

Abstract
The chronic inflammatory bowel diseases (IBD), Crohn’s disease and ulcerative colitis, are recognized as important causes of gastrointestinal disease in children and adults. Insight into IBD is advancing rapidly owing to a plethora of investigations into intestinal inflammation in animal models, advances in the interrogation of diseases inherited as complex genetic traits, and the development of methods to define the composition of the intestinal microbiota. These advances offer a better understanding of the genetically determined interplay between the commensal microbiota, intestinal cells and the immune system, and the manner in which this interaction might be modified by environmental factors in the pathogenesis of IBD. The present review highlights recent advances in IBD research.
Keywords: IBD, genetic susceptibility, NOD2, innate immunity, autophagy
Introduction
The two clinically distinguished forms of inflammatory bowel diseases (IBD), Crohn’s disease (CD) and ulcerative colitis (UC), are chronic remittent or progressive inflammatory diseases, which affect the gastrointestinal tract or only the colonic mucosa respectively. It is believed that both genetic and environmental factors contribute to the pathophysiology of IBD. Studies in the early 1990s proved that interleukin (IL)-2, IL-10 and T-cell receptor (TCR) mutant mice develop IBD-like enterocolitis [1] whereas blockage of tumor necrosis factor (TNF)-α ameliorates the severity of inflammation. Recent studies focus on the interactions between human intestinal microbiota and the mucosal immune system and the manner in which a variety of environmental and genetic factors modify these relationships [2,3]. Another important aspect in the pathophysiology of IBD is the role of the innate immune system and its relationship with the commensal microbiota and the adaptive immune system. Finally it is believed that IBD is likely the result of a continuum of diseases that range from monogenically inherited familial forms to sporadic forms which are polygenic in origin and strongly influenced by environmental factors. Recent studies on the genetic influence of IBD revealed a variety of immunological pathways critical in the pathogenesis of these disorders. These include autophagy and unfolded protein response which play an important role in the interactions between host and highly complex microbial communities within the intestine. It seems that the interaction between intestinal microbiota and particularly some components of it, and the immune system are critical in the pathogenesis of IBD, but are influenced by a variety of environmental factors and the genetic background. This review concludes with the most recent discoveries concerning genetics and innate immune interactions in the pathophysiology of IBD (Fig. 1).
Figure 1.
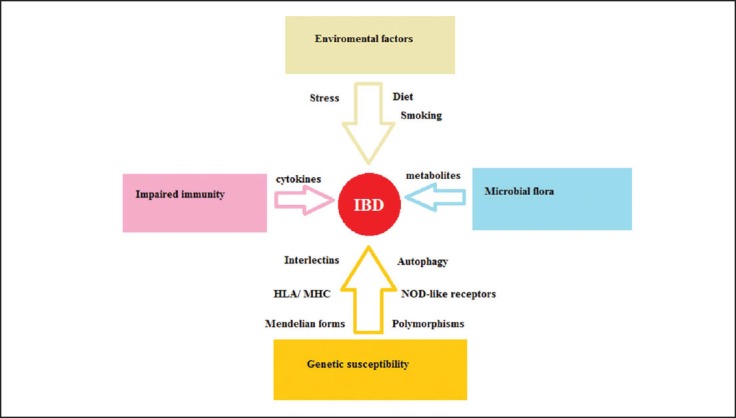
Factors implicated in the pathophysiology of inflammatory bowel disease (IBD).
Genetic basis of IBD
The first person who mentioned that IBD tended to occur within families was Crohn in the 1930s [4]. It was also known that IBD exhibits pathologic similarities to intestinal infections such as those derived from Mycobacterium Mycobacterium tuberculosis, suggesting a potential environmental and immunological component [4]. Today IBD is believed to be a polygenic disorder and is familial in 5–10% of individuals and sporadic in the remainder [5]. It is known that phenotypic concordance between monozygotic twins is more frequent in CD (50–75%) than in UC (10–20%) suggesting that hereditability is more important, the relevant environmental exposure is more common or that copy number variations and epigenetic differences between twins are less frequent in CD [5]. Although the relative risk for UC in monozygotic twins is lower than that observed in CD this does not rule out a critical role for genetic factors in the pathophysiology of UC as well.
Recent studies suggest that mutations in genes, important for the maintenance of intestinal homeostasis between the immune system and commensal microbiota, are critical to the pathophysiology of IBD. However, despite the existence of different genotypes among various populations there is a similarity in the clinicopathologic phenotype and in the response to various treatments suggesting that the variety of risk loci contribute to a limited number of phenotypic pathways in the pathophysiology of IBD. It is also known that the loci identified to date represent only 10% of the overall variance of potential disease risk [6], suggesting that the phenotype of IBD may be due to the interaction between hundreds or thousands of common single nucleotide polymorphisms of minor biological impact [7] or/ and that the phenotype is attributed to the effects of rare variants with profound impact [7]. In other words, we believe that IBD is a syndrome of overlapping phenotypes that involves variable influences of genetic and environmental factors. Considering this model, at the two polar extremes are the polygenic disease, in which the environmental exposure is of central importance relative to genetic factors and the monogenic disease. This latter interpretation explains the existence of some “more” Mendelian forms of IBD like a familial form of early-onset CD, due to homozygous mutations in either IL10RA or IL10RB which encode subunits of the IL-10 receptor [8].
