

Abstract
We present a comparative analysis of predicted highly expressed (PHX) genes in the low G+C Gram-positive genomes of Bacillus subtilis, Bacillus halodurans, Listeria monocytogenes, Listeria innocua, Lactococcus lactis, Streptococcus pyogenes, Streptococcus pneumoniae, Staphylococcus aureus, Clostridium acetobutylicum, and Clostridium perfringens. Most enzymes acting in glycolysis and fermentation pathways are PHX in these genomes, but not those involved in the TCA cycle and respiration, suggesting that these organisms have predominantly adapted to grow rapidly in an anaerobic environment. Only B. subtilis and B. halodurans have several TCA cycle PHX genes, whereas the TCA pathway is entirely missing from the metabolic repertoire of the two Streptococcus species and is incomplete in Listeria, Lactococcus, and Clostridium. Pyruvate-formate lyase, an enzyme critical in mixed acid fermentation, is among the highest PHX genes in all these genomes except for C. acetobutylicum (not PHX), and B. subtilis, and B. halodurans (missing). Pyruvate-formate lyase is also prominently PHX in enteric γ-proteobacteria, but not in other prokaryotes. Phosphotransferase system genes are generally PHX with selection of different substrates in different genomes. The various substrate specificities among phosphotransferase systems in different genomes apparently reflect on differences in habitat, lifestyle, and nutrient sources.
Recently sequenced complete genomes of the low G+C group of Gram-positive bacteria ( ) include Lactococcus lactis (LACLA), Streptococcus pyogenes (STRPY), Streptococcus pneumoniae (STRPN), Listeria monocytogenes (LISMO), Listeria innocua (LISIN), Staphylococcus aureus (STAAU), Bacillus halodurans (BACHA), Bacillus subtilis (BACSU), Clostridium acetobutylicum (CLOAC), and Clostridium perfringens (CLOPE). Metabolic strategies among this group vary widely: the CLOAC and CLOPE bacteria are strictly anaerobic; LACLA, STRPY, STRPN prefer an anaerobic environment but not absolutely, whereas BACSU, BACHA, LISMO, LISIN, and STAAU are facultative aerobes.
) include Lactococcus lactis (LACLA), Streptococcus pyogenes (STRPY), Streptococcus pneumoniae (STRPN), Listeria monocytogenes (LISMO), Listeria innocua (LISIN), Staphylococcus aureus (STAAU), Bacillus halodurans (BACHA), Bacillus subtilis (BACSU), Clostridium acetobutylicum (CLOAC), and Clostridium perfringens (CLOPE). Metabolic strategies among this group vary widely: the CLOAC and CLOPE bacteria are strictly anaerobic; LACLA, STRPY, STRPN prefer an anaerobic environment but not absolutely, whereas BACSU, BACHA, LISMO, LISIN, and STAAU are facultative aerobes.
Various strains of STRPY (1) are responsible for a wide variety of diseases in humans, including scarlet fever and rheumatic fever. At present, STRPY is usually found in opportunistic infections of wounds, in some cases causing necrotizing fasciitis. STRPN (2) is the leading cause of bacterial pneumonia and can also induce bacteremia, meningitis, and inner ear infections in children. By contrast, LACLA (3) is a nonpathogenic bacterium that facilitates milk fermentation, and in nature can be found on plant and animal surfaces. These three closely related species are all nonmotile, are coccoid in shape, and are oxygen-tolerant anaerobes.
LISMO and its close relative, LISIN (4), are facultatively aerobic motile bacilli found in soil and water that are particularly abundant in rotting plant matter. They are frequently consumed by animals and can survive under conditions of extreme temperature, salt concentrations, and pH encountered in the food chain. LISMO is pathogenic in humans and cattle, whereas LISIN is nonpathogenic (4). Subsequent to human infection, LISMO replicates in the cytoplasm of host cells, including macrophages and hepatocytes, and is capable of crossing the placental and blood–brain barriers to cause spontaneous abortions and meningitis.
Bacillus spp. are also rod-shaped soil organisms that share significant gene synteny with Listeria (4). BACSU (5) is the most extensively studied among all Gram-positive bacteria. It has been widely used as a model organism for understanding gene regulation, cell division, quorum sensing, and sporulation. BACHA (6) is a recently identified member of the genus that grows best in alkalic environments at pH 9.5 or higher.
STAAU (7) is a nonmotile  bacterium. It is facultatively aerobic with principal habitat in nasal membranes and on the skin of warm-blooded mammals in which it causes a range of infections based on many toxins, ranging from food poisoning, boils and sties, to life-threatening septicemia and toxic shock, and develops resistance to many antibiotics.
bacterium. It is facultatively aerobic with principal habitat in nasal membranes and on the skin of warm-blooded mammals in which it causes a range of infections based on many toxins, ranging from food poisoning, boils and sties, to life-threatening septicemia and toxic shock, and develops resistance to many antibiotics.
