

Abstract
Aim
To develop a reliable murine model for fulminant liver failure (FLF).
Material and Methods
We treated three groups of male C57BL/6 mice:as controls, with azoxymethane (AOM), and with galactosamine (Gal) and tumor necrosis factor-alpha (TNFα). Effects of body temperature (BT) control on survival, in all three groups were investigated. Using BT control, survival, histopathological findings and biochemical/coagulation profiles were compared between the experimental groups. Effects of hydration on international normalized ratios of prothrombin time (PT-INR) were also checked. Dose-dependent survival curves were made for both experimental groups. Neurological behaviors were assessed using a coma scale.
Results
No unexpected BT effects were seen in the control group. The AOM group, but not the Gal+TNFα group, showed significant differences in survival curves between those with and without BT care. Histopathological assessment showed consistent FLF findings in both experimental groups with BT care. Between the experimental groups, there were significant differences in aspartate aminotransferase levels and PT-INR; and significant differences in PT-INRs between sufficiently- and insufficiently-hydrated groups. There were significant differences between FLF models, in the duration of each coma stage, with significant differences in stages 1 and 3 as percentages of the diseased state (stages 1-4). The two FLF models with BT care showed different survival curves in the dose-dependent survival study.
Conclusion
Azoxymethane can provide a good FLF model, but requires a specialized environment and careful BT control. Other FLF models may also be useful, depending on research purpose. Thoughtful attention to caregiving and close observation are indispensable for successful FLF models.
Keywords: Animal model, acute liver failure, azoxymethane, galactosamine, tumor necrosis factor-alpha
Introduction
Fulminant liver failure (FLF) has a mortality rate of 50-90% if liver transplantation is unavailable; it is responsible for approximately 5% of liver-related deaths [1-3]. Although liver transplantation improved FLF patients’ outcomes over the last two decades [4,5], this option is limited by donor shortages among deceased donors and donor safety among living donors, although auxiliary partial orthotopic liver transplantation is currently available for FLF [6,7]. Therefore, development of effective alternative therapies is needed to improve survival of FLF patients. When liver grafts are not immediately available, therapies to assist liver function until an allograft becomes available might also have clinical applications in maintaining FLF patients within suitable status as liver transplantation candidates. Development of such therapeutic strategies is indispensable for the improvement in FLF outcomes. Such research requires appropriate models, but this field lacks a suitable animal model for FLF [8-12].
Ideally, an animal FLF model would offer reversibility, reproducibility, a therapeutic window, liver-related deaths, suitably sized subject animals, and minimal hazard to research personnel [8,11]. Although researchers have introduced many toxins (e.g., azoxymethane (AOM), galactosamine (Gal), tumor necrosis factor-alpha (TNFα), acetaminophen, carbon tetrachloride, paracetamol and thioacetamide) to several animals [8,9,11,13-22], all are far from ideal. Here, we introduce our FLF model, using the mouse, with AOM, and with Gal and TNFα, and discuss the advantages and disadvantages of each, with a review of earlier literature.
Material and Methods
Animals
Inbred C57BL/6 mice (male, 8–12 weeks of age, approximately 25 g) were used (C57BL/6NHsd; Harlan Laboratories, Indianapolis, IN, USA). All mice were maintained under specific pathogen-free conditions. Body weight (bw) was measured before experiment. Mice were kept well hydrated prior to drug injection. All experimental protocols were approved by the ethics committees of the Mayo Clinic (IACUC 33307 and 24907). Mice were kept in 12-h light/dark cycles in a temperature-controlled, air-conditioned room, and received food and water. Animal handling and care met requirements of our institutional animal welfare guidelines.
AOM
AOM (Azoxymethane, A5486; Sigma-Aldrich Co., St. Louis, MO) was given via intraperitoneal injection (i.p.), 0.25 mL/mouse, using fine needles (30 gauge, 0.5 inch, BD Precision Single-use Needles; Becton Dickson and Company, Franklin Lakes, NJ, USA). AOM is carcinogenic and hepatotoxic for humans; its fumes are toxic after volatilization in room air. Experiments with AOM require prepared environments. To prevent the spread of vaporized AOM into the room air, AOM should be kept in a bottle with a tight rubber cap. All handling should be performed in biohazard rooms in which negative atmospheric pressure prevents diffusion of vaporized AOM to outside areas (Fig. 1A). Detailed experimental procedures and closed observation after AOM injection should be performed under a fume hood with evacuation system (Fig. 1B). Unless otherwise described (as in the dose-dependant study), this study used doses of 100 µg/bw of AOM.
Figure 1.

Safe environments for experiments with azoxymethane (AOM). A. Biohazard room. AOM is very hazardous, for both mice and humans. All handling should therefore be performed in a biohazard room where controlled ambient pressure restricts diffusion of vaporized AOM from outside areas. B. Fume hood with evacuation system. Detailed experimental procedures and closed observation after AOM injections should be performed at a fume hood with an evacuation system. C. Heating pads for body temperature (BT) care after AOM injection. Heating pads were used to maintain BT after AOM injection. Surface temperature of the cage bottom was maintained at 37.1-37.3 °C. D. Bushed cage with food and water. Solid food was placed in cage. Food gel for rodents was also prepared with water in feeding dishes.
Gal followed by TNFα
A total of 1.0 mg bw of Gal (D-(+)-galactosamine hydrochloride, G1639; Sigma-Aldrich Co.) was initially given via i.p. injection, 0.50 mL/mouse, using 28-gauge needles. Mice then received second i.p. injections, 0.50 mL/mouse, of TNFα (Recombinant Mouse TNFα aa 80235, 410MT; R&D Systems, Inc., Minneapolis, MN, USA), 2 hours after an initial Gal injection. Unless otherwise described (as in the dose-dependant study), doses of 0.10 µg/g bw of TNFα were used in this study.
Biochemical assays and coagulation profiles in the blood samples
Serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT) and total bilirubin (T-Bil) were assessed by quantitative determination kits (AST, ALT and T-Bil Reagent; Biotron, Hemet, CA, USA), respectively. The microplate reader (Spectra Max M5e; Molecular Devices, Sunnyvale, CA, USA) was set at 540 nm for wavelength measurement. Plasma and serum were taken in separator tubes (BD Microtainer; Becton Dickson). Plasma was used to measure international normalized ratios of prothrombin time (PT-INR), using a blood analyzer (i-STAT System, Abbott, Princeton, NJ, USA).
Caregiving after injections
Changes in body temperature (BT) offer a means of assessing the health of subject mice, especially after AOM injections [14]; BT was periodically measured with rectal probes (Diqi-Sense, Type T Thermocouple Thermometer; Eutech Instruments Pte Ltd., Ayer Rajah Crescent, Singapore). Surface temperatures of cage bottoms were maintained at 37.1–37.3 °C using heating pads (high level, RightTemp, RTHS-SM; Kent Scientific Co., Torrington, CT, USA) (Fig. 1C) [14]; murine BT was kept at approximately 36 °C. After AOM injections, mouse BT was measured every 2 h until 12 h after injection; thereafter BTs were checked every 1 h. For Gal+TNFα-treated mice, BT was checked every 1 h after TNFα injections.
