

Abstract
Caroli’s disease is a rare congenital disorder characterized by cystic dilatation of the large in-trahepatic bile ducts. The most frequent complications due to biliary stasis are cholelithiasis, cholangitis and sepsis as well as an increased risk of cholangiocarcinoma. Patients may have a history of intermittent abdominal pain, pruritus and/or symptoms of cholangitis. It is rarely diagnosed in childhood. A 12-year-old boy with isolated Caroli’s disease is described. This child presented at the age of 2 years, with 4 episodes of recurrent bacterial infections. Interestingly he remained asymptomatic for over 10 years, between the second and third episode. During the 4th episode, when he presented with fever and slight abdominal pain, the diagnosis was made on the basis of radiological findings: U/S, CT, MRI and especially with MRCP, in relation with a more typical picture, resembling cholangitis. Since then he has been followed-up systematically for ten years and remains in good clinical condition without further relapses and with unchanged radiological findings. This atypically benign course of Caroli’s disease, with intermittent asymptomatic periods, without any treatment, is very rare.
Keywords: Caroli’s disease, recurrent bacterial infections, childhood
Introduction
Caroli’s disease and Caroli’s syndrome are rare congenital disorders characterized by cystic dilatation of the intrahepatic biliary tree [1,2]. Caroli’s disease is sporadic, its prevalence is one case per 1,000,000 of the population [3,4] and is less common than Caroli’s syndrome, mainly inherited in an autosomal recessive manner [5,6], although other modes of inheritance (autosomal dominant) were also described [5,6]. Loss of the distal 3p chromosome and gain of the 8q chromosome is of pathogenic importance [7]. As with congenital hepatic fibrosis Caroli’s syndrome is often associated with autosomal recessive polycystic kidney disease (ARPKD) (8, 9) and less frequently with autosomal dominant polycystic kidney disease (ADPKD) [10,11].
The illness has usually a long-lasting course, with recurrent episodes of cholangitis, which may be complicated by intrahepatic calculi and hepatic abscess formation. There is also good evidence that Caroli’s disease confers an approximately 7% risk of malignancy [12].
The diagnosis rests on demonstrating that the cystic liver lesions are in continuity with the biliary tree. Modern imaging techniques (initially abdominal ultrasound and then with magnetic cholangio-pancreatographic study: MRCP), facilitate diagnosis without the need of invasive imaging of the biliary tree (such as endoscopic cholangio-pancreatographic study: ERCP) [10,13].
We describe this case report of a 12-year-old boy with Caroli’s disease, an extremely rare condition in childhood, because of its atypical presentation and its unexpected benign course, before and after the diagnosis was made.
Case report
A 12-year-old boy was admitted to the Pediatrics Department of our Hospital, because of high fever (40.5°C) with shivers and moderate pain in the right upper quadrant of the abdomen, for the previous two days. The pain increased during the rise in fever. Before his admission a course of antibiotics (amoxicilline-clavulanic acid) had preceded without any response. On physical examination the liver and spleen were just palpable, liver (+1 cm) and spleen (+1cm) under costal margins. Interestingly a slight tenderness in the right hypochondrium was found only as the fever rose. No jaundice, ascites, splenomegaly or venous dilatation of abdominal wall were observed. The physical examination of the other systems was normal. Blood pressure was repeatedly between 100-110 / 60-70 mmHg.
