

Abstract
Purpose:
The transcription factor Forkhead box M1 (FOXM1) plays important roles in the formation of several human tumors, including pancreatic cancer. However, the molecular mechanisms by which FOXM1 promotes pancreatic tumor epithelial-to-mesenchymal transition (EMT) and metastasis are unknown.
Experimental Design:
The effect of altered expression of FOXM1 and urokinase plasminogen activator receptor (uPAR) on EMT and metastasis was examined using animal models of pancreatic cancer. Also, the underlying mechanisms of altered pancreatic cancer invasion and metastasis were analyzed using in vitro molecular biology assays. Finally, the clinical relevance of dysregulated FOXM1/uPAR signaling was investigated using pancreatic tumor and normal pancreatic tissue specimens.
Results:
Pancreatic tumor specimens and cell lines predominantly overexpressed the FOXM1 isoform FOXM1c. FOXM1c overexpression promoted EMT in and migration, invasion, and metastasis of pancreatic cancer cells, whereas downregulation of FOXM1 expression inhibited these processes. The level of FOXM1 expression correlated directly with that of uPAR expression in pancreatic cancer cell lines and tumor specimens. Moreover, FOXM1c overexpression upregulated uPAR expression in pancreatic cancer cells, whereas inhibition of FOXM1 expression suppressed uPAR expression. Furthermore, transfection of FOXM1c into pancreatic cancer cells directly activated the uPAR promoter, whereas inhibition of FOXM1 expression by FOXM1 small interfering RNA suppressed its activation in these cells. Finally, we identified an FOXM1-binding site in the uPAR promoter and demonstrated that FOXM1 protein bound directly to it. Deletion mutation of this site significantly attenuated uPAR promoter activity.
Conclusions:
Our findings demonstrated that FOXM1c contributes to pancreatic cancer development and progression by enhancing uPAR gene transcription and, thus, tumor EMT and metastasis.
Keywords: Progression, invasion, angiogenesis, transcription factor, biomarkers
Introduction
Pancreatic cancer is one of the leading causes of cancer deaths in industrialized countries, and its incidence appears to be increasing (1). Despite improvements in early diagnosis, surgical techniques, and chemotherapy, the majority of pancreatic cancer patients die of the physiological effects of invasion and metastasis to the regional lymph nodes and/or distant organs (2). Unfortunately, little is known concerning the reasons for the aggressiveness of and dismal prognosis for pancreatic cancer. Therefore, improved understanding of the molecular mechanisms underlying pancreatic cancer progression is an urgent need (3, 4).
Forkhead box M1 (FOXM1), previously known as HNF-3, HFH-11, MPP2, Win, and Trident, is a transcription factor in the FOX protein superfamily containing a conserved winged helix DNA-binding domain (5). The human FOXM1 gene consists of 10 exons, two of which—exon Va (A1) and exon VIIa (A2)—are alternatively spliced. These splices give rise to three distinct isoforms of FOXM1: FOXM1a, FOXM1b, and FOXM1c (6-8). FOXM1a, which harbors both exon Va and exon VIIa, is transcriptionally inactive owing to disruption of its transactivation domain (TAD) by the latter exon. In comparison, both FOXM1b, which contains neither of these two exons, and FOXM1c, which has only exon Va, are transcriptionally active and can activate expression of their target genes via different mechanisms (7). Because FOXM1 is an essential transcription factor for many genes key to regulation of multiple aspects of tumor cell survival, growth, epithelial-to-mesenchymal transition (EMT), angiogenesis, and metastasis, abnormal FOXM1 expression may contribute to human cancer development and progression (9, 10).
Several lines of evidence demonstrate that overexpression of FOXM1 occurs frequently in a wide variety of human tumors, including lung, cervical, and gastric cancer (11-13), as well as in nervous system tumors, such as glioblastoma and medulloblastoma (14, 15). Also, recent studies suggested that FOXM1 plays important roles in pancreatic cancer development and progression (16-18). For example, downregulation of FOXM1 expression inhibits pancreatic cancer cell growth, migration, and invasion by decreasing cyclin B, cyclin D1, Cdk2, matrix metalloproteinase (MMP)-2, MMP-9, and vascular endothelial growth factor expression (16). In addition, FOXM1 may regulate the EMT phenotype of pancreatic cancer cells by activating mesenchymal cell markers (17). Moreover, we found that FOXM1–caveolin-1 signaling plays an important role in EMT in and metastasis of pancreatic tumors (18). However, the molecular mechanisms by which each FOXM1 isoform promotes pancreatic cancer progression are unknown, and how FOXM1-targeted gene expression is regulated by each isoform is unclear.
Tumor cell invasion and metastasis depend on the coordinated and temporal expression of proteolytic enzymes to degrade the extracellular matrix surrounding the tumor. The tumor cell-associated urokinase plasminogen activator (uPA) system, which consists of the serine protease uPA, membrane-bound uPA receptor (uPAR), and uPA inhibitors plasminogen activator inhibitor (PAI)-1 and PAI-2, plays important roles in these pericellular processes (19-21). uPAR, which is a glycosyl phosphatidylinositol-anchored protein, not only is bound to the cell surface but also has a soluble form (suPAR). Accumulating clinical and experimental evidence has demonstrated that uPAR plays a central role in angiogenesis in and metastasis of many human tumors, including pancreatic cancer (22-24). Furthermore, inhibition of uPAR expression by a monoclonal antibody or small interfering RNA (siRNA) has inhibited human pancreatic tumor growth, angiogenesis, and metastasis both in vitro and in vivo (25, 26). However, little is known about the molecular mechanisms underlying dysregulated expression and function of uPAR in pancreatic cancer cells.
In the present study, we sought to determine the role of the FOXM1 isoforms in pancreatic cancer EMT, invasion, and metastasis and their regulatory functions regarding uPAR expression and function. We discovered that pancreatic cancer cell lines had high levels of expression of FOXM1c and that FOXM1b and FOXM1c promoted EMT in and metastasis of pancreatic cancer cells via transcriptional regulation of the expression of uPA and uPAR.
Materials and Methods
Details regarding our study animals, and experimental procedures are described in the Supplementary Materials and Methods section.