Accurate survival curves for an FLF model require close follow-up. After AOM injection, mice were checked visually every 2 h until 12 h after injections were given, and hourly thereafter. Mice treated with Gal+TNFα injections were checked hourly after TNFα injections.
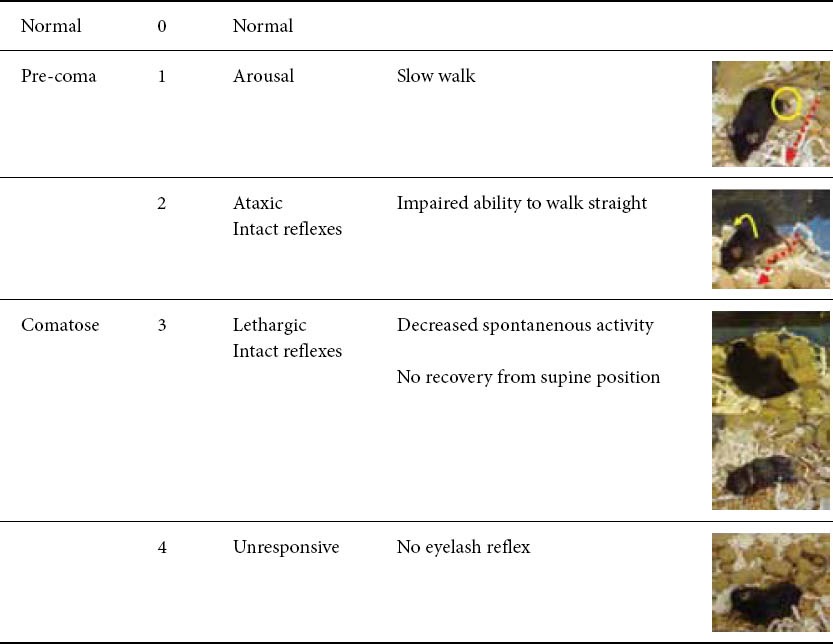
The coma scale for mice, which was slightly modified from the previous scale, is summarized in Table 1 [8,23,24]. To obtain a reliable coma stage, close and/or continuous observation of murine behavior was required. Detailed evaluation about coma status of the mice was performed after their AOM or Gal+TNFα injections.
Table 1.
Coma scale for mice
Bushed cages were used. Solid food was placed in the cage. Food gel for rodents was also prepared in feeding dishes with water (Fig. 1D).
Hydration supplements
Sufficient hydration is important for successful FLF models, because spontaneous drinking activity will be decreased. For animal models in our laboratory, we used supplements with 10% dextrose solution (10% Dextrose Injection USP, 505 mOsm/L, pH 4.0, Baxter, Deerfield, IL, USA), 5% dextrose solution (5% Dextrose Injection USP, 252 mOsm/L, pH 4.0, Baxter), Ringer’s solution (Ringer’s solution USP, 310 mOsm/L, pH 5.8, B. Braun Medical, Inc., Irvine, CA, USA), isotonic saline (0.9% Sodium Chloride Injection USP, 308 mOsm/L, pH 5.6, Hospira, Inc., Lake Forest, IL, USA), and lactated Ringer’s solution (Lactated Ringer’s solution USP, 275 mOsm/L, pH 6.2, B. Braun Medical, Inc.). Lactated Ringer’s solution was warmed to approximately 37 °C using warm water. In AOM-treated mice, 0.5 mL/mouse, i.p. was given every 1-2 h, starting 6 h after AOM injection. In Gal+TNFα-treated mice, 0.5 mL/mouse, i.p. was given every 1 h, starting 2 h after TNFα injections.
Histopathological assessments
Liver tissue was fixed in 10% neutral-buffered formalin (Protocol, 032-059, Fisher Scientific, Inc., Kalamazoo, MI, USA), embedded in paraffin, and sliced into 4 µm-thick sections. Morphological characteristics were evaluated after standard hematoxylin-eosin (H-E) staining.
Statistical analysis
Data are presented as mean ± standard deviation. Univariate and multivariate analyses were used for the between-group comparisons as follows: Student’s t test and X2 test for unpaired continuous or discontinuous variables; repeated-measure ANOVA for changes over time; Kaplan-Meier method for survival curves; and log-rank test for survival rates. Statistical calculations were performed using SPSS Software Version 16.0 (SPSS Inc., Chicago, IL, USA). The P values < 0.05 and ≥ 0.05 were considered statistically significant and not significant (NS), respectively.
Results
Effects of BT care in controls
First, the effect of BT care itself was investigated in mice receiving saline injections. There were two groups of mice: those who were given BT care after saline injections (0.50 mL/ mouse, i.p.; n = 10), and mice who received no such care after saline injections (n = 10). Hydration supplements were given every 6 h after saline injections. Survival and BT were checked every 2 h. Liver and blood samples were taken 30 h after initial injections. There were no significant differences between the two control groups in survival curves (Fig. 2A), changes in BT over time (Fig. 2B), or levels of AST, ALT, T-Bil and PTINR (Fig. 2C) (P ≥ 0.05 for all categories). Histopathological assessment showed normal findings in mice with BT care (Fig. 2D). Hence, BT care had no effects on control mice.
Figure 2.
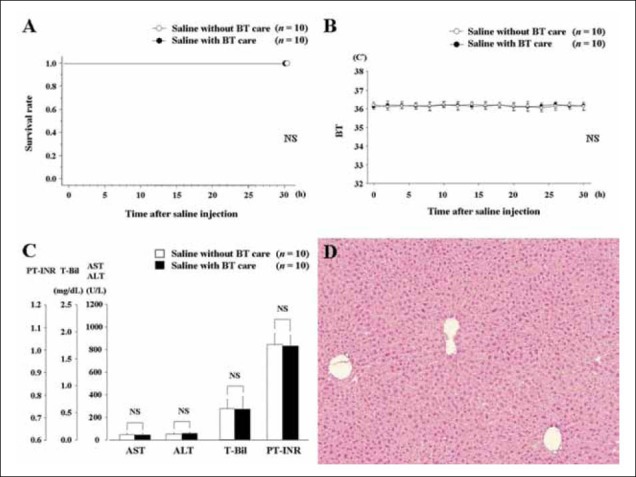
Effects of body temperature (BT) care in mice after saline injection. A. Survival curves in mice with and without BT care. B. Changes of BT in mice with and without BT care. C. Biochemical parameters and coagulation profile in mice with or without BT care. D. Histopathological findings in mice with BT care (hematoxylin-eosin, ×100). Normal findings were confirmed in mice with BT care.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; NS, not significant; PT-INR, international normalized ratio of prothrombin time; T-Bil, total bilirubin
Effects of BT care on survival in FLF models
As described above, BT care had no relevant effects on control mice. However, hypothermia in mice after AOM injection has been previously described[14,25]. We therefore investigated the effects of BT care and changes in BT in mice injected with AOM (n = 10), and in mice injected with Gal+TNFα (n = 10). Hydration supplements were given.
The AOM-treated mice who received BT care had significantly different survival curves and BT changes over time from AOM-treated mice who received no such care (P = 0.0002 and P = 0.0003, respectively; Fig. 3A and 3B, respectively). In contrast, among mice treated with Gal+TNFα, there were no significant differences between groups with or without BT care in survival curves (Fig. 3C) and BT changes over time after TNFα injections (Fig. 3D) (P ≥ 0.05, for both survival and BT changes).