Recent studies have helped to define a variety of pathways involved in IBD pathogenesis suggesting a variety of risk-conferring loci, most of them involved in regulation of innate and adaptive immunity (IL-23R, IL-10, STAT, JAK2), regulation of inflammation (CCR6, MST1) and regulation of endoplasmic reticulum (ER) stress and autophagy [X box binding protein 1 (XBP1), ATG16L1, IRGM] [9–11]. All these pathways seem to affect the regulation of the immune system and its response to commensal bacteria and particularly the function of Paneth cells and the presentation of peptides by innate immune cells to adaptive immune cells such as T lymphocytes [12]. Interestingly, autophagy genes (e.g., ATG16L1), NOD-like receptors (e.g., NOD2) and intelectins (e.g., ITLN1) have been related to CD whereas loci related to regulatory pathways (e.g., IL-10, ARPC2) and intestinal epithelial cell function (e.g., ECM1) are more specific for UC.
Autophagy is the most characteristic example of a genetically mediated pathway which is abnormal in IBD. This highly evolutionary conserved procedure represents the digestion organelles and extracellular bacteria in lysosomes [13]. It is known that the degradation of the above materials requires the formation of an autolysosome from the fusion of ER-derived membranes or autophagosome with a lysosome. This fusion is known to be regulated from a group of autophagy proteins like ATG. A number of genes, linked to the pathogenesis of CD including ATG16L1 and IRGM, are related to autophagy [14]. Recent studies support that the loss of ATGL1 function in mice is associated with CD and abnormal Paneth cell structure and function [15]. These structural abnormalities in Paneth cells seem to be related with exogenous and environmental factors like the presence of non-viral infections. This example shows how a particular genetic risk factor is related to the presence of an environmental factor in the pathophysiology of IBD.
NOD2 (nucleotide-binding oligomerization domain containing 2), also known as the caspase recruitment domain family, member 15 (CARD15), is an intracellular pattern recognition receptor and recognizes molecules containing the specific structure known as muramyl dipeptide (MDP) found in certain bacteria like Mycobacterium Tuberculosis [16]. The C-terminal portion of the protein contains a leukine rich repeat domain known to be critical in protein – protein interactions. The middle part of the protein is characterized by a NOD involved in protein self-oligomerization whereas the N-terminal portion contains two caspase recruitment domains related to apoptosis and nuclear factor-kappa B (NF-κΒ) activation pathways [17]. This protein is primarily expressed in the peripheral blood leukocytes and plays a critical role in the immune response by recognizing the bacterial molecules which possess the MDP pathway and activating the NF-κB protein and finally producing TNFα (Fig. 2). The NOD2 gene, located on the long arm (q) of chromosome 16 in humans, is a member of the NOD1/ Apaf-1 family and has been related to inflammatory diseases such as IBD and especially CD and graft versus host disease [18,19]. It is believed that NOD2 is a negative regulator of TLR2 receptors, which recognize intestinal bacteria. Based on this observation, the loss of function of NOD2 due to mutations can lead to lack of mucosal tolerance to the intestinal bacteria [20]. It is also known that NOD2 is a regulator of autophagy, a very important procedure in the pathophysiology of IBD, since this protein can activate ATG-5,7 and 16L1 [21]. Recent studies support that NPD2-deficient mice exhibit decreased α-defensin expression with normal Paneth cell structure [22], suggesting that NOD2 may regulate Paneth cell function as well. Finally, it is clear that NOD2 is a very important sensor of bacteria, especially in intestinal epithelial cells and structural changes of this protein may influence the innate immune response to intestinal bacteria and contribute to the pathogenesis of IBD.
Figure 2.