Cell lines and culture conditions
The human pancreatic adenocarcinoma cell lines AsPC-1, CaPan-1, CaPan-2, MiaPaca-2, BxPC-3, Hs766T, PANC-1, and PL45 and human embryonic kidney 293 (HEK293) cells were purchased from the American Type Culture Collection. The pancreatic cancer cell lines MDA Panc-28 and MDA Panc-48 were gifts from Dr. Paul J. Chiao (The University of Texas MD Anderson Cancer Center). The human pancreatic adenocarcinoma metastasis cell line COLO357 and its fast-growing variant FG and liver-metastatic variants L3.3 and L3.7 in nude mice as well as the murine ductal adenocarcinoma cell line Panc02 and its highly metastatic variant Panc02-H7 were described previously (18). All of these cell lines were maintained in plastic flasks as adherent monolayers in Eagle's minimal essential medium supplemented with 10% fetal bovine serum, sodium pyruvate, nonessential amino acids, L-glutamine, and vitamin solution (Flow Laboratories). The immortalized normal human pancreatic ductal epithelial cell line HPDE (provided by Dr. Tsao, Ontario Cancer Institute) was maintained in keratinocyte serum-free medium supplemented with epidermal growth factor and bovine pituitary extract (Invitrogen). The cell lines were obtained directly from ATCC that conducts cell line characterizations or authentication by the short tandem repeat profiling and passaged in our laboratory for less than 6 months after receipt
Human tissue specimens and immunohistochemical analysis
Expression of FOXM1, uPAR, uPA, and PAI-1 in pancreatic cancer cells was analyzed using a human pancreatic tumor and normal pancreatic tissue microarray (TMA) (US Biomax). Use of the tissue specimens was approved by The University of Texas MD Anderson Cancer Center Institutional Review Board. Standard immunohistochemical procedures were performed using anti-FOXM1 (Santa Cruz Biotechnology), anti-uPAR (American Diagnostica), anti-uPA, and anti-PAI-1 (Santa Cruz Biotechnology) antibodies. The specificities of those antibodies have been validated in prior reports (22-26). The staining results were scored by two investigators blinded to the clinical data as described previously (27, 28).
Plasmids and siRNAs
The plasmid pcDNA3.1-FOXM1b and control vector pcDNA3.1 were described previously (14). To generate pcDNA3.1-FOXM1a and pcDNA3.1-FOXM1c plasmids, full-length human FOXM1a and FOXM1c were released via EcoRI and XbaI digestion of human cytomegalovirus. FOXM1a (7) and FOXM1c (8) cDNA expression vectors were subcloned into pcDNA3.1. An siRNA sequence targeting FOXM1 was CUCUUCUCCCUCAGAUAUAdTdT (13).
Transfection of pancreatic cancer cells
Transfection of plasmids and siRNAs into pancreatic cancer cells was performed using Lipofectamine LTX (Invitrogen) and Lipofectamine 2000 CD (Invitrogen) reagent, respectively. The cells were transfected with plasmids or siRNA at different doses as indicated for 48 hours before performance of functional assays. Pancreatic cancer cells treated with transfection reagent alone were included as mock-treated controls.
Construction of reporter plasmids and mutagenesis
A 1.103-kb fragment containing 5' uPAR sequences from −873 to +230 bp relative to the transcription initiation site was subcloned into the Asp718 and Xho1 sites of the pGL3-basic vector (Promega). The resulting full-length reporter plasmid, designated puPAR1103, contained two FOXM1-binding sites: ACAAACAA and TAATCA. Its deletion mutation reporter (puPAR627) was generated by subcloning a 0.627-kb fragment containing 5' uPAR sequences from −397 to +230 bp relative to the transcription initiation site into the Asp718 and Xho1 sites of pGL3-basic, which did not contain the FOXM1-binding site ACAAACAA. All reporter plasmid constructs were verified by sequencing the inserts and flanking regions of the plasmids.
Promoter reporter and dual luciferase assays
Pancreatic cancer cells were transfected with the pGL3-uPAR plasmids, siRNAs, or specific gene expression plasmids. The uPAR promoter activity in the cells was the normalized via co-transfection with a β-actin/Renilla luciferase reporter containing a full-length Renilla luciferase gene (29). The luciferase activity in the cells was quantified using a dual luciferase assay kit (Promega) 24 hours after transfection.
Real-time reverse transcription-polymerase chain reaction
Total RNA was extracted from pancreatic tumor cells using TRIzol reagent (Invitrogen). Next, 2 μg of total RNA was reverse-transcribed using a First Strand cDNA Synthesis Kit (Promega) to synthesize cDNA specimens. Quantitative polymerase chain reaction (PCR) analysis of expression of the FOXM1 gene in the cells was performed using 2 μL of cDNA and SYBR Green Master Mix (Bio-Rad) as recommended by the manufacturer for the FOXM1 primers 5'-acgtccccaagccaggctc-3' (forward) and 5'-ctactgtagctcaggaataa-3' (reverse), FOXM1a primers 5'-tggggaacaggtggtgtttgg-3' (forward) and 5'-gctagcagcactgataaacaaag-3' (reverse), FOXM1b primers 5'-ccaggtgtttaagcagcaga-3' (forward) and 5'-tcctcagctagcagcaccttg-3' (reverse), FOXM1c primers 5'-caattgcccgagcacttggaatca-3' (forward) and 5'-tcctcagctagcagcaccttg-3' (reverse), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense strand 5'-caccattggcaatgagcggttc-3' and antisense strand 5'-aggtctttgcggatgtccacgt-3'. GAPDH was used as an internal control. Each PCR was run in triplicate for the target and internal control genes.
Reverse transcription-PCR
Total RNA was extracted from pancreatic tumor cells using TRIzol reagent (Invitrogen). Two micrograms of total RNA was then reverse-transcribed using a First Strand cDNA Synthesis Kit (Promega) to synthesize cDNA specimens. Subsequently, 2 μL of a cDNA product was subjected to PCR amplification with Taq DNA polymerase (Qiagen) using a thermal cycler with the following PCR primers to detect each factor: total FOXM1 sense strand 5'-acgtccccaagccaggctc-3' and antisense strand 5'-ctactgtagctcaggaataa-3', GAPDH sense strand 5'-caccattggcaatgagcggttC-3' and antisense strand 5'-aggtctttgcggatgtccacgt-3', uPA sense strand 5'-ttgctcaccacaacgacatt-3' and antisense strand 5'-attttcagctgctccggata-3', uPAR sense strand 5'-aggccccat gaatcaatgt-3' and antisense strand 5'-gcaggagacatcaatgtggtt-3', and PAI-1 sense strand 5'-tgatggctcagaccaacaag-3' and antisense strand 5'-ataaggggcagcaatgaaca-3' (30). The PCR products were loaded onto 2% agarose gels and visualized via staining with ethidium bromide under ultraviolet light.
Chromatin immunoprecipitation assay
Pancreatic tumor cells (2 × 106) were prepared for a chromatin immunoprecipitation (ChIP) assay using a ChIP assay kit (Millipore) according to the manufacturer’s protocol. The resulting immunoprecipitated DNA specimens were analyzed using PCR to amplify a 231-bp region of the uPAR promoter with the primers 5'-gccacctcatctgacctcttc-3' (sense) and 5'-cattgtcgtaacagtgatatc-3' (antisense) and a 627-bp region of the uPAR promoter with the primers 5'-tgcaatgcctggaatagctgc-3' (sense) and 5'-cacaggagctgccctcgcgac-3' (antisense). The PCR products were resolved electrophoretically on a 2% agarose gel and visualized using ethidium bromide staining.