Figure 3.

Survival curves and body temperature (BT) changes over time in the mice after azoxymethane (AOM) or galactosamine (Gal) + tumor necrosis factor (TNF)α injection, with or without BT care. A. Survival curves in mice with AOM injection with or without BT care. B. Changes over time of BT after AOM injection with or without BT care. C. Survival curves in mice with Gal+TNFα injection with or without BT care. D. Changes in BT over time after Gal+TNFα injection with or without BT care.
NS, not significant
Histopathological findings, biochemical parameters and coagulation profiles in FLF models with BT care
Histopathological findings, biochemical parameters and coagulation profiles were investigated in mice after AOM treatment (n = 10) or Gal+TNFα treatment (n = 10), with both groups receiving BT care. Mice in both treatment groups were given hydration supplements. Liver and blood samples were taken at death, or at 30 h after initial injections.
Histopathological findings of liver-related deaths in both FLF models are shown in Fig. 4 (H-E, ×100 and ×200). In both FLF models given BT care, histopathological assessment showed consistent FLF findings, such as massive necrosis, intrahepatic bleeding, vacuolization and inflammatory cells infiltrations. Levels of AST, ALT, T-Bil and PT-INR are shown in Fig. 5. While there were no significant differences in ALT (949.1 ± 77.7 vs. 814.1±200.3 U/L, P ≥ 0.05) and T-Bil (1.68±0.55 vs. 1.31±0.28 mg/dL, P ≥ 0.05) between the FLF models, there were significant differences in AST (896.1±108.8 vs. 727.2±219.7 U/L, P = 0.0429) and PT-INR (0.77±0.05 vs. 0.87±0.09, P = 0.0135).
Figure 4.
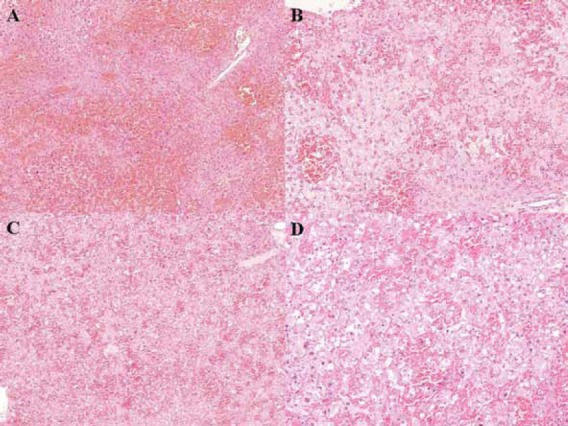
Histopathological findings for liver-related deaths in both fulminant liver failure (FLF) models with body temperature (BT) care. Consistent findings of FLF, such as massive necrosis, intrahepatic bleeding, vacuolization and inflammatory cells infiltrations, were confirmed. A. Histopathological findings in mice treated with azoxymethane (AOM) with BT care (hematoxylin-eosin, ×100). B. Histopathological findings in mice treated with AOM with BT care (hematoxylin-eosin, ×200). C. Histopathological findings in mice treated with galactosamine (Gal) + tumor necrosis factor (TNF)α with BT care (hematoxylin-eosin, ×100). D. Histopathological findings in mice treated with Gal+TNFα with BT care (hematoxylin-eosin, ×200).
Figure 5.
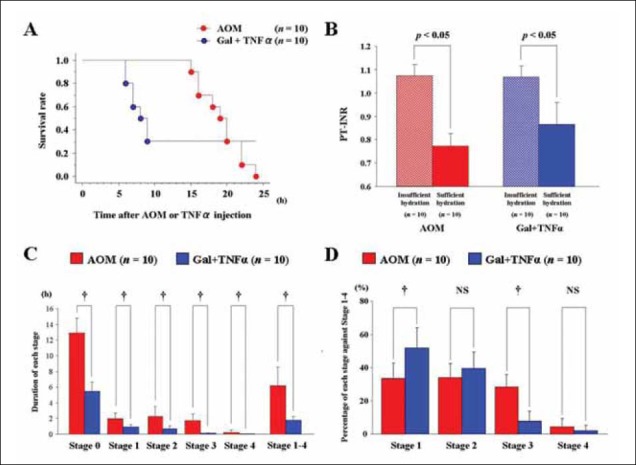
Survival curves, effects of hydration on coagulation profiles and differences in the intervals of coma stages between fulminant liver failure (FLF) models. A. Survival curves after azoxymethane (AOM) or galactosamine (Gal) + tumor necrosis factor (TNF)αinjection with body temperature (BT) care. Our laboratory used doses of 100 µg/g bw for AOM and 0.10 µg/g bw for TNFα, with routine BT care (n = 10 for both groups). B. Effects of hydration on coagulation profile. Mice PT-INR was investigated after AOM or Gal+TNFα injection with BT care. Mice were classified into two groups; mice with and without sufficient hydration (n = 10 for both groups); sufficient hydration was defined as hydration supplements (lactated Ringer’s solution, 0.5 mL/mouse, i.p) hourly from 6 h after AOM injections and from 2 h after TNFα injections. Insufficient hydration was defined as hydration supplements were given every 4 h after AOM or Gal+TNFα injections. Blood samples were taken at death or 30 h after initial injection. C. Durations of each coma stage in FLF models. The durations of each coma stage were measured in mice after AOM or Gal+TNFα injections with BT care (n = 10 for both groups). D. Length of each stage as a percentage of total stage-1-4 time. The period between stages 1-4 was considered to be the diseased state; lengths of each stage as a percentage of the diseased state were calculated in the mice after AOM or Gal+TNFα injections with BT care (n = 10 for both groups).
bw, body weight; NS, not significant
Effects of hydration on coagulation profiles and comastage intervals in FLF models with BT care
Survival curves of mice given BT care after AOM (n = 10) or Gal+TNFα (n = 10) injections are shown in Figure 5A. Mice treated with AOM clearly showed liver-related death in autopsy findings.
Sufficient hydration was also found to be important, as were close observation and BT care, because spontaneous activity decreased after AOM and Gal+TNFα treatments [8]. Coagulation profiles were investigated in mice with BT care after AOM or Gal+TNFα injections. Mice were classified into two groups: with or without sufficient hydration (n = 10 for both groups). Sufficient hydration was as described above, as hydration supplements (lactated Ringer’s solution, 0.5 mL/mouse, i.p) given every hour, starting 6 h after AOM injection or 2 h after TNFα injection. Insufficient hydration consisted of hydration supplements given every 4 h after AOM or Gal+TNFα injection. Blood samples were taken at death or 30 h after initial injection. There were significant differences in PT-INRs between sufficiently- and insufficiently-hydrated mice after AOM injections (1.07±0.05 vs. 0.77±0.05, P < 0.0001) and after Gal+TNFα injection (1.07±0.05 vs. 0.87±0.09, P <0.0001) (Fig. 5B). Hydration has clear effects on PT-INR in both FLF models.