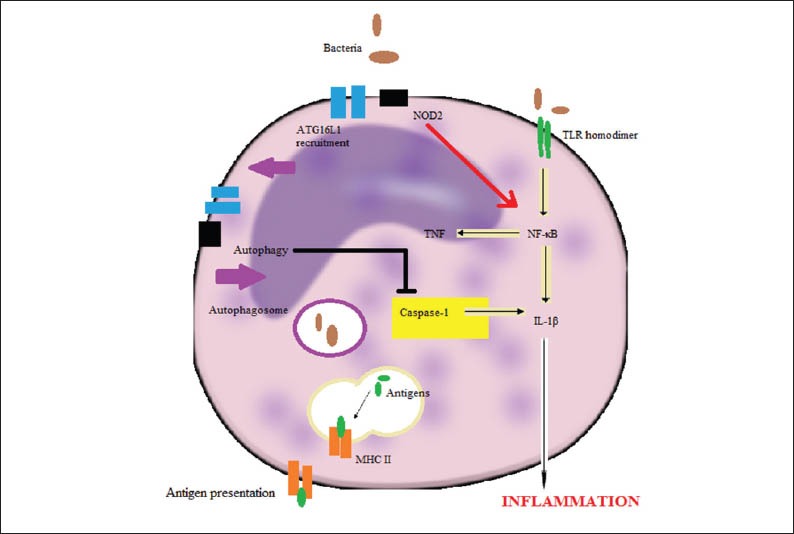
The interactions between NOD2 and bacterially induced autophagy mediators in a macrophage upon intracellular infection.
Recent studies support that the unfolded protein response (UPR), a response that occurs as a result of the accumulation of misfolded or unfolded protein in ER, plays a critical role in the development of IBD. The highly secretory intestinal cells respond to the ER stress through a variety of mechanisms for the maintenance of homeostasis [23]. These mechanisms of UPR include the induction of chaperons enhancing the secretion of proteins, chaperons involved in assisting protein folding and the induction of autophagy [23]. If the ER stress remains unabated the UPR initiates apoptosis (Fig. 3). It is believed that there are three distinct pathways for the regulation of UPR. It seems that the sensing of misfolded proteins in lumen of the ER leads to the activation of the transcription factors ATF4 and ATF5. The second pathway involves the cleavage of the cytoplasmic tail of the ATFp90 protein whereas the third pathway involves the recognition of misfolded proteins by inositol requiring enzyme 1 (IRE1) leading to alternative splicing of XBP1 mRNA forming a transcriptionally active protein which enhances the transcription of a variety of genes related to UPR [23].
Figure 3.
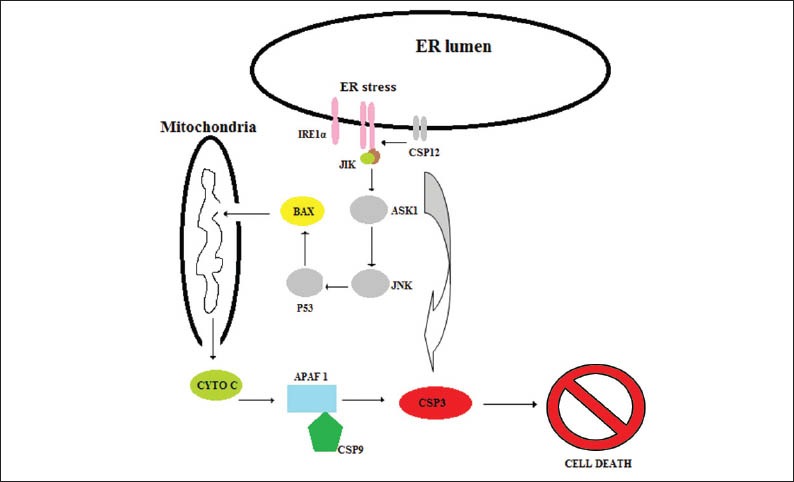
The intracellular consequences of endoplasmic reticulum (ER) stress.
XBP1 has been related to UC and CD in a variety of studies as a genetic risk for IBD [24]. It is known that XBP1-deficient epithelia lack Paneth cells due to apoptosis associated with spontaneous enteritis and susceptibility to colitis (41x) supporting that an accurate IRE1-XBP1 system is critical in the maintenance of homeostasis. Interestingly, it is believed that the activation of IRE1 in the context of deficient XBP1 leads to increased inflammatory tone of the epithelium in response to bacterial antigens and finally to Paneth cell death [24].
On the other hand, environmental factors, like antibiotics, appendectomy, nonsteroidal anti-inflammatory agents and smoking are known to have an important effect on the regulation of the mucosal immune system and on the composition and function of the commensal microbiota [25]. It can be hypothesized that the genetic susceptibility is modified by these factors mainly through epigenetic changes but genetic susceptibility can also modify these environmental factors [26].
In conclusion, IBD is believed to be a polygenic complex of diseases with a variety of phenotypes. It is known that a genetically susceptible individual in the proper environment is at risk for the development of the disease. Therefore, it is important to understand the connection between the metagenome of the host and microbes and the environmental factors in the pathogenesis of IBD.
Role of the innate immune system in the development of IBD
The important efficacy of biological factors like TNF-α, suggest the important role of the innate immune system in the pathophysiology of IBD. It is believed that IBD is characterized by impaired immune responses to the intestinal microbiota and by chronic inflammation of the intestine. Recent studies underline the important role of innate microbial recognition by immune and non immune cells in the gut. Interestingly either diminished or exacerbated innate immune signaling may trigger the disturbance of intestinal immune homeostasis leading to IBD and colitis-associated cancer. This phenomenon may reflect the variety of functional roles of the immune or non-immune intestinal cells.