Statistical analysis
The significance of the patient specimen data was determined using the Pearson correlation coefficient. The significance of the in vitro and in vivo data was determined using the Student t-test (two-tailed), Mann-Whitney test (two-tailed), or one-way analysis of variance. P values less than 0.05 were considered significant.
Results
FOXM1 overexpression and its direct association with clinicopathological parameters and expression of genes related to metastasis in pancreatic cancer cells
We first detected the expression of FOXM1 protein in pancreatic cancer cells using immunohistochemical staining with a specific anti-FOXM1 antibody in a TMA containing 70 primary pancreatic tumor and 10 normal pancreatic tissue specimens. We found FOXM1-positive staining in the nuclei and/or cytoplasm of the tumor cells and FOXM1-negative or weakly FOXM1-positive staining in the cytoplasm of normal pancreatic tissue cells adjacent to the tumor cells or normal pancreatic tissue cells (31). We then analyzed the relationship between clinicopathological parameters and FOXM1 expression in pancreatic tumor specimens. We observed a correlation between increased FOXM1 expression and decreased tumor differentiation and a significant difference in FOXM1 expression and tumor differentiation between well (grade 1) and poorly (grade 3) differentiated tumors (P < 0.05). In addition, FOXM1 expression was positively correlated with disease stage, indicating that FOXM1 expression was upregulated in late-stage pancreatic tumors. This association was significant for stage I and IV tumors (P < 0.05). Moreover, FOXM1 expression levels in lymph node and distant metastasis specimens were significantly higher than those in nonmetastasis specimens (P < 0.05). These results demonstrated that FOXM1 expression plays critical roles in pancreatic cancer development and progression and is a valuable biomarker for this disease.
To determine the molecular basis for the effect of altered FOXM1 expression on pancreatic cancer EMT, angiogenesis, and metastasis, we sought to detect the expression of uPA and uPAR in the same TMA using immunohistochemical staining and quantified the staining for FOXM1, uPA, and uPAR in the pancreatic tumor specimens. We found that the levels of FOXM1, uPA, and uPAR expression were significantly higher in specimens obtained from patients with metastatic disease than in those obtained from patients without it (Fig. 1A-B). We observed immunostaining for uPA and uPAR mainly in the cytoplasm of the pancreatic cancer cells. Moreover, we observed significant direct correlations between FOXM1 and uPAR expression, between FOXM1 and uPA expression, and between uPA and uPAR expression (P < 0.05) (Fig. 1C). However, we did not find a significant correlation between FOXM1 and PAI-1 expression (data not shown). These findings suggested that FOXM1, uPA, and uPAR expression was closely interrelated and correlated with pancreatic cancer metastasis.
Figure 1.
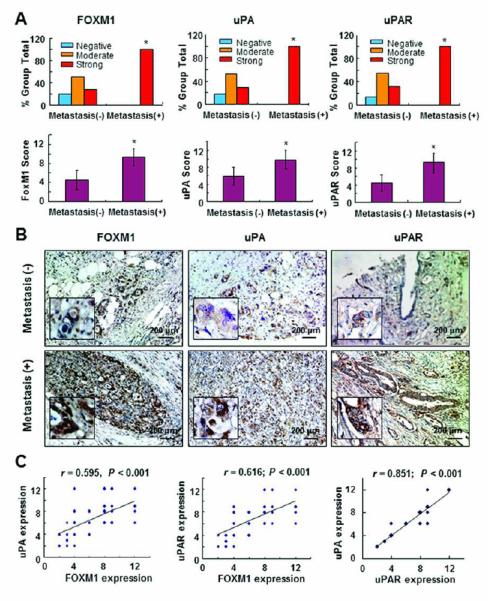
Expression of FOXM1, uPAR, and uPA and its correlation with metastasis in pancreatic tumors. Staining of 70 primary pancreatic tumor specimens and 10 normal pancreatic tissue specimens for FOXM1, uPAR, and uPA protein was performed using a TMA. A, FOXM1, uPAR, and uPA expression was much higher in primary pancreatic tumor specimens obtained from patients with lymph node and/or distant metastasis (Metastasis [+]) than in those obtained from patients without metastasis (Metastasis [-]). *P < 0.05. B, representative photos of uPA, uPAR, and FOXM1 expression in primary pancreatic tumor specimens obtained from patients with or without metastasis at a magnification of ×200 (inserts, ×400). C, positive correlation of FOXM1 expression levels with uPA and uPAR expression levels in pancreatic tumor specimens (Pearson correlation test). Note: some of the dots on the graphs represent more than one specimen (overlapping scores).
FOXM1 isoform expression in pancreatic cancer cells and tumors
Real-time PCR analysis using pancreatic cancer, human pancreatic ductal epithelial (HPDE), and HEK293 cells revealed that FOXM1c is the predominant isoform in HPDE and pancreatic cancer cells, whereas FOXM1b is the predominant isoform in HEK293 cells (Fig. 2A). Moreover, expression of FOXM1c in highly metastatic L3.7 human pancreatic adenocarcinoma cells was significantly higher than that in poorly metastatic COLO357 cells (P < 0.05), suggesting that FOXM1c expression correlated directly with metastatic ability. We further examined the FOXM1 isoform expression in human pancreatic tumor and paired adjacent normal pancreatic tissue specimens using real-time PCR. We observed that expression of FOXM1c in the tumors was significantly higher than that in adjacent normal tissue (P < 0.05). These findings demonstrated that FOXM1c may be the most important FOXM1 isoform to pancreatic cancer development and progression.
Figure 2.