To evaluate the therapeutic window in each coma stage, durations of each stage were studied. Using the murine coma scale (Table 1), the intervals of each coma stage were measured after treatment with AOM (n = 10) or Gal+TNFα (n = 10), with BT care. Times of each coma stage in both FLF models are shown (Fig. 5C). The AOM model and the Gal+TNFα model showed significant differences in their respective durations of stage 0 (12.90±1.91 vs. 5.45±1.14 h, P < 0.0001), stage 1 (1.95±0.76 vs. 0.90±0.32 h, P = 0.0008), stage 2 (2.25±1.25 vs. 0.70±0.35 h, P = 0.0014), stage 3 (1.75±0.82 vs. 0.12±0.08 h, P < 0.0001) and stage 4 (0.25±0.26 vs. 0.03±0.05 h, P < 0.0001). Mice treated with AOM had longer intervals in each stage than did mice treated with Gal+TNFα. The period during stages 1–4 was considered to be a diseased state; there were significant differences between the FLF models in the duration of the diseased state (6.20±2.35 vs. 1.75±0.50 h, P < 0.0001). The percentage of each stage against the entire diseased state was also calculated (Fig. 5D). Although stage 2 (33.9±8.5 vs. 39.2±9.9 %, P ≥ 0.05) and stage 4 (4.3±4.9 vs. 1.9±3.2 %, P ≥ 0.05) did not show significant differences, there were significant differences in stage 1 (33.3±9.3 vs. 51.4±12.2%, P = 0.016) and stage 3 (28.4±7.6 vs. 7.5±6.0%, P < 0.0001).
Dose-dependent survival in BT-treated mice given AOM or Gal+TNFα
While mice were usually dosed at 100 µg/g bw for AOM or 0.10 µg/g bw for TNFα, with routine BT care, dose-dependent survival studies in both FLF models with BT care were performed.
For AOM, doses of 50 or 100 µg/g bw have been used in many previous studies[8,14,23,25-29]. Survival curves for BT-treated mice receiving one of these two doses (n = 10 for each dose) are shown in Fig. 6. Survival ranged from within 19 h for mice dosed at 50 µg/g bw and within 7 h for mice dosed at 100 µg/g bw, though all mice died of liver-related causes.
Figure 6.
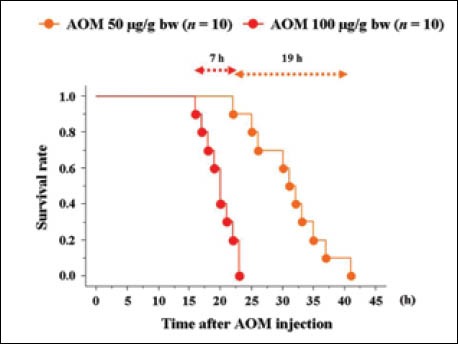
Dose-dependent survival curves after azoxymethane (AOM) injections, with body temperature (BT) care.
For TNFα, researchers have used doses ranging from 0.001 to 0.1 µg/g bw [15,30-34]. We used TNFα doses of 0.01, 0.02, 0.05, 0.10, 0.20, 0.50 and 1.00 µg/g bw, (n = 10 for each dose) because all mice receiving TNFα ≤ 0.02 µg/g bw in the preliminary study survived. Survival curves for mice after Gal+TNFα injections with BT care are shown in Fig. 7.
Figure 7.
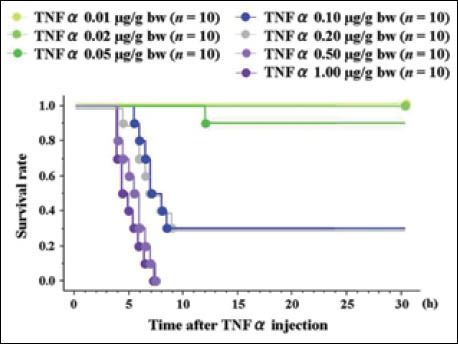
Dose-dependent survival curves after galactosamine (Gal) + tumor necrosis factor (TNF)αinjections, with body temperature (BT) care.
Coma scale based on murine behaviors
The coma scale for mice, which was slightly modified from the previous scale, is summarized in Table 1 [8,23,24]. To obtain a reliable coma stage, close and/or continuous observation of murine behavior was required. Detailed evaluation about coma status of the mice was performed by close observation every 30 min until Stage 2 and continuous monitoring after Stage 2. Important findings for staging were shown in Fig. 8. Note that continuous observation is crucial for reliable staging, especially after Stage 2, because periodic observation never provides an accurate assessment for disease progression and reliable staging for encephalopathy.
Figure 8.
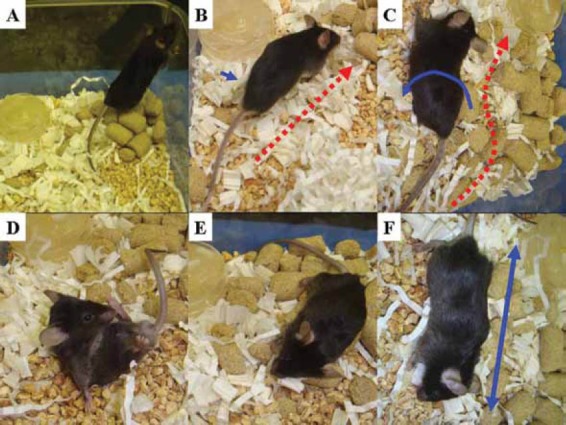
Neurological findings for coma staging in fulminant liver failure (FLF)models. 6To obtain a reliable coma stage, close and/or continuous observation of murine behavior was required. Detailed evaluation about coma status of the mice was performed by the close observation of every 30 min until Stage 2 and the continuous monitor after Stage 2. Note that the continuous observation is crucial for a reliable staging, especially after Stage 2, because the periodic observation never provide an accurate assessment for disease progression and a reliable staging for encephalopathy. A. A mouse at Stage 0 stands straight and runs fast. B. Mouse at Stage 1 walks slow but straight (red arrow). The movement of the feet is slightly impaired (blue arrow). C. At Stage 2, an ataxic finding is enhanced, but any reflexes are kept. The totter is observed (blue arrow), and mouse can not walk straight (red arrow). D. Mouse at Stage 3 is lethargic, and all spontaneous activities are lost. Subsequently, the mouse cannot recover from supine position. E. Eyelash reflex is disturbed at the late phase of Stage 3. F. Mouse becomes unresponsive and grows complacent at Stage 4 (blue arrow).
Discussion
Animal FLF models for dogs, pigs, rabbits, rats and mice have been previously documented [8]. Although different models may be required to evaluate the various types of liver failure seen in humans, mice are particularly suitable for laboratory assays due to the growing availability of gene-altered or knock-out strains and the development of specific agents and antibodies. Essential criteria for animal FLF models should include high reproducibility, liver failure-related death, long therapeutic window, suitably sized subject animals and minimal hazard to personnel [8,11]. Since pre-coma and comatose status have therapeutic values [23], many studies target stages 2 and 3. AOM has enough windows in each stage, even at the dose of 100 µg/g bw. Our results suggested that murine FLF model with AOM seemed to fulfill these criteria, except for a biohazard issue.