It is known that, despite the beneficial roles of intestinal bacteria in the host’s development, metabolism and defense, the relationship between them and the host can break down leading to chronic inflammation. The use of antibiotics which disrupt the intestinal flora has been related to disease attenuation in some CD patients [27]. It is also interesting that CD clinical symptoms tend to develop in the terminal ileum and colon; the parts of the intestine with the highest bacterial load [28]. As mentioned above, genetic association studies have linked pattern recognition receptors (PRR) including NOD2, NLRP3 and Toll-like Receptors (TLR) [29] that mediate microbial sensing with the development of IBD. Recent studies demonstrate the critical role of these innate sensors in the intestinal homeostasis and in the pathogenesis of intestinal inflammation and carcinogenesis.
A characteristic histological hallmark of IBD is the disruption of intestinal epithelium. The processes of differentiation of intestinal stem cells, proliferation, migration and finally apoptosis is known to be disrupted in IBD patients resulting in hyperplasia, goblet cells depletion, enhanced apoptosis and finally ulceration, dysplasia and carcinogenesis. It is believed that intestinal epithelial cells (IECs) and intestinal leukocytes express PRRs and the downstream signaling has the potential to affect epithelial homeostasis at several levels.
Rakoff-Nahoum et al, in a landmark study published in 2004 proved that mice deficient in TLR, IL-1 receptor and IL-18 receptor developed significantly enhanced dextran sodium (DSS) colitis compared with wild-type mice [30]. It can be hypothesized that innate signals can be protective through a variety of mechanisms including the induction of antiapoptotic and cytoprotective factors and the promotion of tight junction formation between IECs [31]. It is also suggested that the innate sensing of intestinal microbiota induce the recruitment of stromal and myeloid cells to the lamina propria facilitating the regeneration of the injured epithelium [32]. Infected trl-/-, Il18r-/- and Il1r-/- mice exhibit delayed recruitment of neutrophils and enhanced bacterial overgrowth in the intestinal mucosa with profound ulceration, bleeding and mortality [33]. Moreover, the innate sensing plays an important role in the limitation of bacterial translocation by inducing the production of antimicrobial peptides like defensins and RegIIIγ by IECs [34]. Recent studies suggest that the induction of the production of these antimicrobial agents is mediated by the activation of NF-κB and PI3-Akt pathways in IECs. However, it is suggested that a variety of PRR have a pathological and not a protective role in the pathogenesis of inflammation. For example Tlr4-/- mice develop protective immunity to Citrobacter rodentium [35].
NOD2 (CARD15), as previously analyzed, is a macrophage specific protein, also expressed in IECs, Paneth cells and T cells, containing two CARD domains, a large nucleotide binding domain and leukine-rich repeats. Human genetic studies have linked polymorphisms in NOD2/CARD15 with CD, whereas no correlation to UC has been reported. It is also known that 93% of these polymorphisms are located in the leukine-rich repeat region [36], which is involved in the binding of N-acetyl MDP, part of bacterial peptidoglycan and N-glycol MDP from mycobacteria. NOD2 intracellular signaling is mediated by receptor-interacting serine-threonine kinase 2 (RIP2), TGF-β activated kinase, IκΒ and NF-κΒ [37] resulting in cytokine and chemokine release. Macrophages from NOD2-/- mice presented decreased IL-6 and IL-12p40 secretion, whereas NOD2-/- APCs induce heightened interferon (INF)-γ responses in antigen-specific CD4+ cells [38]. On the other hand, NOD2-/- mice presented diminished humoral immune responses [38] and active inhibition of IL-10 transcription resulting in decreased TLR ligand - induced IL-10 production by monocytes [39]. Finally, it is believed that other altered functions of NOD2 in IECs and Paneth cells contribute to impaired interactions between intestinal bacteria and gut epithelium, observed in IBD [38,39].
Recent studies support a role of NOD-like receptor (NLR) mediated inflammasome activation in maintaining epithelial integrity [40]. Nlrp3-/- mice which lack the inflammasome signaling component Nlrp3 exhibit enhanced DSS-induced colitis compared with wild-type mice [41]. These findings are consistent with a protective role of NRL signaling concerning the inflammatory mucosal damage. Microbial stimuli and host danger signals like uric acid, extracellular ATP and reactive oxygen species in the microbe-rich gut all activate the inflammasome [42] and induce mucosal damage. However there are findings that do not support the protective role of inflammasome activation in IECs or immune cells [43].