The Clostridiae are a diverse group of rod-shaped, anaerobic, endospore-forming, organotropic bacteria, that ferment carbohydrates, amino acids, or nucleic acids, yielding products such as H2, CO2, and low molecular weight fatty acids and alcohols. These bacteria include several toxin-producing pathogens. They are commonly found in soil and sewage environments and in the intestines of mammals. CLOPE (8) was widely recognized as an important causal organism of gas gangrene. Among toxigenic clostridial species, CLOPE is the paradigm species for genetic studies because of its oxygen tolerance and fast growth rate (≈10-min generation time under optimal conditions) and capacity for easy genetic manipulation. Isolates of CLOAC (9) were first identified during World War I and these were used to develop an industrial starchbased acetone, butanol, and ethanol fermentation process to produce primarily acetone for gunpowder production.
Our approach to ascertaining gene expression levels relates to codon usage differences between gene classes, indicating that codon usage contributes importantly to setting the level of expression of the gene. It is generally recognized that in most prokaryotic cells during fast growth, ribosomal proteins (RPs), and translation/transcription processing factors (TFs) are highly expressed. Also the major chaperone and degradation (CH) genes functioning in protein folding, proteolysis, trafficking, and secretion are highly expressed. The three gene classes RP, CH, and TF are consonant in that they exhibit high codon biases compared to the average gene, whereas the codon usage differences among these three gene classes are low (10). The data support the proposition that each genome has evolved a codon usage pattern accommodating “optimal” gene expression levels for most circumstances of its habitat, energy sources, and lifestyle. We have taken the three gene classes RP, CH, and TF as representative of highly expressed genes. Qualitatively, a gene is predicted highly expressed (PHX) if its codon usage is rather similar to that of the RP genes, the principal TFs, and to that of the major CH genes, but deviates strongly from the average gene of the genome. This approach is robust with respect to specific choice of parameters (11), and the results are in agreement with available 2D gel evaluations for most proteins (10, 12).
Orthologous genes from different species could have very different expression levels if the concentration of the gene product is highly relevant for one species to thrive, but not decisive for another species. In this paper, we analyze and compare collections of PHX genes in the 10 available complete genomes of  . Some genes are PHX in all these genomes. Differences among the PHX genes in different genomes can be interpreted with respect to differences in the species' habitat and nutrition sources.
. Some genes are PHX in all these genomes. Differences among the PHX genes in different genomes can be interpreted with respect to differences in the species' habitat and nutrition sources.
Methods
Theoretical Measures of Gene Expression.
Let G be a group of genes with average codon frequencies g(x,y,z) for the codon triplet (x,y,z) such that Σ g(x,y,z) = 1 for each amino acid family. Similarly, let {f(x,y,z)} indicate the codon frequencies for the gene group F, normalized to 1 in each amino acid codon family. The codon usage difference of F relative to G is calculated by the formula
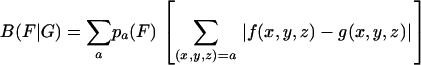 |
[1] |
where {pa(F)}are the average amino acid frequencies of the genes of F. The assessments of Eq. 1 can be made for any two gene groups from the same genome or from different genomes. We refer to the gene collection G as the standard to which different gene groups F(1), F(2),... F(r) are compared. G could be a specific gene class, or an average gene. Let B(g|G) denote the codon usage difference of the gene g relative to the gene class G, and let C be the collection of all genes encoded in the genome. A gene g is PHX if B(g|C) is high, whereas B(g|RP), B(g|CH), and B(g|TF) are low, i.e., the codon usage of g is very different from the codon usage of an average gene but similar to the codon usage of the gene classes RP, CH, and TF. Definition of PHX genes with respect to individual standards will be based on the ratios ERP (g) = B(g|C)/B(g|RP), ECH (g) = B(g|C)/B(g|CH) and ETF (g) = B(g|C)/B(g|TF). In combination, we use the expression measure
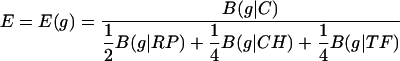 |
[2] |
The three gene classes (RP, CH, and TF) serve as representatives of highly expressed genes, and our method designates genes with similar codon usage as PHX genes. The analyses and results do not qualitatively change when different weights in formula (2) are used.
Definition. A gene is PHX if the following two conditions are satisfied: at least two among the three expression ratios ERP (g), ECH (g), and ETF (g) exceed 1.05, and the measure E (g)is ≥1.00. A gene is designated Putative Alien (PA) if all four values B(g|C), B(g|RP), B(g|TF), and B(g|CH) exceed a threshold M(g) + 0.1, where M(g) is the median codon bias of B(g|C) among all genes of similar length as g.
PA genes are mostly ORFs of unknown function but also include genes encoding transposases, cryptic prophage sequences, restriction or modification enzymes (which are often conjugatively transferred by plasmids), genes associated with lipopolysaccharide biosynthesis and fimbrial-like genes (13, 14).