AOM is the active metabolite of cycasin, found in cycad palm nuts, only on the island of Guam, USA [35]. In 1963, Laqueur et al [36] discovered that cycad palm nuts induced various cancers of gastrointestinal and colorectal tracts. Interestingly, they described anecdotally that AOM also caused liver injury [36]. Thereafter, AOM has been widely used for the research in the Oncology and Hepatology fields. Although an AOM dose of 20 µg/bw g i.p. is not hepatotoxic in mice [8], doses of 50-200 µg/g bw i.p. clearly resulted in liver failure with identical histopathological findings [8]. Previously, many researchers used doses of 50 or 100 µg/g bw for liver failure in mice, because AOM has the advantages of reversibility and reproducibility [8,14]. Our institution has also used these doses for murine FLF models [23,27,28]; a dose of 100 µg/g bw is currently used [26].
Systemic inflammatory response syndrome plays a role in the pathophysiology of FLF [37], and is associated with a poorer prognosis [38]. Systemic inflammatory response syndrome is characterized by the presence of proinflammatory cytokines and interferons secreted from activated macrophages and Kuppfer cells, leading to hepatic necrosis [39-41]. Fulminant liver failure is also associated with accumulation of brain cytokines due to hepatic devascularization or toxic liver injury [42,43]. Inflammation with FLF is associated with more rapid progression of hepatic encephalopathy and intracranial hypertension [14,37,44,45] and proinflammatory cytokines contribute to the pathogenesis of the neurological complications of FLF [14]. Circulating cytokines may enter the brain in regions lacking a blood-brain barrier, or by activation of the endothelial vasculature, where receptors for these cytokines are localized [46]. Activation of these receptors may then trigger activation of the endothelium, leading to an inflammatory cascade involving activation of nitric oxide synthase, synthesis of reactive oxygen species, and microglial activation. Circulating cytokines may also alter blood-brain barrier permeability, promoting increased brain uptake of toxins such as ammonia [47,48].
Mild hypothermic changes during disease progression were documented in the FLF models with AOM or acetaminophen [14,25,43,49]. Mild hypothermia is effective in reducing liver injury caused by a hepatotoxin; this protective effect is mediated by mechanisms involving improved antioxidant status, together with modulation of inflammation [14,43]. Reduction in specific proinflammatory cytokines in plasma and brain results in improvement in FLF prognosis, including neurological complications [14]. Mild hypothermia resulted in reduced hepatic damage, improvement in neurological function, normalization of glutathione levels, and selective attenuation in expression of circulating proinflammatory cytokines [14,29]. In our own results, hypothermic mice showed prolonged survival, although this hypothermia was more drastic rather than mild hypothermia, around 35 °C [14,29]. A possible explanation is the differences in age, experimental conditions and intentional hydration. Our results suggest that the AOM FLF model is also useful for investigation of beneficial effects of hypothermic antioxidant and anti-inflammatory mechanisms in FLF prognosis.
Coagulopathy and encephalopathy are important definitive factors for FLF. Our results suggest that blood inspissation due to severe dehydration may mask coagulopathy, and may make coagulation profiles unreliable. Based on our experiences, dehydration is very severe in the diseased state, especially in the AOM FLF model, and blood sampling with enough volume is often technically difficult. With regard to reliable coagulation profiles and stable blood sampling, sufficient hydration is indispensable.
Hydration is maintained by dishes of gel and water during disease progression, because eating and drinking decrease as disease progresses. Spontaneous activity and intake of food/ water will start to decrease from 6 hours after AOM injection [8]. As reported in the literature [8] and confirmed by our own observations, at the dose of 100 µg/g bw, approximately 50% of spontaneous behaviors are lost by 6 h after AOM injections, and almost all spontaneous activity disappeared at 12 h after injections. Initially, we had tried to feed the mice orally, using a pipette, but failed to maintain oral feeding because of the risk of aspiration. Supplements via i.p. injection are a reasonable and reliable form of caregiving. Decrease in serum glucose was confirmed from 4 h after AOM injection in the FLF model [8]. However, administration of exogenous glucose and maintenance of euglycemia did not improve the coma status in the AOM FLF model. This is similar to what has been previously reported in other species, where hypoglycemia is commonly observed to complicate FLF in humans [50-52] and in other animal models of FLF [53-56]. In no instance does correction of hypoglycemia have any impact on improving neurological status in humans or other species suffering from FLF. Finally, edema complicates progression of FLF [3], with up to 25% of humans reported to suffer edema as a late complication [57]. Previously, we found that FLF mice at stage 3 died immediately after i.p. injection of supplement with 10% dextrose solution. A possible explanation is rapid change of osmolarity and subsequent hemodynamic disorder due to injection of high osmolarity solution under severe hydration. If so, disease course and death cause may be affected by unexpected issues.
Encephalopathy and brain edema are also important complications in the FLF model. Matkowskyj et al [8] had described murine behavior in detail. They classified progressive encephalopathy after the prodromal phase due to hepatic failure into four stages based on neurological behaviors: stage I (lethargic), stage II (ataxic), stage III (loss of righting reflex) and stage IV (coma) [8]. They also showed FLF was to be associated with progressive encephalopathy, and by a prodromal period of decreased eating and drinking lasting approximately 15 h before development of stage I encephalopathy (loss of scatter reflex); and that late encephalopathy, already irreversible, was associated with increased arterial ammonia, decreased serum glucose, and evidence of brain edema (astrocyte swelling) [8]. We also observed progressive encephalopathy in FLF model, and suggested that the continuous observation is crucial for a reliable staging, especially after Stage 2. Note that the periodic observation never provides an accurate assessment for disease progression and a reliable staging for encephalopathy.
We have shown that AOM-induced FLF is highly reproducible, without evidence of lot-to-lot variability, and is dose-dependent. These findings therefore suggest that AOM is an excellent agent for the study of FLF. Hepatic encephalopathy is a syndrome characterized by altered neurological status that may rapidly progress to stupor and coma [29]. Brain edema resulting in increased intracranial pressure is a second major complication of FLF [29]. Brain edema leads to intracranial hypertension and brain herniation. This is a common cause of mortality in FLF [29]. A cytotoxic mechanism and changes in the permeability of the blood-brain barrier cause brain edema in FLF [58,59]; many researchers have focused on the subtle blood-brain barrier disruption in FLF [23,26,27,59]. Osmolarity is an important factor as the disease progresses to organ failure and brain edema. Overall, to obtain relevant findings regarding subtle blood-brain barrier disruption we gave sufficient hydration every 1-2 h from 6 h after AOM injection with lactated Ringer’s solution, which has the lowest osmolarity pressure and highest pH, though our previous protocol was every 8-12 h after AOM injection (50 µg/g bw) with 10% dextrose solution [23,28].
An ideal animal FLF model should fulfill the requirements of reversibility, reproducibility, liver-related death, a therapeutic window, suitably sized subject animals, and minimal hazard to personnel [8,11]. AOM is the first toxin to satisfy all these criteria, and is also associated with the development of hepatic encephalopathy [8,11], mice treated with AOM provide the best model for FLF, except for possible biohazard to personnel. Although work with this model requires a specialized environment to prevent toxic effects to personnel, and also requires intensive caregiving with regard to BT and hydration to avoid confounding influences, our results show that many researchers can cope with these conditions. The murine FLF model with AOM is highly reproducible, causes death from liver failure, has a long therapeutic window, and generates coagulopathy, liver-associated encephalopathy and brain edema in end-stage liver disease.