A critical effector function of inflammasome – mediated activation of caspase-1 is the production of active IL-1β and IL-18. It is known that Il1r-/- and Il18r-/- mice exhibit enhanced DSS-induced colitis compared with wild-type controls [44] whereas recombinant IL-18 can attenuate the inflammation supporting the critical role of inflammasome-mediated mediator in mucosal repair. However, earlier studies support a pathogenic role of these cytokines in intestinal inflammation [45].
These protective for gut TLR and inflammasome signals in acute intestinal inflammation contrast starkly with the pathogenic roles of these signals in the Il10-/- chronic colitis models. These findings suggest that the failure to regulate TLR/ IL-1R activation of myeloid cells in the presence of a triggering microbiota can precipitate chronic intestinal inflammation. The difference in chronic inflammation is that the epithelial damage is induced by microbiota-driven inflammatory response mediated by tissue leukocytes. It is clear that PRR and cytokine signaling play a protective role in the acute phase of inflammation promoting restoration of epithelial barrier and a return to homeostasis whereas these mechanisms seem to induce chronic inflammation and tissue damage.
Interestingly, it is reported that there is increased IL-1β, TNF-α and IL-6 expression in macrophages and IECs cells related to increased NF-κΒ activation in UC, CD and diverticulitis but not in uninflamed mucosa [46]. Consistent with this, A20 (tnfaip3); a potent inhibitor of NF-κB signaling, has been associated with CD as well as other immune-related diseases [47]. Another component of TLR and NOD2 intracellular signaling, IκB kinases (IKKβ) seem to protect from spontaneous colitis when expressed in IECs cells probably increasing thymic stromal lymphopoietin expression induced by intestinal microbiota. It is also reported that deletion of both IKKα and ΙΚΚβ results in severe colitis secondary to apoptosis of colonic IECs supporting the critical role of these cells as part of the physical barrier and source of antimicrobial peptides. On the other hand, deletion of IKKβ in macrophages and neutrophils improves colitis in IL-10-/- mice supporting an inflammatory role of IKKβ when expressed in myeloid cells [46].
Recent studies support the idea that the ER stress in intestinal cells such as goblet cells, IECs cells and Paneth cells caused by unfolded protein response and accumulation of misfolded proteins is implicated in the pathophysiology of IBD [48] (Fig. 2). Genetic and environmental pathways (mainly intestinal microbiota) that increase ER stress appear to be an important pathway for development of UC and CD [49]. Inositol-requiring enzyme (IRE1) is believed to activate XBP1 resulting in decreased ER stress, intestinal cells survival and appropriate function. XBP1 also regulates Paneth cell function whereas deletion of XBP1 gene results in apoptotic depletion of Paneth cells, reduction in goblet cells and increased inflammatory responsiveness of IECs cells to TLR and cytokine signals [50]. Consistent with these findings, a candidate gene study revealed significant associations of the complex XBP1 locus with both CD and UC [50]. It is also known that misfolding form of human HLA-B27 heavy chain induces ER stress in certain tissues (e.g., stomach, liver, skin, intestine, joints), correlated with the severity of inflammation [51]. These observations may explain the high prevalence of frequently asymptomatic ileitis in patients with seronegative spondyloarthropathies. Autophagy, as previously reported, is the major intracellular degradation system delivering cytoplasmic components to lysosomes, and it accounts for degradation of most long-lived proteins and some organelles. Cytoplasmic constituents, including organelles, are sequestered into double membraned autophagosomes, which subsequently fuse with lysosomes. ATG16L1 is a component of a large protein complex essential for autophagy which is a highly conserved procedure related to cancer, inflammation and aging [52]. ATG16L1 has been proposed as a genetic risk factor only for CD and not for UC. Stimulation of Atg16l1-/- mice with LPS leaded to high production of IL-1β and IL-18 suggesting that ATG16L1 is related to LPS-induced inflammation [53], whereas deletion of ATG16L1 in bone marrow resulted in high susceptibility to severe colitis [53]. It is believed that impaired ATG16L1 function is related to abnormal granule exocytosis, increased PPARγ expression and increased leptin and adipocytokines expression in Paneth cells implicating them in the development of intestinal inflammation. It is known that ER stress may activate autophagy through interactions between IRE1, TNF receptor-associated factor 2 (TRAF2) and Jun Kinase (JNK) activation [54]. One the other hand autophagic response can be triggered by NOD1 and NOD2 inducing the recruitment of ATG16L1 to plasma membrane, upon intracellular infection, bacterially-induced autophagy and consequent induction of antigen specific T-cells [55]. These observations support the convergence of several genetic risk factors for IBD (NOD2, XBP1 and ATG16L1) on the function of intestinal epithelium and especially in Paneth cells.