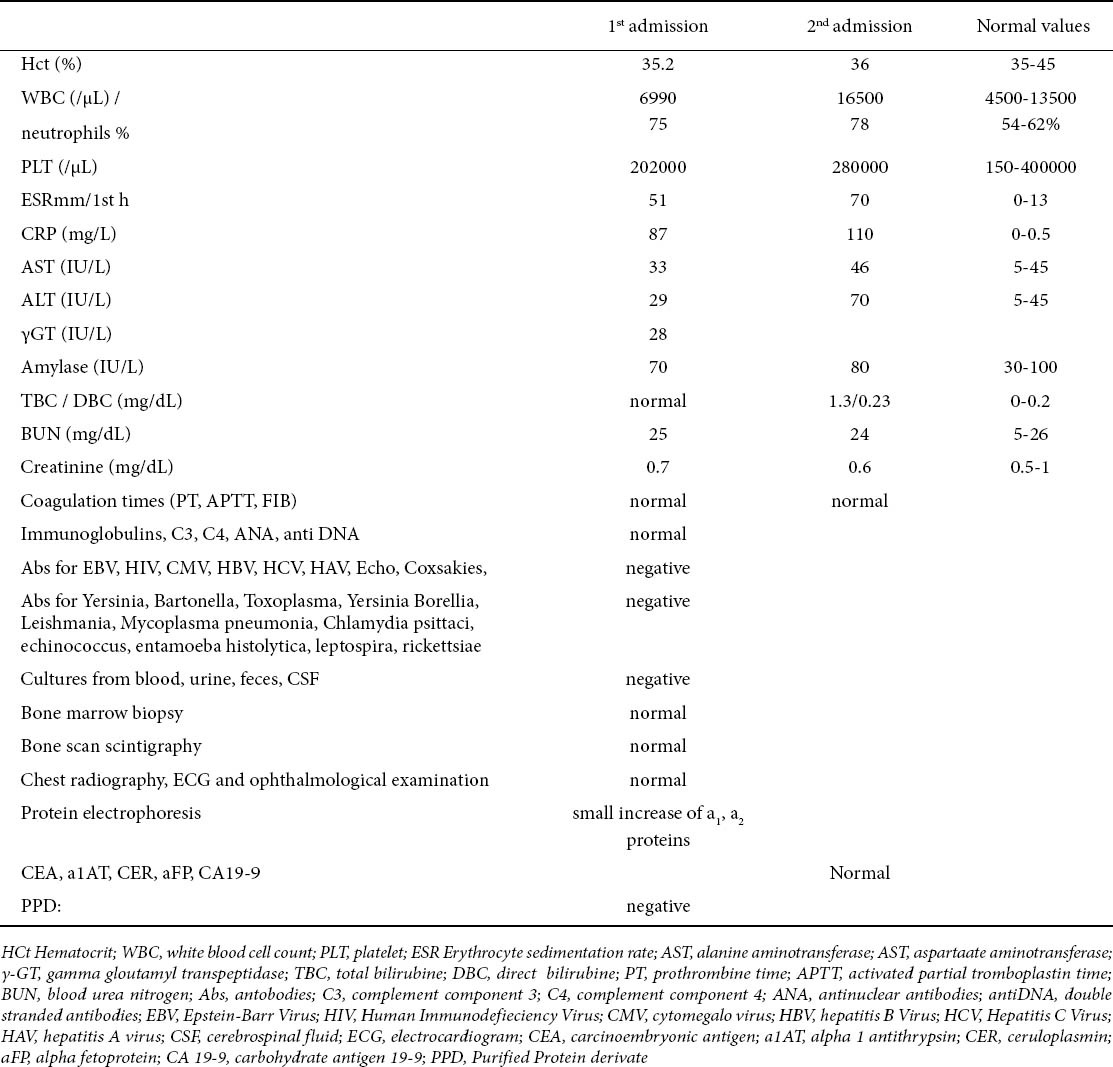
A month before this episode, he had been hospitalized again because of an episode with similar clinical picture (fever for 8 days: 38-40.5°C, with shivers, muscle aches and diffuse abdominal pain) and he was treated for suspected bacteremia for 10 days. The laboratory findings on these two admissions appear on Table 1 and it is noteworthy that during the second admission the laboratory findings of bacterial infection were more obvious (although with negative cultures) in addition to a slight elevation of transaminases. His medical history was extremely interesting in that he had had two consecutive hospitalizations at the age of 2.5 years, because of suspected bacteremia. At that time he presented again with elevated WBC, CRP, ESR, negative cultures, normal immune profile and normal liver function tests, so an ultrasound (U/S) of the abdomen was not considered necessary. In the intermediate period (the last 10 years) he was asymptomatic with normal growth (weight and height: between 50th to 75th percentile, according to our national charts) and without any other chronic illness.