One paradoxical problem may arise: as AOM itself may be directly toxic to extrahepatic cells, research that focuses on extrahepatic phenomena in FLF may see its extrahepatic findings mediated by AOM toxicity rather than FLF. Therefore, another model for FLF is also required.
Tumor necrotic factor-α is a proinflammatory cytokine, mainly produced by activated macrophages [15]. It is a critical early mediator of organ injury [60-62], and is an important mediator in triggering lethal hepatic injury [13,62]. It is also available to induce an FLF model [30,34]. Carbon tetrachloride is problematic because it gives inconsistent results between experiments and across species, and depending on species, it requires concomitant enzyme induction or partial hepatectomy, because this drug by itself does not induce deep coma [55,63,64]. Acetaminophen and paracetamol also produce inconsistent toxicity from animal to animal and between experiments [21,54,65-67]. Hepatotoxicity of Gal was first determined in the rat [68], and Gal is metabolized only in the liver [69]. The FLF model with Gal has some disadvantages; Gal is associated with lot-to-lot variability and has no known human correlate as a hepatotoxin [53,56,70]. Furthermore, whereas most toxins cause a centrilobular pattern of injury, Gal produces diffuse rather than zonal injury [53,56,70]. Use of thioacetamide to produce an FLF model requires multiple administrations and supportive therapy [8,71]. Thus, FLF models based on Gal, TNFα, acetaminophen, carbon tetrachloride, paracetamol and thioacetamide are far from ideal.
Injection of TNFα in animals causes severe liver cell toxicity, especially when Gal is co-administered [30]. Tumor necrotic factor-α is a dominant and terminal mediator of specific failure in Gal-sensitized mice [72,73]. Some researchers have used an FLF model based on Gal followed by TNFα [13,15,72,73], or a TNFα-dependent FLF model with galactosamine followed by lipopolysaccharide [13,74,75]. However, many researchers used various doses of these toxins and intervals between injections [13,15,72,73]. Although many cytokines are involved in inflammatory organ failure, TNFα is sufficient to induce hepatic apoptosis in Gal-sensitized mice [13]. We performed a preliminary dose-dependent study beforehand in our facility, because FLF models with Gal and/or TNFα have low reversibility, poor reproducibility and low rates of liver-related death compared to the AOM FLF [8,14]. Depending on the purpose of an investigation, FLF models using Gal followed by TNFα (0.10 µg/g bw) can be used as a second FLF model [26].
AOM-induced FLF is the best murine model for research in the fields of liver failure and hepatic encephalopathy, although specialized environments and caregiving are required. A second FLF model is also needed for investigations requiring negation of some of AOM’s effects (i.e., extrahepatic toxicity). In any case, thoughtful attention to caregiving and close observation are indispensable for reliable data with successful FLF models.
Summary Box.
What is already known:
Ideally, an animal model for fulminant liver failure (FLF) would offer reversibility, reproducibility, a therapeutic window, liver-related deaths, suitably sized subject animals, and minimal hazard to research personnel.
Although researchers have introduced many toxins to several animals, all are far from ideal.
What the new findings are:
Azoxymethane (AOM)-induced FLF is the best murine model for research in the fields of liver failure and hepatic encephalopathy, although specialized environments and caregiving are required.
A second FLF model is also needed for investigations requiring negation of some of AOM’s effects (i.e., extrahepatic toxicity).
In any case, thoughtful attention to caregiving and close observation are indispensable for reliable data with successful FLF models.
Acknowledgments:
We are grateful to Dennis W. Dickson, Monica Castanedes-Casey, Virginia R. Phillips, Linda G. Rousseau and Melissa E. Murray (Department of Neuroscience, Mayo Clinic in Florida, Jacksonville, FL 32224, USA) for their technical support in histopathological evaluations, and to Satoshi Yamamoto (Division of Digestive and General Surgery, Niigata University Hospital, Niigata, Japan), Naoki Shimojima (Department of Pediatric Surgery, Keio University Hospital, Tokyo, Japan) and Norifumi Ohashi (Department of Surgery II, Nagoya University Hospital, Aichi, Japan) for their supports.
Biography
Mayo Clinic in Florida, USA
Footnotes
Conflict of Interest: None
Financial disclosures: This work was partially supported by grants to J.H. Nguyen from the Deason Foundation (Sandra and Eugene Davenport, Mayo Clinic CD CRT-II), AHA (0655589B) and NIH (R01NS051646-01A2), and by a grant to T. Hori from the Uehara Memorial Foundation (200940051).
References
- 1.Hoofnagle JH, Carithers RL, Jr, Shapiro C, Ascher N. Fulminant hepatic failure: summary of a workshop. Hepatology. 1995;21:240–252. [PubMed] [Google Scholar]
- 2.Lee WM. Acute liver failure. N Engl J Med. 1993;329:1862–1872. doi: 10.1056/NEJM199312163292508. [DOI] [PubMed] [Google Scholar]
- 3.Munoz SJ. Difficult management problems in fulminant hepatic failure. Semin Liver Dis. 1993;13:395–413. doi: 10.1055/s-2007-1007368. [DOI] [PubMed] [Google Scholar]
- 4.Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology. 2010;52:2065–2076. doi: 10.1002/hep.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farmer DG, Anselmo DM, Ghobrial RM, et al. Liver transplantation for fulminant hepatic failure: experience with more than 200 patients over a 17-year period. Ann Surg. 2003;237:666–675. doi: 10.1097/01.SLA.0000064365.54197.9E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azoulay D, Samuel D, Ichai P, et al. Auxiliary partial orthotopic versus standard orthotopic whole liver transplantation for acute liver failure: a reappraisal from a single center by a case-control study. Ann Surg. 2001;234:723–731. doi: 10.1097/00000658-200112000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubernatis G, Pichlmayr R, Kemnitz J, Gratz K. Auxiliary partial orthotopic liver transplantation (APOLT) for fulminant hepatic failure: first successful case report. World J Surg. 1991;15:660–665. doi: 10.1007/BF01789221. [DOI] [PubMed] [Google Scholar]
- 8.Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am J Physiol. 1999;277:G455–462. doi: 10.1152/ajpgi.1999.277.2.G455. [DOI] [PubMed] [Google Scholar]
- 9.Shito M, Balis UJ, Tompkins RG, Yarmush ML, Toner M. A fulminant hepatic failure model in the rat: involvement of interleukin-1beta and tumor necrosis factor-alpha. Dig Dis Sci. 2001;46:1700–1708. doi: 10.1023/a:1010653504568. [DOI] [PubMed] [Google Scholar]