CD as a multifactorial disease is related both to environmental and nutritional factors. Latest studies suggest that impaired β-oxidation in IECs cells is also implicated in the development of CD [56]. OCTN2 is a Na+- dependent, high-affinity L-carnitine transporter encoded by Slc22a5 gene having an important role for transport of long chain fatty acids Genetics and innate immunity in IBD 169
into mitochondria for β-oxidation in IECs cells. The deletion of Scl22a5 gene in mice was associated with IEC apoptosis, abnormal villous structure and inflammatory infiltration in the mucosa [57]. Consistent with this, pharmacological inhibition of β-oxidation also results in spontaneous colitis. It is hypothesized that the decreased expression of Scl22a5 results in highly susceptible IEC cells in situations with increased energy needs, like alterations in microbiota and inflammation.
Natural killer T cells (NKT) responding to phospholipids and glycolipids, presented by CD1d on an APC, secrete a variety of Th1, Th2 and Th17 cytokines that trigger almost all the immune system functions [58]. NKT cells can be activated by presentation of self- or microbial-derived lipids by the nonclassical MHC class I molecule CD1d and via IL-12 and IL-18 secretion [58]. Recent studies revealed the increased presence of NKT cells expressing CD161 in inflamed lamina propria of UC but not CD patients [58]. CD1d is known to be expressed in DCs, macrophages, B cells and IECs. Recent studies suggest that the expression of CD1d in IEC is protective upon inflammation while CD1d expression in hematopoietic cells is believed to play a pathogenic role in IBD development. Consistent with this, IECs of IBD patients present decreased CD1d expression [59] whereas CD1d expression in lamina propria was increased probably due to increased infiltration of inflammatory cells [60].
The efficacy of anti-TNF antibodies as a treatment for IBD indicates that TNF is a major factor in the pathogenesis of these diseases. TNF is located only 1 MB apart from the MHC locus, associated mainly with UC [61]. TNF is known to be a transmembrane or soluble protein, which stimulates cellular activation, proliferation, cytotoxicity and apoptosis as well, through two receptors, TNFR1 and TNFR2 [62]. Recent studies suggest that TNF action can lead only to superficial injury in the absence of B and T cells, required for the development of transmural inflammation with granulomas [63]. This superficial pathway of tissue inflammation, common in CD and UC, involves TNF produced by parechymal and innate immune cells and action upon TNFR1. It is believed that the action of TNF on TNFR1 of mesechymal cells plays an important role in the regulation of balance between matrix metalloproteinases (MMP) and inhibitor of MMP (TIMP) leading to inflammation and intestinal tissue destruction [64]. Another important role of TNF in superficial inflammation is the stimulation of IEC apoptosis, which seems to be decreased with the use of anti-TNF antibody.
For the development of transmural inflammation regulated by IL-12 and INF-γ CD8+ and CD4+ T cells are required [65]. TNF secreted by macrophages, Th1 cells and colonic epithelial cells has also an important role in this type of inflammation, whereas other cells like adipocytes and Paneth cells contribute to inflammation within their microenvironments [66]. These suggestions highlight the importance of TNF secreted from a variety of cells upon the regulation of T cells for the development of transmural inflammation. It is also believed that the retrograde signaling via transmembrane TNF may distinguish effective (anti-TNF) and ineffective (TNFR2-Fc) TNF-targeted therapies in CD [67]. TNF ligand-related molecule 1 (TL1A), a TNF-related family member signaling through NF-κB is produced by monocytes and DCs enhancing INF-γ production by T cells and NK cells [68]. Interestingly, polymorphisms in TNFSF15 encoding TL1A were found to increase the genetic risk for CD in Japanese and Korean cohorts [69] making TL1A an interesting therapeutic target in Asians with IBD.
IL-6 secretion by non-T cells and subsequent action on T cells and macrophages is increased in IBD. IL-6 signals through IL-6R on cell surface and soluble IL6R, via binding of the sIL-6/ IL-6 complex to the receptor β subunit gp130 [70]. The last transmembrane receptor signals either through JAK1, JAK2 and STAT3 activation, or STAT1 and NF-κB activation involving src-homology tyrosine phosphate (SHP2) stimulation and activation of the Ras-ERK pathway. Recent studies suggest that IL-6 increases the survival of T cells and macrophages and deviates T cells from a Treg fate toward an inflammatory-like Th17 phenotype [71]. Consistent with these observations a placebo-controlled trial reported benefit of anti-IL6R in CD whereas STAT3 and JAK2 promoter polymorphisms have been associated with CD and UC [72]. On the other hand, IECs express the IL-6R on the basal surface and produce intracellular signaling via NF-κΒ activation, which protects intestinal epithelium from spontaneous ulcerations [73]. It is also believed that in IBD there is a loss of protective function of IL-6 in IECs due to impaired STAT1/ STAT3 signaling whereas there is an increased promotion of inflammatory pathways in immune cells leading to inflammation and tissue destruction.