Results
Distribution and Types of PHX Genes Among Available  Genomes. Table 1 displays the statistics of PHX genes and the maximum E(g) value for each of the currently available
Genomes. Table 1 displays the statistics of PHX genes and the maximum E(g) value for each of the currently available  genomes. The percentage of PHX genes varies from 4.2% and 4.4% for the two Bacillus genomes, to 10.1% for STRPN. The counts of PA genes are generally <4%. Table 2 lists the 10 genes with the highest predicted expression levels for each of the
genomes. The percentage of PHX genes varies from 4.2% and 4.4% for the two Bacillus genomes, to 10.1% for STRPN. The counts of PA genes are generally <4%. Table 2 lists the 10 genes with the highest predicted expression levels for each of the  genomes (expanded to the top 20 genes in Table 4, which is published as supporting information on the PNAS web site). The genes are segregated into functional categories. Almost all RPs attain high PHX values in most bacteria and the RP S1 is often among the top PHX genes. However, the S1 predicted expression levels are reduced in most
genomes (expanded to the top 20 genes in Table 4, which is published as supporting information on the PNAS web site). The genes are segregated into functional categories. Almost all RPs attain high PHX values in most bacteria and the RP S1 is often among the top PHX genes. However, the S1 predicted expression levels are reduced in most 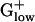 genomes (excepting Streptococcus and Lactococcus species) and in 5 of the 10 genomes the S1 gene does not qualify as PHX. The RP S1 also has a reduced size of 377–410 amino acids in the
genomes (excepting Streptococcus and Lactococcus species) and in 5 of the 10 genomes the S1 gene does not qualify as PHX. The RP S1 also has a reduced size of 377–410 amino acids in the 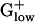 genomes (excepting CLOAC), compared with >500 amino acids in most Gram-negative bacteria (Table 3 and ref. 12), possibly conveying a lesser role for S1 in most
genomes (excepting CLOAC), compared with >500 amino acids in most Gram-negative bacteria (Table 3 and ref. 12), possibly conveying a lesser role for S1 in most 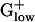 genomes. The S1 gene in CLOAC is fused with a segment encoding a penicillin tolerance domain (LytB) and has an aggregate length of 641 codons. Unlike most RPs, the S1 protein is only loosely attached to the ribosome. It acts during initial recognition and binding to the mRNA and contains multiple copies of an RNA-binding domain (15).
genomes. The S1 gene in CLOAC is fused with a segment encoding a penicillin tolerance domain (LytB) and has an aggregate length of 641 codons. Unlike most RPs, the S1 protein is only loosely attached to the ribosome. It acts during initial recognition and binding to the mRNA and contains multiple copies of an RNA-binding domain (15).
Table 1. General statistics of PHX and PA genes.
| Characteristic | BACSU | BACHA | LISIN | LISMO | LACLA | STRPY | STRPN | STAAU | CLOAC | CLOPE |
|---|---|---|---|---|---|---|---|---|---|---|
| Genome size (kb) | 4,215 | 4,202 | 3,011 | 2,945 | 2,366 | 1,852 | 2,161 | 2,878 | 3,941 | 3,031 |
| No. of genes with ≥80 codons | 3,795 | 3,724 | 2,811 | 2,732 | 2,122 | 1,579 | 1,788 | 2,469 | 3,426 | 2,504 |
| PHX genes, No. | 166 | 155 | 251 | 256 | 154 | 109 | 181 | 153 | 299 | 203 |
| PHX genes, % | 4.4 | 4.2 | 8.9 | 9.4 | 7.3 | 6.9 | 10.1 | 5.7 | 8.7 | 8.1 |
| Highest E(g) value* | 2.34(fus) | 1.79(groEL) | 2.40(pfl) | 2.49(pfl) | 2.46(fus) | 2.32(pfl) | 2.88(glgP) | 2.64(fus) | 1.52(rpL5) | 2.19(dnaK) |
| PA genes, No. | 179 | 107 | 90 | 101 | 70 | 34 | 65 | 47 | 75 | 29 |
| PA genes, % | 4.7 | 2.9 | 3.2 | 3.7 | 3.3 | 2.2 | 3.6 | 1.9 | 2.2 | 1.2 |
The E(g) value is used as a measure of expression level of a gene g; see Methods for details. The gene with the highest E(g) value is also indicated in parentheses. See also Table 2
Table 2. Top 10 PHX genes from genomes of 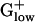 bacteria.
bacteria.