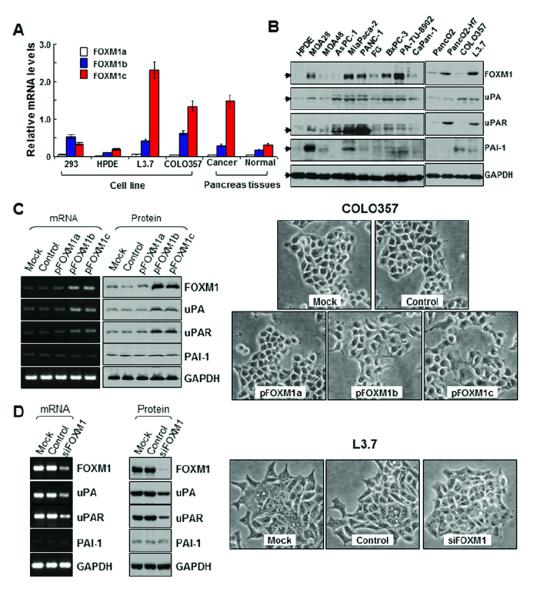
Expression of FOXM1a, FOXM1b, and FOXM1c and its influence on EMT in pancreatic cancer cells. A, real-time PCR analysis using specific primers for FOXM1a, FOXM1b, and FOXM1c with pancreatic cancer cell lines and tumor specimens. FOXM1c was the predominant isoform expressed in HPDE cells and all pancreatic cancer cell lines, whereas FOXM1b was the predominant isoform expressed in HEK293 cells. The expression of FOXM1c in the highly metastatic L3.7 cells was much higher than that in the poorly metastatic COLO357 cells. Also, the expression of FOXM1c in pancreatic tumor specimens was much higher than that in adjacent normal pancreatic tissue specimens. B, Western blot analysis of FOXM1, uPA, uPAR, and PAI-1 protein expression in the pancreatic cancer cell lines and HPDE cells. C, COLO357 cells were transfected with pcDNA3.1-FOXM1a (pFOXM1a), -FOXM1b (pFOXM1b), or -FOXM1c (pFOXM1c) or with pcDNA3.1 alone and incubated for 48 hours. Forced expression of FOXM1b or FOXM1c in COLO357 cells greatly increased FOXM1, uPA, and uPAR mRNA and protein expression (left). Phase-contrast photomicrographs demonstrated that COLO357 cells underwent morphological changes typical of EMT after FOXM1b or FOXM1c overexpression (right). D, L3.7 cells were transfected with siFOXM1 or control siRNA and incubated for 48 hours. Forced siFOXM1 expression in these cells markedly inhibited FOXM1, uPA, and uPAR mRNA and protein expression (left). Phase-contrast photomicrographs demonstrated that L3.7 cells underwent morphological changes typical of mesenchymal-to-epithelial transition after knockdown of FOXM1 expression (right).
To determine the roles of the FOXM1 isoforms in pancreatic cancer metastasis, we measured the expression of FOXM1, uPA, uPAR, and PAI-1 in nine pancreatic cancer cell lines and one transformed HPDE cell line (Fig. 2B). uPA and uPAR expression correlated directly with FOXM1 expression in the pancreatic cancer cell lines. Furthermore, the levels of FOXM1 and uPAR expression were higher in highly metastatic human and murine pancreatic cancer cell lines (L3.7 and Panc02-H7) than in poorly metastatic cell lines (COLO357 and Panc02), suggesting that FOXM1 and uPAR expression correlated directly with metastatic ability.
To obtain causal evidence of a direct correlation between expression of FOXM1 and that of uPA and uPAR, we sought to determine the impact of altered expression of FOXM1 on expression of uPA and uPAR in human pancreatic cancer cell lines with either low (COLO357) or high (L3.7) levels of FOXM1 expression. To that end, we transfected the FOXM1 expression vectors pcDNA3.1-FOXM1a, pcDNA3.1-FOXM1b, and pcDNA3.1-FOXM1c and the control vector pcDNA3.1 into COLO357 cells, which typically have an epithelial phenotype (18). We found that increased expression of FOXM1b or FOXM1c in COLO357 cells led to markedly increased uPA and uPAR mRNA and protein expression (Fig. 2C) and that the cells underwent morphological changes typical of EMT in that they scattered from cell clusters and acquired a spindle-shaped and fibroblast-like phenotype. Conversely, we transfected FOXM1 siRNA (siFOXM1) or a control siRNA into L3.7 cells, which have a mesenchymal phenotype (18). This transfection greatly inhibited FOXM1 mRNA and protein expression (Fig. 2D). Also, the L3.7 cells underwent a morphological change typical of mesenchymal-to-epithelial transition, going from an elongated, spindle-shaped, mesenchymal phenotype to a more rounded, epithelial-like phenotype. These results indicated that altered FOXM1b and FOXM1c expression affected the epithelial and mesenchymal phenotypes of pancreatic cancer cells.
Effect of altered FOXM1 expression on pancreatic cancer cell migration and invasion in vitro and tumor growth and metastasis in vivo
To determine the effect of altered FOXM1 expression on migration of pancreatic cancer cells, we transfected COLO357 and L3.7 cells with pcDNA3.1-FOXM1a, pcDNA3.1-FOXM1b, pcDNA3.1-FOXM1c, and siFOXM1 for 48 hours. We then wounded the transfected cells via scratching and maintained them at 37°C for an additional 12 hours. We observed that overexpression of FOXM1b and FOXM1c strongly promoted the flattening and spreading of COLO357 cells, whereas knockdown of expression of FOXM1 attenuated the flattening and spreading of L3.7 cells (Supplementary Fig. S1A). Also, cell migration assay results indicated that overexpression of FOXM1b and FOXM1c promoted the migratory ability of COLO357 cells, whereas knockdown of expression of FOXM1 attenuated this ability of L3.7 cells (Supplementary Fig. S1B). Similarly, overexpression of FOXM1b and FOXM1c promoted the invasiveness of COLO357 cells, whereas knockdown of expression of FOXM1 attenuated the invasiveness of L3.7 cells (Supplementary Fig. S1C).
Consistent with the impact of altered FOXM1 expression on the migration and invasion of pancreatic cancer cells in vitro, transfection of COLO357 and L3.7 cells with pcDNA3.1-FOXM1b or pcDNA3.1-FOXM1c significantly promoted pancreatic tumor growth (Fig. 3A-B) and increased liver metastasis of COLO357 cells (P < 0.05) in nude mice (Fig. 3C). However, transfection with siFOXM1 or siFOXM1a/c significantly inhibited pancreatic tumor growth (P < 0.05) (Fig. 4A-B) and inhibited liver metastasis of L3.7 cells (Fig. 4C-D) in the mice. These findings clearly indicated that FOXM1b and FOXM1c are oncogenic and promote invasion and metastasis of pancreatic tumors.
Figure 3.
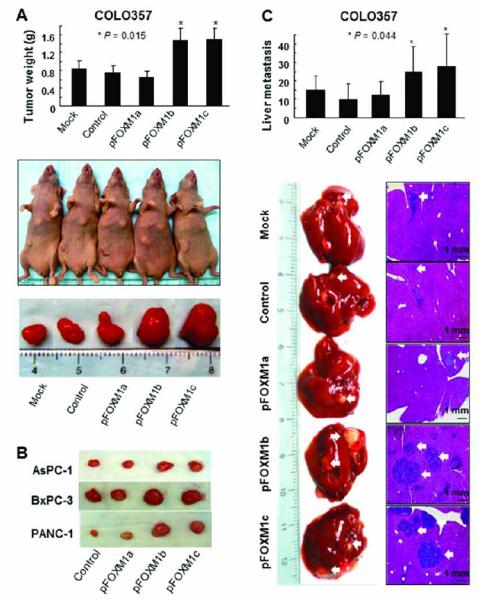
Influence of FOXM1a, FOXM1b, and FOXM1c overexpression on pancreatic tumor growth and metastasis. COLO357 cells were transfected with pcDNA3.1-FOXM1a (pFOXM1a), -FOXM1b (pFOXM1b), or -FOXM1c (pFOXM1c) expression vectors or the control pcDNA3.1 vector. A, tumor growth in nude mice. COLO357 cells were injected subcutaneously into the right scapular region (1 × 106 per mouse). Shown are the resulting tumor weights (top), gross tumors (middle), and resected tumors (bottom). *P < 0.01 in a comparison of the pFOXM1b- and pFOXM1c-treated groups with the mock-treated or control group. B, experiments similar to those in A were performed using AsPC-1, BxPC-3, and PANC-1 cells. Shown are representative tumors resected from mice injected with each cell type. C, pancreatic tumor metastasis in the study mice. COLO357 cells (1 × 106 per mouse) were injected into the ileocolic veins of five nude mice. Shown are the numbers of liver metastases (top), gross liver metastases (bottom left), and hematoxylin and eosin-stained liver metastasis sections (bottom right). *P < 0.01 in a comparison of the pFOXM1b- or pFOXM1c-treated group with the mock-treated or control group.