Table 1.
Laboratory investigations during the last two admissions (3nd and 4th episode)
During the last hospitalization (4th episode), because of the localization of moderate pain in the right upper quadrant of abdomen during the rise in fever, an U/S of the abdomen was performed and revealed two small cystic formations in the superior part of right upper lobe of the liver.
According to the clinical picture and to the above U/S findings, more extensive radiological investigation was performed. CT scan of abdomen revealed several small cystic formations in the VII segment of the liver. MRI of the liver showed several well-defined round lesions (maximum diameter 2 cm), hyperintense on T2 and hypointense on T1 -weighted images. There was no contrast enhancement after intravenous contrast (Gadolinium DTPA) injection, therefore these findings were thought to be cysts [Fig. 1a, 1b].
Figure 1.
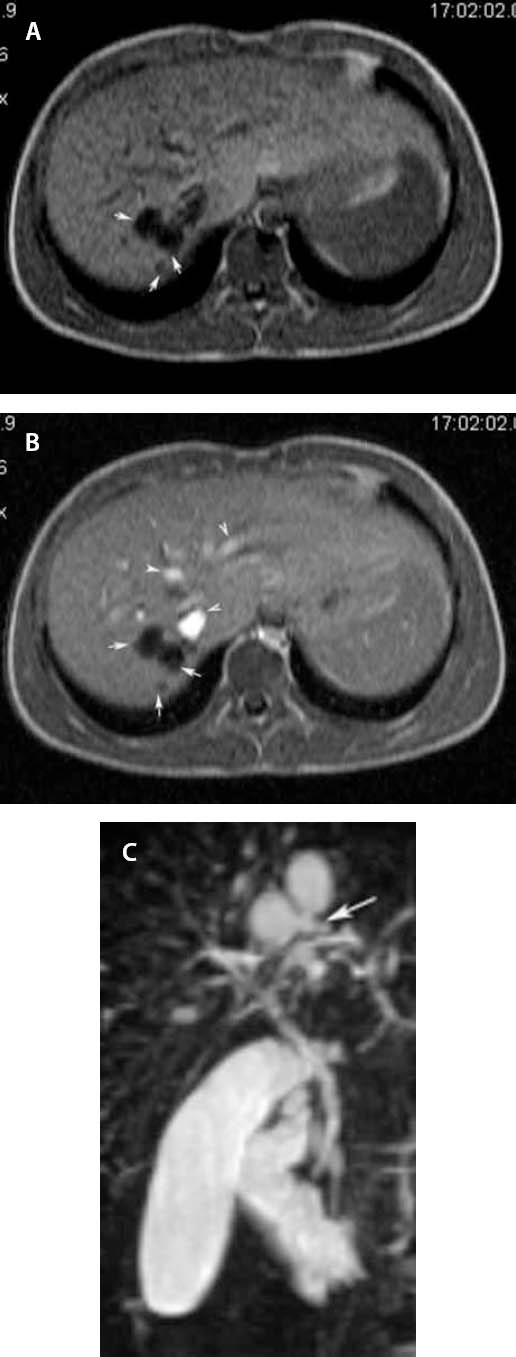
a Plain T1-weighted axial scan demonstrates, in segment VII of the liver, three well defined round lesions of low signal intensity (arrows), b Contrast-enhanced T1-weighted axial scan shows no enhancement of the lesions (arrows); the enhancing structures represent vascular branches (arrowheads), c MRCP depicts the cysts communicating with the biliary tree (arrow).