|
E(g)*
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | BACSU | BACHA | LISIN | LISMO | LACLA | STRPY | STRPN | STAAU | CLOAC | CLOPE |
| RPs | ||||||||||
| L1 (rplA) | 1.91 | 1.55 | 2.05 | 2.03 | 2.00 | 1.95 | 2.11 | 2.12 | 1.18 | 1.71 |
| L2 (rplB) | 2.02 | 1.69 | 1.92 | 1.85 | 2.09 | 2.16 | 2.33 | 1.70 | 1.40 | 1.52 |
| L3 (rplC) | 1.78 | 1.60 | 1.44 | 1.42 | 1.71 | 1.73 | 1.87 | 1.59 | 1.30 | 1.85 |
| L4 (rplD) | 1.98 | 1.67 | 1.69 | 1.73 | 1.70 | 1.71 | 1.91 | 1.46 | 1.23 | 1.24 |
| L5 (rplE) | 1.92 | 1.35 | 1.34 | 1.36 | 1.79 | 1.69 | 2.17 | 1.79 | 1.52 | 1.72 |
| L14 (rplN) | 1.98 | 1.43 | 1.30 | 1.37 | 1.51 | 1.69 | 1.89 | 1.70 | 1.08 | 1.24 |
| L17 (rplQ) | 1.87 | 1.51 | 1.69 | 1.72 | 2.31 | — | 1.83 | 2.03 | 1.18 | 1.23 |
| L19 (rplS) | 1.84 | 1.62 | 1.54 | 1.60 | 1.90 | 1.76 | 1.99 | 1.71 | 1.38 | 1.55 |
| L20 (rplT) | 1.57 | 1.54 | 1.38 | 1.42 | 1.57 | 1.72 | 2.15 | 1.80 | 1.24 | 1.96 |
| S1 (rpsA) | 1.20 | (1.01) | (0.90) | 1.17 | 2.14 | 2.32 | 2.31 | (0.80) | (0.74) | (0.76) |
| S2 (rpsB) | 1.84 | 1.47 | 1.72 | 1.67 | 2.37 | 2.03 | 2.30 | 2.09 | 1.33 | 1.58 |
| S3 (rpsC) | 1.87 | 1.48 | 1.69 | 1.70 | 2.17 | 1.95 | 2.10 | 1.83 | 1.21 | 1.99 |
| S4 (rpsD) | 1.94 | 1.61 | 1.84 | 1.82 | 2.09 | 2.12 | 2.33 | 2.11 | 1.19 | 1.67 |
| S9 (rpsI) | 1.51 | 1.53 | 1.86 | 1.87 | 1.52 | 1.92 | 1.87 | 2.36 | 1.42 | 1.46 |
| S13 (rpsM) | 2.02 | 1.38 | 1.93 | 1.86 | 1.59 | 1.72 | 1.95 | 1.86 | 1.27 | 1.23 |
| TFs | ||||||||||
| Translation elongation factor G (fus) | 2.34 | 1.77 | 2.34 | 2.31 | 2.46 | 2.16 | 2.47 | 2.64 | 1.45 | 2.08 |
| Translation elongation factor Ts (tsf) | 1.75 | 1.44 | 1.98 | 2.33 | 1.41 | 1.87 | 2.35 | 2.11 | 1.31 | 1.62 |
| Translation elongation factor Tu (tuf) | 1.97 | 1.50 | 2.00 | 1.92 | 1.91 | 1.96 | 2.06 | 2.09 | 1.14 | 1.42 |
| 1.42 | ||||||||||
| Translation initiation factor IF-2 (infB) | (0.79) | (0.77) | 1.06 | 1.23 | 1.65 | (0.56) | 1.04 | 1.00 | (1.04) | 2.04 |
| RNA polymerase β-subunit (rpoB) | 1.49 | 1.19 | 2.37 | 2.19 | 1.87 | 1.72 | 1.83 | 1.67 | (0.98) | 1.62 |
| RNA polymerase β′-subunit (rpoC) | 1.76 | 1.54 | 2.39 | 2.26 | 2.02 | 1.72 | 1.77 | 1.55 | 1.19 | 1.53 |
| GTP-binding protein TypA/BipA | 1.11 | (0.78) | 1.91 | 2.09 | 1.95 | 1.45 | 1.98 | (0.97) | 1.18 | 1.45 |
| Chaperones | ||||||||||
| HSP 60 (groEL) | 1.87 | 1.79 | 1.91 | 1.89 | 1.23 | (0.86) | 1.08 | 1.19 | 1.45 | (0.71) |
| HSP 70 (dnaK) | 1.83 | 1.14 | 2.25 | 2.17 | 2.08 | 2.25 | 2.43 | 2.21 | 1.38 | 2.19 |
| Trigger factor (tig) | 1.85 | 1.53 | 2.02 | 1.81 | 1.86 | 1.55 | 2.55 | 2.02 | 1.42 | 1.60 |
| Glycolysis | ||||||||||
| Glucose-6-phosphate isomerase (pgi) | (0.92) | (0.93) | 1.67 | 1.77 | 2.13 | 1.57 | 2.46 | 1.33 | 1.28 | 1.74 |
| Fructose-1,6-bisphosphate aldolase (fba) | 1.99 | 1.20 | 1.71 | 1.70 | 2.08 | 2.07 | 2.15 | 2.00 | 1.20 | 1.52 |
| (0.59) | (0.83) | (0.67) | (0.59) | (0.86) | (0.71) | |||||
| (etc.) | (etc.) | |||||||||
| Glyceraldehyde-3-phosphate dehydrogenase | 1.80 | 1.53 | 1.77 | 1.72 | 1.95 | 2.13 | 2.08 | 2.12 | 1.27 | 1.48 |
| (gap) | (0.48) | (0.70) | 1.04 | (0.69) | ||||||
| Phosphoglycerate kinase (pgk) | 1.34 | 1.09 | 2.08 | 2.02 | 2.30 | 2.18 | 2.24 | 1.86 | 1.34 | 1.65 |
| Phosphoglycerate mutase (gpmA) | (0.70) | — | (0.93) | (1.04) | 2.27 | 1.88 | 1.94 | (0.79) | (0.95) | — |
| Phosphoglycerate mutase (2.3-bisphosphoglycerate-independent) (pgm) | 1.08 | (0.88) | 1.97 | 1.98 | — | — | — | 1.15 | 1.29 | 1.80 |
| Enolase (eno) | 1.92 | 1.61 | 1.83 | 1.92 | 1.96 | 2.17 | 2.24 | 2.12 | 1.26 | 1.51 |
| (0.49) | ||||||||||