It is known that the anti-inflammatory activity of IL-10 is also mediated by STAT3 signaling in macrophages and neutrophils. The deletion of exon 22 in STAT3 in myeloid cells was related to spontaneous colitis [74] whereas deletion of 18-20 exons was related to formation of granuloma-like structures and crypt abscesses [75]. These observations suggest that the inability of macrophages to respond to IL-10 increases responses to microbial signals (e.g., via LPS) and consequently leads to severe intestinal inflammation.
Another important cytokine of the innate immune system is IL-1β produced mainly by macrophages. NALP3 directs the conversion of procaspase-1 to caspase-1 and generates secretory IL-1β [76]. Decreased IL-1β secretion from myeoloid cells upon MDP stimulation has been associated with NALP3 and NOD2 polymorphisms and IBD [77]. These studies suggest that the impaired innate IL-1β activity could be a risk factor for development of CD and UC. On the other hand, IL-1β expression is increased in IBD intestinal tissues and the administration of anti-IL-1β ameliorates the severity of colitis [78]. It can be hypothesized that impaired innate IL-1 response increases the risk for IBD through an adaptive immune response as IL-1 is known to be implicated in the expression of Th17 pathways [79].
Conclusions
The latest studies confirm the complexity of IBD and the importance of interaction between a variety of factors in their pathophysiology.
It is known that the development of intestinal inflammation requires impaired interaction between intestinal microbiota, the host’s immune system and environmental factors.
Microbial metabolites, inflammatory cytokines and exogenous factors are implicated in a variety of genetically impaired pathways.
Genetic studies, supporting the concept of familial and sporadic forms of IBD, involve a wide range of biological pathways like innate immunity, ER stress, adaptive immunity, regulation of inflammation and autophagy.
Latest studies also highlight the importance of impaired innate immune functions of hematopoietic and non-hematopoietic cells related to the commensal microbiota and adaptive immune system.
Functional analyses of PRR pathways in intestinal homeostasis have revealed a complex picture with evidence for protective and pathogenic roles.
PRR signaling should protect the homeostasis whereas tissue repair and host defense must be preserved.
It is supported that immune pathology can result from either impaired or exuberant PRR receptors.
It is believed that PRR signaling is mainly protective in acute inflammation and tissue damage whereas it can be pathological in chronic inflammation.
Finally, the efficacy of biological therapies like anti-TNF antibodies supports the above theories and proposes IL6, IL-12, IL-23 and other mediators as future potential therapeutic targets. Careful examination of PRR signaling networks in different cellular compartments and in acute and chronic inflammation models is very important in designing the best therapeutic strategies to restore intestinal homeostasis in IBD.
Biography
University of Athens, Greece
Footnotes
Conflict of Interest: None
References
- 1.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 2.Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenstein RJ. Is Crohn's disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis and Johne's disease. Lancet Infect Dis. 2003;3:507–514. doi: 10.1016/s1473-3099(03)00724-2. [DOI] [PubMed] [Google Scholar]
- 5.Halme L, Paavola-Sakki P, Turunen U, Lappalainen M, Farkkila M, Kontula K. Family and twin studies in inflammatory bowel disease. World J Gastroenterol. 2006;12:3668–3672. doi: 10.3748/wjg.v12.i23.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 8.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn's disease. Annu Rev Genomics Hum Genet. 2009;10:89–116. doi: 10.1146/annurev-genom-082908-150013. [DOI] [PubMed] [Google Scholar]
- 10.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 11.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 12.Petnicki-Ocwieja T, Hrncir T, Liu YJ, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 15.Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kufer T, Banks D, Philpott D. Innate immune sensing of microbes by Nod proteins. Ann NY Acad Sci. 2006;1072:19–27. doi: 10.1196/annals.1326.020. [DOI] [PubMed] [Google Scholar]
- 17.Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF kappaB. Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 18.Radford-Smith G, Pandeya N. Associations between NOD2/CARD15 genotype and phenotype in Crohn's disease--Are we there yet? World J Gastroenterol. 2006;12:7097–7103. doi: 10.3748/wjg.v12.i44.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim T, Payne U, Zhang X, et al. Altered host:pathogen interactions conferred by the Blau syndrome mutation of NOD2. Rheumatol Int. 2007;27:257–262. doi: 10.1007/s00296-006-0250-0. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Asano N, Murray PJ, et al. Muramyl dipeptide activation of nucleotidebinding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 23.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 24.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakatos PL. Enviromental factors affecting inflammatory bowel disease: have we made progress? Dig Dis. 2009;27:215–225. doi: 10.1159/000228553. [DOI] [PubMed] [Google Scholar]
- 26.Beaudet AL. Epigenetics and complex human disease: is there a role in IBD? J Pediatr Gastroenterol Nutr. 2008;46(suppl 1):E2. doi: 10.1097/01.mpg.0000313815.73649.37. [DOI] [PubMed] [Google Scholar]
- 27.Perencevich M, Burakoff R. Use of antibiotics in the treatment of inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:651–664. doi: 10.1097/01.MIB.0000225330.38119.c7. [DOI] [PubMed] [Google Scholar]