Figure 4.
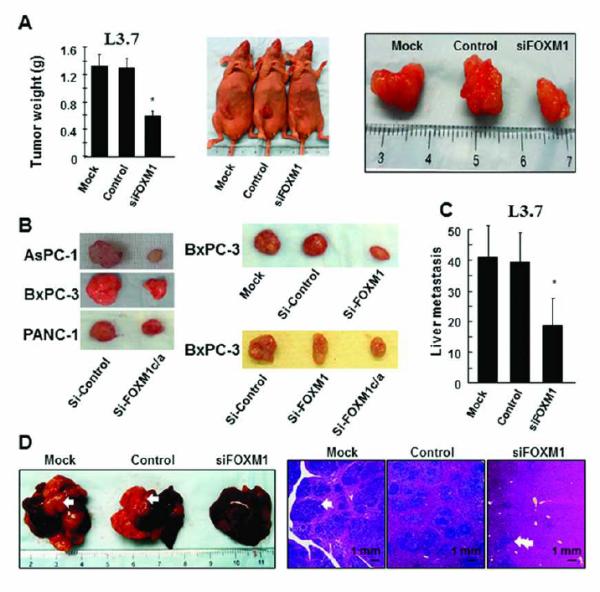
Influence of knockdown of FOXM1a, FOXM1b, and FOXM1c expression on pancreatic tumor growth and metastasis. L3.7 cells were transfected with siFOXM1 or control siRNA and injected into nude mice. The tumor-bearing mice were killed when they became moribund or on day 21 after tumor-cell injection. A, tumor growth in the mice. The cells were injected subcutaneously (1 × 106 per mouse) into the right scapular region. Shown are the resulting tumor weights (left), gross tumors (middle), and resected tumors (right). *P < 0.01 in a comparison of the siFOXM1-treated group with the mock-treated and control groups. B, experiments similar to those in A were performed using AsPC-1, BxPC-3, and PANC-1 cells and siFOXM1 or FOXM1a and FOXM1c siRNA (siFOXM1a/c). Shown are representative tumors resected from mice injected with each cell type. C, pancreatic tumor metastasis in the study mice. L3.7 cells (1 × 105 per mouse) were injected into the ileocolic veins of five mice. Shown are the numbers of liver metastases. D, the numbers of gross liver metastases (bottom left) and hematoxylin and eosin-stained liver metastasis sections (bottom right) in the mice in C. *P < 0.01 in a comparison of the siFOXM1-treated group with the mock-treated and control groups.
Transcriptional activation of uPAR expression by FOXM1 in pancreatic cancer cells
To determine whether FOXM1 regulates uPAR expression at the transcriptional level, we analyzed the uPAR promoter sequence for the presence of potential FOXM1-binding elements by using the FOXM1 consensus sequences 5'-A(C/T)AAA(C/T)AA-3', 5'-AGATTGAGTA-3', and 5'-TAATCA-3' (29, 32, 33)). We identified two putative FOXM1-binding elements (referred to as DNA sequences #1 and #2) in the uPAR promoter sequence (Fig. 5A). We then co-transfected a uPAR promoter luciferase reporter construct (puPAR1103) into COLO357 cells with pcDNA3.1-FOXM1a, pcDNA3.1-FOXM1b, pcDNA3.1-FOXM1c, or pcDNA3.1. Co-transfection with pcDNA3.1-FOXM1b or pcDNA3.1-FOXM1c activated the luciferase activity driven by the uPAR promoter (Fig. 5B). Conversely, we knocked down FOXM1 expression in L3.7 cells by co-transfecting them with siFOXM1 and the uPAR promoter. We observed that siFOXM1 inhibited the luciferase activity driven by the uPAR promoter (Fig. 5B). Furthermore, transfection with puPAR627 (lacking DNA sequence #1) attenuated the luciferase activity driven by the uPAR promoter in COLO357 and L3.7 cells (Fig. 5C).
Figure 5.

Direct binding of FOXM1 to the uPAR promoter. A, sequences and positions of putative FOXM1-binding elements on the uPAR promoter puPAR1103 (sites #1 and #2) and a deletion mutation of the uPAR promoter puPAR627 (site #2). B, COLO357 cells were co-transfected with 0.8 mg of the uPAR promoter luciferase construct puPAR1103 and 0-0.8 mg of pcDNA3.1-FOXM1a (pFOXM1a), pcDNA3.1-FOXM1b (pFOXM1b), pcDNA3.1-FOXM1c (pFOXM1c), or pcDNA3.1 (left), whereas L3.7 cells were co-transfected with 0.8 μg of puPAR1103 and 50 nmol/L siFOXM1 or control siRNA (siControl; right). The promoter activity in the cells was measured using a dual luciferase assay kit. *P < 0.01. C, COLO357 and L3.7 cells were co-transfected with pGL3-basic, puPAR1103, or puPAR627. The promoter activity in the cells was assessed using a dual luciferase assay kit. D, ChIP assay. Chromatins were isolated from COLO357 cells transfected with pcDNA3.1-FOXM1a, pcDNA3.1-FOXM1b, pcDNA3.1-FOXM1c, or pcDNA3.1, and binding of FOXM1 to the uPAR promoter was analyzed using a specific anti-FOXM1 antibody and oligonucleotides flanking the uPAR promoter regions containing putative FOXM1-binding sites as described in Materials and Methods (left). Similar ChIP assays were conducted using chromatins isolated from L3.7 and L3.7-siFOXM1 cells (right). Normal IgG was used as a control, and 1% of the total cell lysates was subjected to PCR analysis before immunoprecipitation (input control).