| Pyruvate kinase (pykA) | 1.18 | (0.76) | 2.38 | 2.17 | 2.25 | 2.04 | 2.28 | 1.70 | 1.29 | 1.68 |
| (0.83) | ||||||||||
| 1.29 | ||||||||||
| PTS | ||||||||||
| Phosphotransferase enzyme IIC component | (0.57) | (0.57) | 2.06 | 2.05 | 1.32 | (0.55) | (1.01)† | — | 1.24 | — |
| (cellobiose-specific) | (0.46) | 1.16 | 1.32 | 1.10 | (0.60) | |||||
| (0.45) | (etc.) | (etc.) | (0.47) | (0.49) | ||||||
| PTS component IID (mannose-specific) (manN) | — | — | 1.26 | 1.47 | 1.85 | 2.15 | 1.54 | — | — | 1.27 |
| 1.23 | 1.09 | 1.04 | ||||||||
| PTS system, IIABC components‡ | — | — | — | — | — | (0.60) | 2.43 | — | — | (0.80) |
| Pyruvate dehydrogenase, pyruvate oxidase | ||||||||||
| Dihydrolipoamide acelyltransferase component of pyruvate dehydrogenase complex (pdhC) | 2.05 | 1.48 | 1.79 | 1.81 | (0.94) | — | — | 1.43 | — | — |
| Dihydrolipoamide dehydrogenase E3-subunit of pyruvate dehydrogenase (pdhD) | 2.14 | 1.55 | 1.98 | 1.80 | 1.09 | — | — | 2.05 | — | — |
| Pyruvate oxidase | (0.47) | — | (0.86) | 1.03 | (0.47) | — | 2.48 | (0.66) | — | — |
| Fermentation and anaerobic respiration | ||||||||||
| Pyruvate formate-lyase (formate acetyltransferase) (pflB, pflA) | — | — | 2.40 | 2.49 | 1.78 | 2.32 | 2.87 | 2.09 | (0.98) | 1.92 |
| 1.98 | 2.09 | |||||||||
| l-lactate dehydrogenase (ldh) | (0.76) | (0.69) | 1.63 | 1.53 | 2.26 | 1.72 | 2.30 | (0.90) | (0.94) | (0.78) |
| (0.45) | (0.88) | (0.80) | ||||||||
| (0.44) | ||||||||||
| Alcohol-acetaldehyde dehydrogenase (adhE) | — | — | 2.08 | 2.14 | 1.44 | — | 2.22 | (0.56) | — | 2.00 |
| β-hydroxybutyryl-CoA dehydrogenase, NAD-dependent (CAC2708) | (0.52) | (0.81) | — | — | — | — | — | — | 1.44 | 1.55 |
| (0.71) | ||||||||||
| (0.68) | ||||||||||
| Butyryl-CoA dehydrogenase (CAC2711) | (0.56) | (0.66) | — | — | — | — | — | — | 1.44 | 1.70 |
| (0.48) | (0.63) | (0.66) | ||||||||
| (0.41) | (0.62) | |||||||||
| Pyruvate ferredoxin oxidoreductase (CAC2229, CPE2061) | — | — | (0.51) | (0.45) | (0.40) | — | — | — | 1.43 | 2.05 |
| 1.07 | ||||||||||
| Amino acid blosynthesis | ||||||||||
| Ketol-acid reductoisomerase (ilvC) | 1.03 | 1.59 | 1.16 | 1.27 | (0.79) | — | 2.11 | (1.06) | 1.04 | — |
| Methionine synthase (metE) | (0.72) | (0.88) | (0.56) | (0.61)† | (0.44) | — | 2.44 | 1.34 | — | — |
| Transporters | ||||||||||
| Permease of the Na+:galactoside symporter family (CAC0694, yjmB) | (0.46) | — | — | — | — | — | — | — | 1.47 | — |
| ABC transporter, substrate-binding protein (SP0092) | — | (0.63) | (0.83) | (0.82) | (0.68) | (0.80) | 2.73 | — | — | 1.54 |
| Oligonucleotide ABC transporter (BH0031) | — | 1.78 | — | — | — | — | — | — | — | — |
| (0.66) | ||||||||||
| (etc.) | ||||||||||
| Oligopeptide ABC transporter (CAC3643) | (0.58) | (0.76) | (0.69) | (0.75) | (0.69) | — | — | (0.51) | 1.42 | (0.65) |
| (0.49) | (1.06) | |||||||||
| Glycogen degradation | ||||||||||
| Glycogen phosphorylase (glgP) | (0.39) | (0.52) | — | — | (0.42) | 1.27 | 2.88 | — | (0.90) | (0.57) |
| (0.43) | ||||||||||
| Phosphoglucomutase | (0.54) | (0.64) | (0.44) | (0.56) | — | (0.84) | 2.46 | — | — | — |
| Other | ||||||||||
| P60 extracellular protein, invasion-associated (iap) | — | — | 2.16 | 1.76 | — | — | — | — | — | — |
| Hypothetical protein (CPE1232) | — | — | — | — | — | — | — | — | 1.35 | 2.00 |
| Hypothetical protein (CPE1233) | — | — | — | — | — | — | — | — | 1.30 | 1.90 |
Included are all genes ranking among the top 10 PHX genes in any of the eight genomes (underlined) and their homologs in other genomes even if they are not among the top 10.