In addition, there were no specific findings from other organs (kidneys, pancreas, spleen etc) or signs of portal hypertention.
A subsequent MRCP revealed communication of the cystic formations with the biliary tree [Fig. 1c], compatible with Caroli’s disease.
The child was treated for cholangitis. He was afeverish on the 4th day of treatment. The abdominal pain subsided later (on 9th day).
He was discharged in generally good condition with the advice of frequent follow-up studies, including liver function tests (AST, ALT, γGT, CEA, CA19-9, aFP etc.) and U/S of the liver.
Since, he has been followed-up systematically for ten years and remains asymptomatic, with normal blood pressure, normal growth, normal laboratory findings and unchanged imaging (U/S and MRI) parameters, in follow-up studies.
Discussion
Patients with Caroli’s disease/syndrome suffer from relapsing episodes of cholangitis with the severe danger of bacteremia and sepsis. They report in their history, episodes of intermittent abdominal pain owing mainly to cholestasis and choledocholithiasis. Fever with shivers and pain in the upper right quadrant of the abdomen with or without jaundice are due to the episodes of cholangitis that these patients suffer from. It is rarely present in childhood and the diagnosis is usually made at an advanced age [6,14,15], although there are case reports published of even neonatal presentation of the disease [16]. Caroli’s disease results from an arrest in ductal plate remodeling at the level of the larger intrahepatic bile ducts. In contrast, Caroli’s syndrome results when the full spectrum of bile duct differentiation is affected, such that the smaller interlobular ducts are involved and congenital hepatic fibrosis develops.
Our patient presented initially at the age of 2 years, with 2 episodes of recurrent “suspected” bacterial infections. In these admissions, he presented with fever without focus, elevation of parameters of inflammation (WBC, ESR, CRP) with negative cultures and without any symptom or sign of abdominal pathology. Therefore he was treated as having bacteremia at that time. Interestingly he remained asymptomatic for 10 years and relapsed at the age of 12, when diagnosis was made. During the last hospitalization he showed more specific clinical signs. Clinical and laboratory findings were suggestive of cholangitis and an extended radiological investigation was performed.
There have been reports of isolated sporadic cases of children with Caroli’s disease, who presented with clinical signs of recurrent abdominal pain associated with intermittent crises of cholestatic jaundice [15].
There are also some pediatric cases with unusual presentation, as in one 2-year-old girl with a palpable mass below the right costal margin and displacement of the right colon and in whom the diagnosis was confirmed histologically after the excision of the mass [17]. Another report of two siblings, a 5-year-old girl in whom diagnosis was made by laparotomy because of hepatomegaly and mottled radiopacities shown by cholangiography and her 9-year-old asymptomatic brother who was diagnosed by cholangiography, during his sister’s investigation [6].
The characteristic findings of the disease are the dilated sacculi or cystic spaces, visible by U/S or CT of the abdomen, only in some patients, particularly in the early stages. The findings include saccular or tubular dilatation of intrahepatic bile ducts, intraluminal bulbar protrusions, bridge formation across dilated bile ducts resembling internal septa, within the dilated bile ducts and portal radicles partially or completely surrounded by dilated bile ducts. These portal radicles - if seen in axial projection - appear as tiny, hyperechogenic structures centrally in the dilated bile duct and have been named; the “central dot” signs [12]. Higher density small structures within the dilated ducts represent non-calcified or calcified stones.
In the boy described above, U/S depicted two small cystic formations in the liver, initially thought to represent simple liver cysts. Simple liver cysts commonly seen on U/S seldom cause symptoms, but, occasionally, if they are large may cause pain and some require surgical drainage.
Polycystic liver disease could be a possible diagnosis, but in this condition although large cysts developed from the biliary tree, they did not obstruct the ducts. The clinical picture is also different as cysts may cause pain, but do not affect liver function and in most patients, the kidneys are similarly affected with cysts, which may cause high blood pressure and kidney failure. The tendency to form cysts is probably present at birth, but usually they do not enlarge and cause problems until adulthood [10]. Our patient had normal blood pressure and his kidneys were normal in shape and size, with normal function.