Finally, to prove that FOXM1 is recruited to the endogenous uPAR promoter in pancreatic cancer cells during transcription in vivo, we performed ChIP assays using chromatins prepared from COLO357 and L3.7 cells and two primer sets flanking 231-bp (−873 to −643; site #2) and 627-bp (−397 to +230; site #1) regions of the uPAR promoter. We observed that the 231-bp DNA fragment was amplified in the immunoprecipitates by anti-FOXM1 antibodies but not by a control IgG antibody in both cell types (Fig. 5D), suggesting that endogenous FOXM1 bound to the region from −873 to −643 bp of the uPAR promoter. Additionally, induced FOXM1b or FOXM1c overexpression led to increased FOXM1 recruitment to the uPAR promoter in COLO357 cells, whereas knockdown of FOXM1 expression led to decreased FOXM1 recruitment to the uPAR promoter in L3.7 cells (Fig. 5D). Collectively, these findings demonstrated that FOXM1 bound to the region from −707 to −692 bp of the uPAR promoter and positively regulated uPAR transcription in pancreatic cancer cells.
Discussion
In the present study, we sought to determine the critical roles of the FOXM1 isoforms in pancreatic cancer pathogenesis. We found that FOXM1b and FOXM1c transcriptionally activated the uPAR gene, constituting a novel signaling pathway that directly impacts EMT in and invasion and metastasis of pancreatic cancer cells, and that alterations of these two isoforms modulate the clinicopathological behavior of pancreatic cancer. Collectively, our novel clinical and mechanistic findings strongly suggest that dysregulated FOXM1b or FOXM1c expression causes abnormal uPAR and uPA expression in pancreatic cancer cells and is a critical/an important contributor to the pathogenesis and aggressiveness of this cancer.
The transcription factor FOXM1 is a key regulator of the cell cycle at both the G1/S and G2/M phase (33). Besides its involvement in cell-cycle transition, it plays important roles in tumor angiogenesis, EMT, invasion, and metastasis (9, 10). Lines of evidence demonstrate that FOXM1 is highly expressed in human lung carcinoma (11), cervical cancer (12), gastric cancer (13), and glioblastoma (14) cells and in a number of other types of human tumor cells (34), suggesting that FOXM1 is involved in the development and progression of many different human tumors. In the present study, we found that FOXM1 was highly expressed in human pancreatic tumors and that high expression of FOXM1 was strongly correlated with histological differentiation, advanced disease stage, and lymph node and/or distant metastasis, demonstrating important roles for FOXM1 in pancreatic cancer pathogenesis.
Alternative splicing of exons Va and VIIa in the FOXM1 gene results in formation of the three FOXM1 isoforms. FOXM1a is transcriptionally inactive, whereas FOXM1b and FOXM1c function as transcription activators (35). Many published studies have convincingly demonstrated that FOXM1b is an important regulator of cellular proliferation in tumorigenesis. Specifically, FOXM1b induces expression of cyclin A2, c-Jun N-terminal kinase 1, and activating transcription factor 2, all of which are critical for G1/S transition and DNA replication (36). Also, FOXM1b induces transcription of Skp2 and Cks1, which encode for subunits of the Skp/Cullin-1/F-box ubiquitin ligase complex; this transcription is required for downregulation of expression of the Cdk2 inhibitors p21Cip1 and p27Kip1 during the G1 phase (36). In addition, FOXM1b is a transcriptional activator of several genes critical to EMT, angiogenesis, and metastasis, such as caveolin-1 (18), vascular endothelial growth factor (13, 29), and MMP-2 (32); FOXM1b may contribute to human cancer progression by upregulating the expression of these three genes. Taken together, these results demonstrated that FOXM1b regulates the expression of proteins required for pancreatic tumor growth, EMT, angiogenesis, and metastasis and plays important roles in tumorigenesis.
In comparison, FOXM1c has a very strong C-terminal TAD, but full-length FOXM1c is only a weak transactivator under normal physiological conditions because its TAD is completely inhibited by the autoinhibitory N-terminus. However, a recent study demonstrated that cyclin E/Cdk2, cyclin A/Cdk2, and cyclin A/Cdk1 activate FOXM1c (37). In addition, protein kinases such as casein kinase 2, protein kinase A, c-Src, and Raf-1 can strongly activate FOXM1c via phosphorylation of the isoform’s N-terminus and blockage of FOXM1c inhibition by its N-terminus (38). Because cell cyclins and protein kinases are overactivated in cancer cells during tumorigenesis, these molecules may activate FOXM1c and subsequently promote cancer cell growth and metastasis during tumor development and progression. Recently, Wierstra et al. (39) found that FOXM1c regulates c-Myc, a key modulator of cell proliferation and differentiation, by directly transactivating the c-Myc promoter via the P1 and P2 TATA boxes. Furthermore, studies demonstrated that enforced expression of FOXM1c in cervical and ovarian cancer cells enhances their proliferation, anchorage-independent growth, and migration and invasive ability (40, 41).
In the present study, we found that the role of FOXM1c predominated over those of the other two isoforms in human pancreatic cancer cells and tumors. In addition, the expression of FOXM1c in highly metastatic human pancreatic cancer cells was much higher than that in poorly metastatic cells. Moreover, FOXM1b and FOXM1c promoted EMT in and migration, invasion, and metastasis of pancreatic cancer cells by upregulating uPA and uPAR expression; FOXM1a did not have any such effects. These data demonstrate that FOXM1b and FOXM1c both play important roles in pancreatic cancer pathogenesis and progression.
Authors first described the function of the uPA system in tumor invasion and metastasis by demonstrating pivotal roles of uPA and uPAR during pericellular proteolysis in degradation of the extracellular matrix (23). In addition, via their interactions with extracellular matrix proteins as well as integrins and other transmembrane receptors, uPA and uPAR modulate tumor angiogenesis and growth by regulating the adhesion, migration, and proliferation of cancer cells and/or vascular endothelial cells (42, 43). A large body of experimental evidence from in vitro, in vivo, and clinical studies indicates critical roles for the uPA system in the development of human tumors, including pancreatic cancer (22-26). Recently, a study demonstrated that uPAR induced EMT and promoted breast cancer metastasis by activating diverse cell signaling pathways (44). Moreover, researchers observed that uPAR-initiated cell signaling could be targeted to reverse EMT in cancer cells (45). Our present findings are consistent with these previous results, as they suggest a pivotal role for the uPA system in the progression of pancreatic cancer. In addition, we proved that uPAR expression levels correlated directly with FOXM1 expression levels in pancreatic cancer cells and tumors. Additionally, FOXM1b and FOXM1c increased the expression of both uPA and uPAR in pancreatic cancer cells, whereas knockdown of expression of FOXM1 decreased the expression of uPA and uPAR. Moreover, FOXM1 bound directly to the promoter region of the uPAR gene and positively transactivated its activity, demonstrating that uPAR is a novel downstream target of FOXM1. Given the important effects of uPAR on pancreatic cancer progression and the transcriptional regulation of uPAR by FOXM1, FOXM1 contributes to the pathogenesis and aggressive biology of pancreatic cancer at least in part by increasing uPAR expression, suggesting that dysregulated signaling by this novel FOXM1/uPAR pathway promotes pancreatic cancer development and progression.