Numbers in parentheses indicate the gene is not PHX; —, the gene does not have a homolog in the genome; etc, more than three homologs
PA gene
All genomes have homologs of PTS system IIABC components but of lower similarity (≈30%). Those listed exhibit high mutual similarity (>50%)
Table 3. Giant ribosomal protein S1 gene.
| S1 | BACSU | BACHA | LISIN | LISMO | LACLA | STRPY | STRPN | STAAU | CLOAC | CLOPE |
|---|---|---|---|---|---|---|---|---|---|---|
| Size (codons) | 381 | 382 | 380 | 380 | 407 | 400 | 399 | 390 | 641† | 377 |
| E(g)* | 1.20 | (1.01) | (0.90) | 1.17 | 2.14 | 2.32 | 2.31 | (0.80) | (0.74)† | (0.76) |
No. in parentheses indicates the gene is not PHX
Fused with penicillin tolerance LytB domain
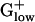 bacteria generally show the highest predicted expression levels for the genes of the RP, TF, and CH gene classes. The highest E(g) values usually are >2.00 (Tables 1 and 2), which has been correlated with a doubling time of <1 h in rich media (10, 12). Among the top 10 PHX genes in most bacterial genomes are the major chaperone proteins DnaK (HSP70), GroEL (HSP60), and Trigger factor. Trigger factor and DnaK cooperate in the folding of newly synthesized proteins (16). The HSP60 chaperonin complex is believed to assist protein folding by providing a cavity in which nonnative polypeptides are enclosed and protected against intermolecular aggregation (17). GroEL carries high PHX values in BACSU, BACHA, LISMO, and LISIN, but attains only moderate PHX levels in LACLA, STRPY, STRPN, and STAAU (Table 2). HSP70 is high PHX in all
bacteria generally show the highest predicted expression levels for the genes of the RP, TF, and CH gene classes. The highest E(g) values usually are >2.00 (Tables 1 and 2), which has been correlated with a doubling time of <1 h in rich media (10, 12). Among the top 10 PHX genes in most bacterial genomes are the major chaperone proteins DnaK (HSP70), GroEL (HSP60), and Trigger factor. Trigger factor and DnaK cooperate in the folding of newly synthesized proteins (16). The HSP60 chaperonin complex is believed to assist protein folding by providing a cavity in which nonnative polypeptides are enclosed and protected against intermolecular aggregation (17). GroEL carries high PHX values in BACSU, BACHA, LISMO, and LISIN, but attains only moderate PHX levels in LACLA, STRPY, STRPN, and STAAU (Table 2). HSP70 is high PHX in all 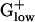 genomes except for BACHA, and Trigger factor is significantly PHX in all
genomes except for BACHA, and Trigger factor is significantly PHX in all 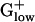 genomes. In most
genomes. In most 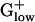 genomes, the asparaginyl tRNA synthetase is PHX. This finding is striking because asparaginyl tRNA synthetase is absent from many bacterial genomes. Generally, asparaginyl tRNA synthetase is present only in
genomes, the asparaginyl tRNA synthetase is PHX. This finding is striking because asparaginyl tRNA synthetase is absent from many bacterial genomes. Generally, asparaginyl tRNA synthetase is present only in 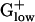 , in γ-proteobacteria, and in cyanobacteria, but missing from other proteobacteria and from high G+C Grampositive species. Processing factors for protein synthesis are emphatically PHX, especially the ATP-dependent DNA-directed RNA polymerase units RpoB and RpoC, and the elongation factors EF-G (fus), EF-Tu (tuf), and EF-Ts (tsf).
, in γ-proteobacteria, and in cyanobacteria, but missing from other proteobacteria and from high G+C Grampositive species. Processing factors for protein synthesis are emphatically PHX, especially the ATP-dependent DNA-directed RNA polymerase units RpoB and RpoC, and the elongation factors EF-G (fus), EF-Tu (tuf), and EF-Ts (tsf).