- 10.Maddison JE, Dodd PR, Johnston GA, Farrell GC. Brain gammaaminobutyric acid receptor binding is normal in rats with thioacetamide-induced hepatic encephalopathy despite elevated plasma gamma-aminobutyric acid-like activity. Gastroenterology. 1987;93:1062–1068. doi: 10.1016/0016-5085(87)90570-1. [DOI] [PubMed] [Google Scholar]
- 11.Terblanche J, Hickman R. Animal models of fulminant hepatic failure. Dig Dis Sci. 1991;36:770–774. doi: 10.1007/BF01311235. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann C, Ferenci P, Pifl C, et al. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats: characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
- 13.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG. Wendel Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 14.Bemeur C, Desjardins P, Butterworth RF. Antioxidant and antiinflammatory effects of mild hypothermia in the attenuation of liver injury due to azoxymethane toxicity in the mouse. Metab Brain Dis. 2010;25:23–29. doi: 10.1007/s11011-010-9186-x. [DOI] [PubMed] [Google Scholar]
- 15.Wielockx B, Lannoy K, Shapiro SD, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- 16.Rahman TM, Selden AC, Hodgson HJ. A novel model of acetaminophen-induced acute hepatic failure in rabbits. J Surg Res. 2002;106:264–272. doi: 10.1006/jsre.2002.6476. [DOI] [PubMed] [Google Scholar]
- 17.Newsome PN, Henderson NC, Nelson LJ, et al. Development of an invasively monitored porcine model of acetaminophen-induced acute liver failure. BMC Gastroenterol. 2010;10:34. doi: 10.1186/1471-230X-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mark AL, Sun Z, Warren DS, Lonze BE, et al. Stem cell mobilization is life saving in an animal model of acute liver failure. Ann Surg. 2010;252:591–596. doi: 10.1097/SLA.0b013e3181f4e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tunon MJ, Alvarez M, Culebras JM, Gonzalez-Gallego J. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol. 2009;15:3086–3098. doi: 10.3748/wjg.15.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Animal models of fulminant hepatic failure: a critical evaluation. Liver Transpl. 2000;6:21–31. doi: 10.1002/lt.500060110. [DOI] [PubMed] [Google Scholar]
- 21.Francavilla A, Makowka L, Polimeno L, et al. A dog model for acetaminophen-induced fulminant hepatic failure. Gastroenterology. 1989;96:470–478. doi: 10.1016/0016-5085(89)91573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda AS, Rodrigues DH, Vieira LB, et al. A thioacetamideinduced hepatic encephalopathy model in C57BL/6 mice: a behavioral and neurochemical study. Arq Neuropsiquiatr. 2010;68:597–602. doi: 10.1590/s0004-282x2010000400022. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen JH, Yamamoto S, Steers J, et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol. 2006;44:1105–1114. doi: 10.1016/j.jhep.2005.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto S, Nguyen JH. TIMP-1/MMP-9 imbalance in brain edema in rats with fulminant hepatic failure. J Surg Res. 2006;134:307–314. doi: 10.1016/j.jss.2005.11.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bemeur C, Vaquero J, Desjardins P, Butterworth RF. N-acetylcysteine attenuates cerebral complications of non-acetaminophen-induced acute liver failure in mice: antioxidant and anti-inflammatory mechanisms. Metab Brain Dis. 2010;25:241–249. doi: 10.1007/s11011-010-9201-2. [DOI] [PubMed] [Google Scholar]
- 26.Chen F, Hori T, Ohashi N, Baine AM, Eckman CB, Nguyen JH. Occludin is regulated by epidermal growth factor receptor activation in brain endothelial cells and brains of mice with acute liver failure. Hepatology. 2011;53:1294–1305. doi: 10.1002/hep.24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen F, Ohashi N, Li W, Eckman C, Nguyen JH. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology. 2009;50:1914–1923. doi: 10.1002/hep.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimojima N, Eckman CB, McKinney M, et al. Altered expression of zonula occludens-2 precedes increased blood-brain barrier permeability in a murine model of fulminant hepatic failure. J Invest Surg. 2008;21:101–108. doi: 10.1080/08941930802043565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belanger M, Cote J, Butterworth RF. Neurobiological characterization of an azoxymethane mouse model of acute liver failure. Neurochem Int. 2006;48:434–440. doi: 10.1016/j.neuint.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 30.Libert C, Wielockx B, Grijalba B, et al. The role of complement activation in tumour necrosis factor-induced lethal hepatitis. Cytokine. 1999;11:617–625. doi: 10.1006/cyto.1998.0462. [DOI] [PubMed] [Google Scholar]
- 31.Song HL, Lv S, Liu P. The roles of tumor necrosis factor-alpha in colon tight junction protein expression and intestinal mucosa structure in a mouse model of acute liver failure. BMC Gastroenterol. 2009;9:70. doi: 10.1186/1471-230X-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wullaert A, Wielockx B, Van Huffel S, et al. Adenoviral gene transfer of ABIN-1 protects mice from TNF/galactosamine-induced acute liver failure and lethality. Hepatology. 2005;42:381–389. doi: 10.1002/hep.20785. [DOI] [PubMed] [Google Scholar]
- 33.Takenaka K, Sakaida I, Yasunaga M, Okita K. Ultrastructural study of development of hepatic necrosis induced by TNF-alpha and D-galactosamine. Dig Dis Sci. 1998;43:887–892. doi: 10.1023/a:1018898905478. [DOI] [PubMed] [Google Scholar]
- 34.Van Lint P, Wielockx B, Puimege L, Noel A, Lopez-Otin C, Libert C. Resistance of collagenase-2 (matrix metalloproteinase8)-deficient mice to TNF-induced lethal hepatitis. J Immunol. 2005;175:7642–7649. doi: 10.4049/jimmunol.175.11.7642. [DOI] [PubMed] [Google Scholar]
- 35.Wright N, Alison M. The Biology of Epithelial Cell Populations. Oxford: Clarendon; 1984. Cell proliferation in gastrointestinal carcinogenesis; pp. 805–841. [Google Scholar]
- 36.Laqueur GL, Mickelsen O, Whiting MG, Kurland LT. Carcinogenic Properties of Nuts from Cycas Circinalis L. Indigenous to Guam. J Natl Cancer Inst. 1963;31:919–951. [PubMed] [Google Scholar]
- 37.Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt LE, Larsen FS. Prognostic implications of hyperlactatemia, multiple organ failure, and systemic inflammatory response syndrome in patients with acetaminophen-induced acute liver failure. Crit Care Med. 2006;34:337–343. doi: 10.1097/01.ccm.0000194724.70031.b6. [DOI] [PubMed] [Google Scholar]
- 39.Andus T, Bauer J, Gerok W. Effects of cytokines on the liver. Hepatology. 1991;13:364–375. [PubMed] [Google Scholar]
- 40.Nagaki M, Iwai H, Naiki T, Ohnishi H, Muto Y, Moriwaki H. High levels of serum interleukin-10 and tumor necrosis factor-alpha are associated with fatality in fulminant hepatitis. J Infect Dis. 2000;182:1103–1108. doi: 10.1086/315826. [DOI] [PubMed] [Google Scholar]