Von Meyenburg complex could be another possible diagnosis, which is a rare condition characterized by multiple small hepatic cysts within the parenchyma at a distance from the peribiliary portal regions and with no communication with the bile ducts [18]. In our patient the cystic formations were in communication with the biliary tree.
On the other hand, in Caroli’s syndrome both CT and U/S show focal medium dilatation of intrahepatic bile ducts (2-3 mm) and in addition the liver shows changes of portal hypertension (shrunken liver, splenomegaly, splenic and esophageal varices and ascites) and intrahepatic biliary calculi are absent [17]. In the presence of portal hypertension with splenomegaly, the expected laboratory findings were leukopenia, thrombocytopenia. These clinical and laboratory findings were absent in our case, therefore the most possible diagnosis was Caroli’s disease.
ERCP and, especially in children today, MRCP is the most specific and non invasive examination [19,20] to depict the multiple ductal dilatation seen in Caroli’s disease, called the “lollipop tree” aspect (T2 and most notably T1 sequences after contrast injection). It shows diverticulum-like sacculi of intrahepatic bile duct ectasiae of varying sizes, shapes and distribution, which communicate freely with the bile duct, which were present in our patient and made the diagnosis. Conversely, in Caroli’s syndrome the cystic bile duct ectasiae are always smaller (<2cm) and periportal hepatic fibrosis is seen on T2 – weighted sequences as high signal areas among the portal veins.
Thus with the help of MRCP, a complete noninvasive investigation, offering similar findings to the ERCP, without the danger of ascending cholangitis and bile duct abscess [13,17,19], the diagnosis of Caroli’s disease was made, resolving the differential diagnosis problems, as it provides an exhaustive investigation of the bile ducts and the liver parenchyma [20].
Liver biopsy for exclusion of associated congenital hepatic fibrosis was not performed in our patient since the clinical picture, with such an extremely benign course of the disease, and imaging studies did not show any evidence of portal hypertension. Moreover, liver biopsy is not indispensable for the diagnosis especially in the absence of chronic cholangitis. Genetic study was also not performed.
The treatment depends on the clinical features and the location of the biliary abnormality. Ursodeoxycholic acid is a useful treatment for primary hepatolithiasis in Caroli’s disease/syndrome [21] and can decrease the frequency of complications from gallbladder stones.
Wide-spectrum antibiotics are indicated in episodes of cholangitis because they can control infection, but the underlying anatomical abnormality may make permanent sterilization impossible [2].
Surgery is indicated when bile stasis exists leading to either calculi or mechanical obstruction or in the presence of cholangiocarcinoma [12,22]. In case of unilobar form of the disease without any concurrent liver fibrosis or cirrhosis, partial hepatectomy achieves satisfactory results by providing a radical solution to the recurrent problems of cholangitis, stone formation and intrahepatic cholangiocarcinoma [22-24]. In diffuse Caroli’s disease, treatment options include conservative or endoscopic therapy, internal biliary bypass procedures and liver transplantation on carefully selected cases [2]. In case of bilobar disease complicated with hepatic fibrosis or cirrhosis, liver transplantation seems to be the treatment of choice [22,25].
In patients with Caroli’s disease, the long-term prognosis is determined mainly by the frequency and the gravity of the episodes of cholangitis that can lead to sepsis and death or creation of hepatic abscesses. Hepatic insufficiency can develop and transplantation of the liver may be required. Malignancies are also possible complications of Caroli’s disease, as cholangiocarcinoma risk is 100-fold in comparison with the general population [2]. Amyloidosis is also described as a complication of Caroli’s disease [12].