PHX Genes in Energy Metabolic Pathways. PHX enzymes of metabolic pathways emphasize five groups: glycolysis, pyruvate metabolism (including variations on mixed acid fermentation), pentose phosphate pathway, TCA cycle, and fatty acid metabolism. The glycolysis genes are predominantly PHX in 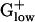 bacteria, which use mainly anaerobic metabolism. Hexokinase or glucokinase perform the first step in glycolysis in most eukaryotes, the ATP-dependent phosphorylation of glucose to glucose-6-phosphate, but the former is rarely found in prokaryotes. However, in many bacteria, glucose-6-phosphate is generated from other hexose phosphates, and glucose phosphorylation is coupled to sugar uptake by means of the phosphotransferase system (PTS). Perhaps the multiplicity of sources allows that glucokinase need not be PHX. In a consistent manner, all
bacteria, which use mainly anaerobic metabolism. Hexokinase or glucokinase perform the first step in glycolysis in most eukaryotes, the ATP-dependent phosphorylation of glucose to glucose-6-phosphate, but the former is rarely found in prokaryotes. However, in many bacteria, glucose-6-phosphate is generated from other hexose phosphates, and glucose phosphorylation is coupled to sugar uptake by means of the phosphotransferase system (PTS). Perhaps the multiplicity of sources allows that glucokinase need not be PHX. In a consistent manner, all 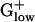 genomes possess 2–18 PHX PTS system enzymes, including duplicated genes. Notably, cellobiose-specific PTS enzymes are mainly PHX in LISIN and LISMO but not in other
genomes possess 2–18 PHX PTS system enzymes, including duplicated genes. Notably, cellobiose-specific PTS enzymes are mainly PHX in LISIN and LISMO but not in other 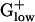 genomes. These bacteria thrive on rotting plant matter where cellobiose is their primary carbohydrate source. A mannose-specific PTS enzyme is PHX in LACLA, STRPY, STRPN, LISIN, and LISMO. This diversity among different organisms may reflect in the variety of their preferred carbon sources during rapid growth.
genomes. These bacteria thrive on rotting plant matter where cellobiose is their primary carbohydrate source. A mannose-specific PTS enzyme is PHX in LACLA, STRPY, STRPN, LISIN, and LISMO. This diversity among different organisms may reflect in the variety of their preferred carbon sources during rapid growth.
Many glycolysis genes are among the top PHX genes in 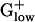 genomes of LACLA, STRPN, STRPY, LISIN, and LISMO (Table 2, and Tables 4 and 5, which are published as supporting information on the PNAS web site). Notably, phosphofructokinase (pfk) is not among the top PHX genes in any of the 10 genomes. This finding is interesting because the pfk reaction is the first committed step of the glycolysis pathway and a major regulation point. There appears to be a correlation between the frequency with which glycolysis genes are PHX in a genome and the characteristic lifestyle of the organism. For example, although BACSU prefers aerobic growth, it preferentially uses glycolysis for energy metabolism during exponential growth in rich nutrients and then shifts to oxidative phosphorylation by using mixed acid fermentation by- products as substrates in stationary phase.
genomes of LACLA, STRPN, STRPY, LISIN, and LISMO (Table 2, and Tables 4 and 5, which are published as supporting information on the PNAS web site). Notably, phosphofructokinase (pfk) is not among the top PHX genes in any of the 10 genomes. This finding is interesting because the pfk reaction is the first committed step of the glycolysis pathway and a major regulation point. There appears to be a correlation between the frequency with which glycolysis genes are PHX in a genome and the characteristic lifestyle of the organism. For example, although BACSU prefers aerobic growth, it preferentially uses glycolysis for energy metabolism during exponential growth in rich nutrients and then shifts to oxidative phosphorylation by using mixed acid fermentation by- products as substrates in stationary phase.
Glycogen phosphorylase exhibits dramatically different expression levels in different 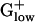 bacteria. It has the highest predicted expression level among all genes of STRPN (E = 2.88) but it is generally not PHX in the other
bacteria. It has the highest predicted expression level among all genes of STRPN (E = 2.88) but it is generally not PHX in the other 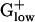 genomes. Among an assortment of prokaryotic complete genomes besides STRPN, glycogen phosphorylase is PHX only in STRPY and in Synechocystis (18) but at reduced E(g) values compared with STRPN. A second gene required for breakdown of glycogen and its utilization by glycolysis, phosphoglucomutase, is also strongly PHX in STRPN but not in the other
genomes. Among an assortment of prokaryotic complete genomes besides STRPN, glycogen phosphorylase is PHX only in STRPY and in Synechocystis (18) but at reduced E(g) values compared with STRPN. A second gene required for breakdown of glycogen and its utilization by glycolysis, phosphoglucomutase, is also strongly PHX in STRPN but not in the other 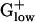 genomes. This result suggests that STRPN cells may be capable of using glycogen at higher rates than other bacteria. Interestingly, the genes for glycogen synthase and other enzymes active in synthesis of glycogen are not PHX in STRPN, perhaps indicating that unlike glycogen degradation, glycogen synthesis is not more efficient in STRPN. Because unusually high expression levels of specific genes in an organism often reflect on environmental influences, we speculate that some aspects of STRPN habitat and lifestyle require a high rate of glycogen utilization. STRPN is considered closely related to oral Streptococci, and a complete genome of Streptococcus mutans has recently become available (19). Whereas phosphoglucomutase is PHX in S. mutans [E(g) = 1.34], glycogen phosphorylase is not, arguing against the efficient glycogen utilization as a general trait of oral Streptococci.