- 28.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 29.Villani AC, Lemire M, Fortin G, et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat Genet. 2009;41:71–76. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;23(118):229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 31.Kim JY, Kajino-Sakamoto R, Omori E, Jobin C, Ninomiya-Tsuji J. Intestinal epithelial-derived TAK1 signaling is essential for cytoprotection against chemical-induced colitis. PLoS One. 2009;4:e4561. doi: 10.1371/journal.pone.0004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown SL, Riehl TE, Walker MR, et al. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest. 2007;117:258–269. doi: 10.1172/JCI29159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibson DL, Ma C, Rosenberger CM, et al. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol. 2008;10:388–403. doi: 10.1111/j.1462-5822.2007.01052.x. [DOI] [PubMed] [Google Scholar]
- 34.Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 35.Khan MA, Ma C, Knodler LA, et al. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun. 2006;74:2522–2536. doi: 10.1128/IAI.74.5.2522-2536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 38.Cario E. Innate immune signaling at intestinal mucosal surfaces: a fine line between host protection and destruction. Curr Opin Gastroenterol. 2008;24:725–732. doi: 10.1097/MOG.0b013e32830c4341. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 40.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci USA. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 42.Franchi L, Warner N, Viani K, Nuñez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen IC, TeKippe EM, Woodford RM, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salcedo R, Worschech A, Cardone M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siegmund B. Interleukin-1beta converting enzyme (caspase-1) in intestinal inflammation. Biochem Pharmacol. 2002;64:1–8. doi: 10.1016/s0006-2952(02)01064-x. [DOI] [PubMed] [Google Scholar]
- 46.Eckmann L, Nebelsiek T, Fingerle AA, et al. Opposing functions of IKKβ during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wellcome Trust Case Control Consort. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 49.Kaser A, Blumberg RS. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010;3:11–16. doi: 10.1038/mi.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turner MJ, Sowders DP, DeLay ML, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 52.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 54.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 56.Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shekhawat PS, Srinivas SR, Matern D, et al. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2-/-) mice. Mol Genet Metab. 2007;92:315–324. doi: 10.1016/j.ymgme.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol. 2007;5:405–417. doi: 10.1038/nrmicro1657. [DOI] [PubMed] [Google Scholar]
- 59.Perera L, Shao L, Patel A, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis. 2007;13:298–307. doi: 10.1002/ibd.20026. [DOI] [PubMed] [Google Scholar]
- 60.Page MJ, Poritz LS, Tilberg AF, Zhang WJ, Chorney MJ, Koltun WA. Cd1d-restricted cellular lysis by peripheral blood lymphocytes: relevance to the inflammatory bowel diseases. J Surg Res. 2000;92:214–221. doi: 10.1006/jsre.2000.5940. [DOI] [PubMed] [Google Scholar]
- 61.Fernando MM, Stevens CR, Walsh EC, et al. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 63.Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205:331–337. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pender SL, MacDonald TT. Matrix metalloproteinases and the gut—new roles for old enzymes. Curr Opin Pharmacol. 2004;4:546–550. doi: 10.1016/j.coph.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 65.Kontoyiannis D, Boulougouris G, Manoloukos M, et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J Exp Med. 2002;196:1563–1574. doi: 10.1084/jem.20020281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Desreumaux P, Ernst O, Geboes K, et al. Inflammatory alterations in mesenteric adipose tissue in Crohn's disease. Gastroenterology. 1999;117:73–81. doi: 10.1016/s0016-5085(99)70552-4. [DOI] [PubMed] [Google Scholar]
- 67.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 68.Papadakis KA, Prehn JL, Landers C, et al. TL1A synergizes with IL-12 and IL-18 to enhance IFN-γ production in human T cells and NK cells. J Immunol. 2004;172:7002–7007. doi: 10.4049/jimmunol.172.11.7002. [DOI] [PubMed] [Google Scholar]
- 69.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14:3499–506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 70.Kishimoto T. Interleukin-6: from basic science to medicine—40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- 71.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 72.Anderson CA, Massey DC, Barrett JC, et al. Investigation of Crohn's disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology. 2009;136:523–529. doi: 10.1053/j.gastro.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ernst M, Inglese M, Waring P, et al. Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J Exp Med. 2001;194:189–203. doi: 10.1084/jem.194.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 75.Welte T, Zhang SS, Wang T, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 77.Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL1β expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2005;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- 78.Cominelli F, Nast CC, Llerena R, Dinarello CA, Zipser RD. Interleukin 1 suppresses inflammation in rabbit colitis. Mediation by endogenous prostaglandins. J Clin Invest. 1990;85:582–586. doi: 10.1172/JCI114476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]