- 41.Sekiyama KD, Yoshiba M, Thomson AW. Circulating proinflammatory cytokines (IL-1 beta, TNF-alpha, and IL-6) and IL-1 receptor antagonist (IL-1Ra) in fulminant hepatic failure and acute hepatitis. Clin Exp Immunol. 1994;98:71–77. doi: 10.1111/j.1365-2249.1994.tb06609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang W, Desjardins P, Butterworth RF. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab. 2009;29:944–952. doi: 10.1038/jcbfm.2009.18. [DOI] [PubMed] [Google Scholar]
- 43.Jalan R, Olde Damink SW, Deutz NE, Hayes PC, Lee A. Moderate hypothermia in patients with acute liver failure and uncontrolled intracranial hypertension. Gastroenterology. 2004;127:1338–1346. doi: 10.1053/j.gastro.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 44.Jalan R, Williams R. The inflammatory basis of intracranial hypertension in acute liver failure. J Hepatol. 2001;34:940–942. doi: 10.1016/s0168-8278(01)00038-1. [DOI] [PubMed] [Google Scholar]
- 45.Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755–764. doi: 10.1016/s0016-5085(03)01051-5. [DOI] [PubMed] [Google Scholar]
- 46.Ericsson A, Liu C, Hart RP, Sawchenko PE. Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J Comp Neurol. 1995;361:681–698. doi: 10.1002/cne.903610410. [DOI] [PubMed] [Google Scholar]
- 47.Duchini A, Govindarajan S, Santucci M, Zampi G, Hofman FM. Effects of tumor necrosis factor-alpha and interleukin-6 on fluid-phase permeability and ammonia diffusion in CNS-derived endothelial cells. J Investig Med. 1996;44:474–482. [PubMed] [Google Scholar]
- 48.de Vries HE, Blom-Roosemalen MC, van Oosten M, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. 1996;64:37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- 49.Vaquero J, Butterworth RF. Mild hypothermia for the treatment of acute liver failure--what are we waiting for? Nat Clin Pract Gastroenterol Hepatol. 2007;4:528–529. doi: 10.1038/ncpgasthep0927. [DOI] [PubMed] [Google Scholar]
- 50.Chapman RW, Forman D, Peto R, Smallwood R. Liver transplantation for acute hepatic failure? Lancet. 1990;335:32–35. doi: 10.1016/0140-6736(90)90150-4. [DOI] [PubMed] [Google Scholar]
- 51.Emond JC, Aran PP, Whitington PF, Broelsch CE, Baker AL. Liver transplantation in the management of fulminant hepatic failure. Gastroenterology. 1989;96:1583–1588. doi: 10.1016/0016-5085(89)90530-1. [DOI] [PubMed] [Google Scholar]
- 52.O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439–445. doi: 10.1016/0016-5085(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 53.Blitzer BL, Waggoner JG, Jones EA, et al. A model of fulminant hepatic failure in the rabbit. Gastroenterology. 1978;74:664–671. [PubMed] [Google Scholar]
- 54.Kelly JH, Koussayer T, He DE, et al. An improved model of acetaminophen-induced fulminant hepatic failure in dogs. Hepatology. 1992;15:329–335. doi: 10.1002/hep.1840150225. [DOI] [PubMed] [Google Scholar]
- 55.van Leenhoff JA, Hickman R, Saunders SJ, Terblanche J. Massive liver cell necrosis induced in the pig with carbon tetrachloride. S Afr Med J. 1974;48:1201–1204. [PubMed] [Google Scholar]
- 56.Sielaff TD, Hu MY, Rollins MD, et al. An anesthetized model of lethal canine galactosamine fulminant hepatic failure. Hepatology. 1995;21:796–804. [PubMed] [Google Scholar]
- 57.Ellis A, Wendon J. Circulatory, respiratory, cerebral, and renal derangements in acute liver failure: pathophysiology and management. Semin Liver Dis. 1996;16:379–388. doi: 10.1055/s-2007-1007251. [DOI] [PubMed] [Google Scholar]
- 58.Traber P, DalCanto M, Ganger D, Blei AT. Effect of body temperature on brain edema and encephalopathy in the rat after hepatic devascularization. Gastroenterology. 1989;96:885–891. [PubMed] [Google Scholar]
- 59.Butterworth RF. Hepatic encephalopathy: a central neuroinflammatory disorder? Hepatology. 2011;53:1372–1376. doi: 10.1002/hep.24228. [DOI] [PubMed] [Google Scholar]
- 60.Tracey KJ, Beutler B, Lowry SF, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 61.Wakabayashi G, Gelfand JA, Jung WK, Connolly RJ, Burke JF, Dinarello CA. Staphylococcus epidermidis induces complement activation, tumor necrosis factor and interleukin-1, a shock-like state and tissue injury in rabbits without endotoxemia. Comparison to Escherichia coli. J Clin Invest. 1991;87:1925–1935. doi: 10.1172/JCI115218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ito H, Ando K, Ishikawa T, Saito K, et al. Role of TNF-alpha produced by nonantigen-specific cells in a fulminant hepatitis mouse model. J Immunol. 2009;182:391–397. doi: 10.4049/jimmunol.182.1.391. [DOI] [PubMed] [Google Scholar]
- 63.Watanabe A, Fujiwara M, Shiota T, Tsuji T. Amino acid neurotransmitters and their receptors in the brain synaptosomes of acute hepatic failure rats. Biochem Med Metab Biol. 1988;40:247–252. doi: 10.1016/0885-4505(88)90125-9. [DOI] [PubMed] [Google Scholar]
- 64.Watanabe A, Shiota T, Takei N, Fujiwara M, Nagashima H. Ammonia detoxification by accelerated oxidation of branched chain amino acids in brains of acute hepatic failure rats. Biochem Med Metab Biol. 1986;35:367–375. doi: 10.1016/0885-4505(86)90095-2. [DOI] [PubMed] [Google Scholar]
- 65.Francavilla A, Makowka L, Polimeno L, et al. A Novel Model of Acute Hepatic Failure in Dogs With Implications for Transplantation Research. Transplant Proc. 1988;20:713–715. [PMC free article] [PubMed] [Google Scholar]
- 66.Gazzard BG, Hughes RD, Mellon PJ, Portmann B, Williams R. A dog model of fulminant hepatic failure produced by paracetamol administration. Br J Exp Pathol. 1975;56:408–411. [PMC free article] [PubMed] [Google Scholar]
- 67.Miller DJ, Hickman R, Fratter R, Terblanche J, Saunders SJ. An animal model of fulminant hepatic failure: a feasibility study. Gastroenterology. 1976;71:109–113. [PubMed] [Google Scholar]
- 68.Keppler D, Lesch R, Reutter W, Decker K. Experimental hepatitis induced by D-galactosamine. Exp Mol Pathol. 1968;9:279–290. doi: 10.1016/0014-4800(68)90042-7. [DOI] [PubMed] [Google Scholar]
- 69.Decker K, Keppler D. Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev Physiol Biochem Pharmacol. 1974;71:77–106. doi: 10.1007/BFb0027661. [DOI] [PubMed] [Google Scholar]
- 70.Watanabe A, Higashi T, Nagashima H. An animal model of fulminant hepatic failure in the rat. Acta Med Okayama. 1979;33:443–450. [PubMed] [Google Scholar]
- 71.Oppong KN, Bartlett K, Record CO, al Mardini H. Synaptosomal glutamate transport in thioacetamide-induced hepatic encephalopathy in the rat. Hepatology. 1995;22:553–558. [PubMed] [Google Scholar]
- 72.Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165:657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tiegs G, Wolter M, Wendel A. Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol. 1989;38:627–631. doi: 10.1016/0006-2952(89)90208-6. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stuart WD, Kulkarni RM, Gray JK, Vasiliauskas J, Leonis MA, Waltz SE. Ron receptor regulates Kupffer cell-dependent cytokine production and hepatocyte survival following endotoxin exposure in mice. Hepatology. 2011;53:1618–1628. doi: 10.1002/hep.24239. [DOI] [PMC free article] [PubMed] [Google Scholar]