Researchers have identified upstream signals for uPAR other than FOXM1, suggesting that the presence of transcriptional factors is essential for regulation of uPAR expression (23). For example, Sp1, a zinc finger transcription factor, promotes the expression of uPAR by binding to the uPAR promoter and subsequently affecting cell biology (46). In fact, investigators have frequently found overexpression of Sp1 in a wide variety of human tumors, including pancreatic cancer, and that Sp1 expression is correlated with aggressive biology and poor clinical outcome of cancer (47). However, whether overexpression of Sp1 contributes to pancreatic cancer progression via regulation of uPAR is unclear. Recent studies clearly demonstrated that the transcription factor hypoxia-inducible factor (HIF)-1α consistently upregulated the expression and activity of uPAR, thus contributing to the invasiveness of human cancer cells (48). Given the roles of HIF-1α in pancreatic cancer growth and metastasis (49), regulation of uPAR expression may be a mechanism whereby HIF-1α signaling affects cancer cells, leading to the development of pancreatic cancer. Thus, we are currently investigating whether HIF-1α signals promote pancreatic cancer development and progression via regulation of uPAR expression. More importantly, researchers demonstrated that transcription factors have many cross-links in tumor signal transduction and that different transcription factors can function cooperatively to activate target gene expression during tumorigenesis (50). Determination of how the interactions of FOXM1 with other transcription factors regulate the expression and function of uPAR and contribute to pancreatic carcinogenesis warrants further investigation. Therefore, further exploration of the molecular mechanisms that result in uPAR overexpression may not only shed more light on abnormal uPAR activation in pancreatic cancer cells but also help improve understanding of the value of uPAR as a prognostic factor and aid in the development of effective pancreatic cancer therapies that target uPAR.
In summary, this study provided critical insight into the role of the FOXM1 isoforms in pancreatic cancer progression and identified a critical role for novel FOXM1/uPAR signaling in the progression of pancreatic cancer. Collectively, our results not only identify a detailed molecular mechanism underlying regulation of uPA/uPAR by FOXM1c in particular in pancreatic cancer progression but also identify aberrant FOXM1c/uPAR signaling as a promising new molecular target for designing novel therapeutic modalities to control pancreatic cancer progression.
Supplementary Material
Translational Relevance
We have used pancreatic cancer tissue microarrays and molecular biology and animal models to evaluate the role and regulation of the urokinase plasminogen activator receptor (uPAR) system as a regulatory target of Forkhead box M1 (FOXM1) in human pancreatic cancer cells. Our clinical and mechanistic findings indicated that uPA/uPAR is a novel target of FOXM1 and that increased uPA/uPAR expression is closely associated with the clinicopathological characteristics of patients with and predicted poor prognosis for pancreatic cancer. Moreover, FOXM1-uPA/uPAR positively regulates pancreatic cancer cell migration, invasion, and growth, suggesting a novel cellular basis for the critical role of FOXM1-uPA/uPAR signaling in pancreatic cancer development and progression. It also suggests that deregulated FOXM1 signaling is a promising new molecular target for designing novel preventive and therapeutic strategies to control this malignancy. Therefore, our findings may have a major effect on clinical management of pancreatic cancer.
Acknowledgments
We thank Don Norwood for editorial comments.
Grant Support: This work was supported in part by grants R01-CA129956, R01-CA148954, R01-CA152309, and R01-CA172233 (to K. Xie) from the National Institutes of Health; grants 81101844 and 81210108027 (to C. Huang) from the National Natural Science Foundation of China; grants 2012040 and 13PJD024 (to C. Huang) from the Shanghai Municipal Human Resources and Social Security Bureau; and grant 13Y068 (to C. Huang) from the Shanghai Health and Family Planning Commission.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Nieto J, Grossbard ML, Kozuch P. Metastatic pancreatic cancer 2008: is the glass less empty. Oncologist. 2008;13:562–76. doi: 10.1634/theoncologist.2007-0181. [DOI] [PubMed] [Google Scholar]
- 3.Pliarchopoulou K, Pectasides D. Pancreatic cancer: current and future treatment strategies. Cancer Treat Rev. 2009;35:431–6. doi: 10.1016/j.ctrv.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 5.Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–20. doi: 10.1038/364412a0. [DOI] [PubMed] [Google Scholar]
- 6.Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997;25:1715–9. doi: 10.1093/nar/25.9.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997;17:1626–41. doi: 10.1128/mcb.17.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao KM, Sha M, Lu Z, Wong GG. Molecular analysis of a novel winged helix protein, WIN. Expression pattern, DNA binding property, and alternative splicing within the DNA binding domain. J Biol Chem. 1997;272:19827–36. doi: 10.1074/jbc.272.32.19827. [DOI] [PubMed] [Google Scholar]
- 9.Raychaudhuri P, Park HJ. FOXM1: a master regulator of tumor metastasis. Cancer Res. 2011;71:4329–33. doi: 10.1158/0008-5472.CAN-11-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koo CY, Muir KW, Lam EW. FOXM1: from cancer initiation to progression and treatment. Biochim Biophys Acta. 2012;1819:28–37. doi: 10.1016/j.bbagrm.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–61. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 12.Chan DW, Yu SY, Chiu PM, Yao KM, Liu VW, Cheung AN, et al. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis. J Pathol. 2008;215:245–52. doi: 10.1002/path.2355. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D, et al. Critical role and regulation of transcription factor FOXM1 in human gastric cancer angiogenesis and progression. Cancer Res. 2009;69:3501–9. doi: 10.1158/0008-5472.CAN-08-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, et al. FOXM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 15.Priller M, Poschl J, Abrao L, von Bueren AO, Cho YJ, Rutkowski S, et al. Expression of FOXM1 is required for the proliferation of medulloblastoma cells and indicates worse survival of patients. Clin Cancer Res. 2011;17:6791–801. doi: 10.1158/1078-0432.CCR-11-1214. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 17.Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, et al. Over-expression of FOXM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112:2296–306. doi: 10.1002/jcb.23150. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD, et al. A novel FOXM1-Caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. 2012;72:655–65. doi: 10.1158/0008-5472.CAN-11-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3:15–32. doi: 10.1023/a:1009095825561. [DOI] [PubMed] [Google Scholar]