The clinical presentation and the outcome of the disease is variable and depends on the extension of the disease, the correct and rapid confrontation of complications, with frequent follow-up examinations of the liver function and U/S control for the existence of stones and the early recognition of the probable associated conditions.
In our patient the diagnosis could be missed because of the absence of special symptoms in the beginning and the extremely benign course of the disease. The fact that he remained asymptomatic for ten years before the diagnosis and ten years afterwards without any medication is extremely rare. The isolated unilobar form of disease, the absence of fibrosis and its complications, as well as the absence of cholelithiasis and the limited episodes of cholangitis could be possible explanations for this atypically benign course of the disease, with intermittent asymptomatic periods. Hypothetically, isolated right lobe partial hepatectomy could be an alternative option of treatment for our patient, if more episodes of cholangitis occur.
In the available literature, there have been only two reported cases of children with a similar benign course, a girl who was diagnosed as having the disease at the age of 5 and remained asymptomatic and in good health for over 21 years, and her brother who after the same period in which he remained asymptomatic, suddenly relapsed with hematemesis due to esophageal varices [6].
Therefore, further studies are needed with special focus on the long-term results, in order to estimate more accurately long-term prognosis in children with this rare disease.
Biography
University Hospital of Ioannina, Greece
Footnotes
Conflict of Interest: None
References
- 1.Caroli JR, Soupault J, Kossakowski L, Plocker M, Paradowska. La dilatation polykystique congenitale des voies biliares intraphepatiques: essai de classification. Semin Hop Paris. 1958;34:488–495. [PubMed] [Google Scholar]
- 2.Taylor AC, Palmer KR. Caroli's disease. Eur J Gastroenterol Hepatol. 1998;2:105–108. doi: 10.1097/00042737-199802000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Veigel MC, Focht JP, Rodriguez MG, et al. Fibrocystic liver disease in children. Ped Radiol. 2009;39:317–327. doi: 10.1007/s00247-008-1070-z. [DOI] [PubMed] [Google Scholar]
- 4.Miwala F, Segev D, Thuluvath P. Caroli's disease and outcomes after liver transplantation. Liver Transpl. 2008;14:11–17. doi: 10.1002/lt.21366. [DOI] [PubMed] [Google Scholar]
- 5.Yoshizawa K, Kiyosawa K, Yabu K, et al. Caroli's disease in three siblings. Gastroent Jpn. 1992;27:780–784. doi: 10.1007/BF02806532. [DOI] [PubMed] [Google Scholar]
- 6.Tsuchida Y, Sato T, Sango K, et al. Evaluation of long-term results of Caroli's disease: 21 years observation of a family with autosomal “dominant” inheritance, and review of the literature. Hepatogastroenterology. 1995;42:175–181. [PubMed] [Google Scholar]
- 7.Parada La Hallen M, Hagestrand I, Tranberg KG, Johansson B. Clonal chromosomal abnormalities in congenital bile duct dilatation (Caroli's disease) Gut. 1999;45:780–782. doi: 10.1136/gut.45.5.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung G, Benz-Bohm G, Kugel H, Keller KM, Querfield U. MR cholangiography in children with autosomal recessive polycystic kidney disease. Pediatr Radiol. 1999;29:463–466. doi: 10.1007/s002470050618. [DOI] [PubMed] [Google Scholar]
