 10.1158/0008-5472.CAN-05-3043. [DOI] [PubMed] [Google Scholar]
- 43.Damdinsuren B, Nagano H, Sakon M, et al. Interferon-beta is more potent than interferon-alpha in inhibition of human hepatocellular carcinoma cell growth when used alone and in combination with anticancer drugs. Ann Surg Oncol. 2003;10:1184–90. doi: 10.1245/aso.2003.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Vitale G, van Eijck CHJ, van Koetsveld PM, et al. Type I Interferons in the treatment of pancreatic cancer - Mechanisms of action and role of related receptors. Ann Surg. 2007;246:259–68. doi: 10.1097/01.sla.0000261460.07110.f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stewart WE, Scott WD, Sulkin SE. Relative Sensitivities of Viruses to Different Species of Interferon. J Virol. 1969;4:147–53. doi: 10.1128/jvi.4.2.147-153.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz RM, Galivo F, Kottke T, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–48. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 47.Huneycutt BS, Bi ZB, Aoki CJ, et al. Central Neuropathogenesis of Vesicular Stomatitis-Virus Infection of Immunodeficient Mice. J Virol. 1993;67:6698–706. doi: 10.1128/jvi.67.11.6698-6706.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomsen AR, Nansen A, Andersen C, et al. Cooperation of B cells and T cells is required for survival of mice infected with vesicular stomatitis virus. Int Immunology. 1997;9:1757–66. doi: 10.1093/intimm/9.11.1757. [DOI] [PubMed] [Google Scholar]
- 49.Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nature Med. 2000;6:821–5. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 50.Ahmed M, Marino TR, Puckett S, et al. Immune response in the absence of neurovirulence in mice infected with M protein mutant vesicular stomatitis virus. J Virol. 2008;82:9273–7. doi: 10.1128/JVI.00915-08. [DOI] [PMC free article] [PubMed] [Google Scholar]