- 20.Ulisse S, Baldini E, Sorrenti S, D'Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009;9:32–71. doi: 10.2174/156800909787314002. [DOI] [PubMed] [Google Scholar]
- 21.Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem. 2009;284:22825–33. doi: 10.1074/jbc.M109.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bock CE, Wang Y. Clinical significance of urokinase-type plasminogen activator receptor (uPAR) expression in cancer. Med Res Rev. 2004;24:13–39. doi: 10.1002/med.10054. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med Res Rev. 2001;21:146–70. doi: 10.1002/1098-1128(200103)21:2<146::aid-med1004>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 24.Harvey SR, Hurd TC, Markus G, Martinick MI, Penetrante RM, Tan D, et al. Evaluation of urinary plasminogen activator, its receptor, matrix metalloproteinase-9, and von Willebrand factor in pancreatic cancer. Clin Cancer Res. 2003;9:4935–43. [PubMed] [Google Scholar]
- 25.Bauer TW, Liu W, Fan F, Camp ER, Yang A, Somcio RJ, et al. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005;65:7775–81. doi: 10.1158/0008-5472.CAN-05-0946. [DOI] [PubMed] [Google Scholar]
- 26.Gorantla B, Asuthkar S, Rao JS, Patel J, Gondi CS. Suppression of the uPAR-uPA system retards angiogenesis, invasion, and in vivo tumor development in pancreatic cancer cells. Mol Cancer Res. 2011;9:377–89. doi: 10.1158/1541-7786.MCR-10-0452. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Wei D, Huang S, Peng Z, Le X, Wu TT, et al. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin Cancer Res. 2003;9:6371–80. [PubMed] [Google Scholar]
- 28.Huang C, Jiang T, Zhu L, Liu J, Cao J, Huang KJ, et al. STAT3-targeting RNA interference inhibits pancreatic cancer angiogenesis in vitro and in vivo. Int J Oncol. 2011;38:1637–44. doi: 10.3892/ijo.2011.1000. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Zhang N, Dai B, Liu M, Sawaya R, Xie K, et al. FOXM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res. 2008;68:8733–42. doi: 10.1158/0008-5472.CAN-08-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith R, Xue A, Gill A, Scarlett C, Saxby A, Clarkson A, et al. High expression of plasminogen activator inhibitor-2 (PAI-2) is a predictor of improved survival in patients with pancreatic adenocarcinoma. World J Surg. 2007;31:493–502. doi: 10.1007/s00268-006-0289-9. [DOI] [PubMed] [Google Scholar]
- 31.Kong X, Li L, Li Z, Le X, Huang C, Jia Z, et al. Dysregulated expression of FOXM1 isoforms drives progression of pancreatic cancer. Cancer Res. 2013;73:3987–96. doi: 10.1158/0008-5472.CAN-12-3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai B, Kang SH, Gong W, Liu M, Aldape KD, Sawaya R, et al. Aberrant FOXM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26:6212–9. doi: 10.1038/sj.onc.1210443. [DOI] [PubMed] [Google Scholar]
- 33.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257–74. doi: 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH. Forkhead box M1 transcription factor: a novel target for cancer therapy. Cancer Treat Rev. 2010;36:151–6. doi: 10.1016/j.ctrv.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laoukili J, Stahl M, Medema RH. FOXM1: at the crossroads of ageing and cancer. Biochim Biophys Acta. 2007;1775:92–102. doi: 10.1016/j.bbcan.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FOXM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wierstra I, Alves J. FOXM1c is activated by cyclin E/Cdk2, cyclin A/Cdk2, and cyclin A/Cdk1, but repressed by GSK-3alpha. Biochem Biophys Res Commun. 2006;348:99–108. doi: 10.1016/j.bbrc.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Wierstra I. The transcription factor FOXM1c is activated by protein kinase CK2, protein kinase A (PKA), c-Src and Raf-1. Biochem Biophys Res Commun. 2011;413:230–5. doi: 10.1016/j.bbrc.2011.08.075. [DOI] [PubMed] [Google Scholar]
- 39.Wierstra I, Alves J. FOXM1c transactivates the human c-myc promoter directly via the two TATA boxes P1 and P2. FEBS J. 2006;273:4645–67. doi: 10.1111/j.1742-4658.2006.05468.x. [DOI] [PubMed] [Google Scholar]
- 40.Chan DW, Yu SY, Chiu PM, Yao KM, Liu VW, Cheung AN, et al. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis. J Pathol. 2008;215:245–52. doi: 10.1002/path.2355. [DOI] [PubMed] [Google Scholar]
- 41.Lok GT, Chan DW, Liu VW, Hui WW, Leung TH, Yao KM, et al. Aberrant activation of ERK/FOXM1 signaling cascade triggers the cell migration/invasion in ovarian cancer cells. PLoS One. 2011;6:e23790. doi: 10.1371/journal.pone.0023790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prager GW, Breuss JM, Steurer S, Olcaydu D, Mihaly J, Brunner PM, et al. Vascular endothelial growth factor receptor-2-induced initial endothelial cell migration depends on the presence of the urokinase receptor. Circ Res. 2004;94:1562–70. doi: 10.1161/01.RES.0000131498.36194.6b. [DOI] [PubMed] [Google Scholar]
- 43.Margheri F, Chilla A, Laurenzana A, Serrati S, Mazzanti B, Saccardi R, et al. Endothelial progenitor cell-dependent angiogenesis requires localization of the full-length form of uPAR in caveolae. Blood. 2011;118:3743–55. doi: 10.1182/blood-2011-02-338681. [DOI] [PubMed] [Google Scholar]
- 44.Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–36. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo M, Lester RD, Montel V, Eastman B, Takimoto S, Gonias SL. Reversibility of epithelial-mesenchymal transition (EMT) induced in breast cancer cells by activation of urokinase receptor-dependent cell signaling. J Biol Chem. 2009;284:22825–33. doi: 10.1074/jbc.M109.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zannetti A, Del VS, Carriero MV, Fonti R, Franco P, Botti G, et al. Coordinate up-regulation of Sp1 DNA-binding activity and urokinase receptor expression in breast carcinoma. Cancer Res. 2000;60:1546–51. [PubMed] [Google Scholar]
- 47.Shi Q, Le X, Abbruzzese JL, Peng Z, Qian CN, Tang H, et al. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 2001;61:4143–54. [PubMed] [Google Scholar]
- 48.Shyu KG, Hsu FL, Wang MJ, Wang BW, Lin S. Hypoxia-inducible factor 1alpha regulates lung adenocarcinoma cell invasion. Exp Cell Res. 2007;313:1181–91. doi: 10.1016/j.yexcr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 49.Chang Q, Qin R, Huang T, Gao J, Feng Y. Effect of antisense hypoxia-inducible factor 1alpha on progression, metastasis,and chemosensitivity of pancreatic cancer. Pancreas. 2006;32:297–305. doi: 10.1097/00006676-200604000-00010. [DOI] [PubMed] [Google Scholar]
- 50.Huang C, Xie K. Crosstalk of Sp1 and Stat3 signaling in pancreatic cancer pathogenesis. Cytokine Growth Factor Rev. 2012;23:25–35. doi: 10.1016/j.cytogfr.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.