- 9.Pirson Y, Lannoy N, Peters D, Geubel A, Gigot JF, Breunning M. Isolated polycystic liver disease as a distinct genetic disease, unlinked to polycystic kidney disease 1 and polycystic kidney disease. Hepatology. 1996;23:249–252. doi: 10.1002/hep.510230208. [DOI] [PubMed] [Google Scholar]
- 10.Itai Y, Ebihara R, Eguchi N, Saida Y, Kurosaki Y, Minami M. Hepatobiliary cysts in patients with autosomal dominant polycystic kidney disease: prevalence and CT findings. AJR. 1995;164:339–342. doi: 10.2214/ajr.164.2.7839965. [DOI] [PubMed] [Google Scholar]
- 11.Mousson C, Rabec M, Cercueil JP, Viret JS, Hillon P, Rifle G. Caroli's disease and autosomal dominant polycystic kidney disease: a rare association? Nephrol Dial Transplant. 1997;12:1481–1483. doi: 10.1093/ndt/12.7.1481. [DOI] [PubMed] [Google Scholar]
- 12.Gupta AK, Gupta A, et al. Caroli's Disease. Indian J Pediatr. 2006;73:233–235. doi: 10.1007/BF02825490. [DOI] [PubMed] [Google Scholar]
- 13.Hussain SZ, Bloom DA, Tolia V. Caroli's disease diagnosed in a child by MRCP. Clin Imag. 2000;24:289–291. doi: 10.1016/s0899-7071(00)00215-1. [DOI] [PubMed] [Google Scholar]
- 14.Keramidas DC, Kapouleas GP, Sakellaris G. Case Report. Isolated Caroli's disease presented as an exophytic mass in the liver. Ped Surg Internat. 1998;13:177–179. doi: 10.1007/s003830050281. [DOI] [PubMed] [Google Scholar]
- 15.Fagundes-Neto U, Schettini ST, Wehba J, Pinus J, Patricio FR. Caroli's disease in childhood: report of two new cases. J Pediatr Gastroenterol Nutr. 1983;2:708–711. [PubMed] [Google Scholar]
- 16.Keane F, Hadzić N, Wilkinson ML, et al. Neonatal presentation of Caroli's disease. Arch Dis Child Fetal Neonatal Ed. 1997;77:F145–146. doi: 10.1136/fn.77.2.f145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernstine J. What is Caroli's disease? Gastroenterology. 1975;68:417–419. [PubMed] [Google Scholar]
- 18.Maher MM, Dervan P, Keogh B, Murray JG. Bile duct hamartomas (Von Meyenburg complexes) value of MR imaging in diagnosis. Abdom Imaging. 1999;24:171–173. doi: 10.1007/s002619900469. [DOI] [PubMed] [Google Scholar]
- 19.Krause D, Cercueil JP, Dranssart M, Cognet F, Piard F, Hillon P. MRI for evaluating congenital bile duct abnormalities. J Comput Assist Tomogr. 2002;26:541–552. doi: 10.1097/00004728-200207000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Guy F, Cognet F, Dranssart M, Cercueil JP, Conciatori L, Krause D. Caroli's disease: magnetic resonance imaging features. Eur Radiol. 2002;12:2730–2736. doi: 10.1007/s00330-002-1471-6. [DOI] [PubMed] [Google Scholar]
- 21.Ros E, Navarro S, Bru C, Gilabert R, Bianchi L, Bruguera M. Ursodeoxycholic acid treatment of primary hepatolithiasis in Caroli's syndrome. Lancet. 1993;342:404–406. doi: 10.1016/0140-6736(93)92817-d. [DOI] [PubMed] [Google Scholar]
- 22.Kassahun WT, Kahn T, Wittekind C, et al. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery. 2005;138:888–898. doi: 10.1016/j.surg.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Espinoza R, San Martin S, Court F, et al. Hepatic resection in localized Caroli disease. Rev Med Chil. 2003;131:183–189. [PubMed] [Google Scholar]
- 24.Mabrut JY, Partensky C, Jaeck D, et al. Congenital intrahepatic bile duct dilatation is a potentially curable disease: long-term results of a multi-institutional study. Ann Surg. 2007;246:236–245. doi: 10.1097/SLA.0b013e3180f61abf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Habib S, Shakil O, Couto OF, et al. Caroli's disease and orthotopic liver transplantation. Liver Transpl. 2006;12:416–421. doi: 10.1002/lt.20719. [DOI] [PubMed] [Google Scholar]