genomes. This result suggests that STRPN cells may be capable of using glycogen at higher rates than other bacteria. Interestingly, the genes for glycogen synthase and other enzymes active in synthesis of glycogen are not PHX in STRPN, perhaps indicating that unlike glycogen degradation, glycogen synthesis is not more efficient in STRPN. Because unusually high expression levels of specific genes in an organism often reflect on environmental influences, we speculate that some aspects of STRPN habitat and lifestyle require a high rate of glycogen utilization. STRPN is considered closely related to oral Streptococci, and a complete genome of Streptococcus mutans has recently become available (19). Whereas phosphoglucomutase is PHX in S. mutans [E(g) = 1.34], glycogen phosphorylase is not, arguing against the efficient glycogen utilization as a general trait of oral Streptococci.
The four genes encoding the pyruvate dehydrogenase complex (pdhABCD) are PHX in BACSU, BACHA, LISIN, LISMO, LACLA, and STAAU whereas pyruvate dehydrogenase is apparently missing from CLOAC, CLOPE, STRPN, and STRPY. However, it is interesting to compare the acetoin dehydrogenase of the two Streptococci with the pyruvate dehydrogenase of the other  bacteria. Acetoin and pyruvate dehydrogenases are enzymes with similar architecture and sequence, but with different substrate specificity. Pyruvate dehydrogenase converts pyruvate to acetyl-CoA and is comprised of three or four polypeptide chains. These proteins are generally encoded in an operon and are PHX in many genomes, including most
bacteria. Acetoin and pyruvate dehydrogenases are enzymes with similar architecture and sequence, but with different substrate specificity. Pyruvate dehydrogenase converts pyruvate to acetyl-CoA and is comprised of three or four polypeptide chains. These proteins are generally encoded in an operon and are PHX in many genomes, including most  bacteria. STRPN and STRPY do not have pyruvate dehydrogenase and use pyruvate-phosphate lyase instead. Acetoin dehydrogenase functions in acetoin catabolism by cleavage of acetoin into acetate and acetaldehyde. The complete acetoin dehydrogenase operon (four genes) is PHX in STRPY and two of the four genes are PHX in STRPN. Among the 10
bacteria. STRPN and STRPY do not have pyruvate dehydrogenase and use pyruvate-phosphate lyase instead. Acetoin dehydrogenase functions in acetoin catabolism by cleavage of acetoin into acetate and acetaldehyde. The complete acetoin dehydrogenase operon (four genes) is PHX in STRPY and two of the four genes are PHX in STRPN. Among the 10  genomes, only BACSU and BACHA, in addition to the Streptococci, possess genes for acetoin dehydrogenase but these are not PHX. Streptococci are fermentative bacteria that convert pyruvate to acetoin. Acetoin dehydrogenase functions in acetoin catabolism and its PHX status suggests that at certain stages (probably in stationary phase) the cells rely on acetoin as carbon source. BACSU stores carbon in the form of acetoin and other fermentation products during exponential growth, which is later used during stationary growth and sporulation (20). Alternatively, due to similarity between acetoin and pyruvate dehydrogenases, the acetoin dehydrogenase of STRPN and STRPY may also possess the activity of pyruvate dehydrogenase. Notably, S. mutans possesses both pyruvate dehydrogenase and acetoin dehydrogenase (19), but neither is PHX (data not shown).
genomes, only BACSU and BACHA, in addition to the Streptococci, possess genes for acetoin dehydrogenase but these are not PHX. Streptococci are fermentative bacteria that convert pyruvate to acetoin. Acetoin dehydrogenase functions in acetoin catabolism and its PHX status suggests that at certain stages (probably in stationary phase) the cells rely on acetoin as carbon source. BACSU stores carbon in the form of acetoin and other fermentation products during exponential growth, which is later used during stationary growth and sporulation (20). Alternatively, due to similarity between acetoin and pyruvate dehydrogenases, the acetoin dehydrogenase of STRPN and STRPY may also possess the activity of pyruvate dehydrogenase. Notably, S. mutans possesses both pyruvate dehydrogenase and acetoin dehydrogenase (19), but neither is PHX (data not shown).
BACHA contains two small clusters (putative operons) of PHX genes functioning in the TCA cycle. One cluster combines three subunits of succinate dehydrogenase sdhC, sdhA, and sdhB. The same cluster occurs in BACSU and in STAAU, but these genes are only PHX in BACHA. The other  genomes do not possess succinate dehydrogenase homologs. The second TCA cluster in BACHA contains the PHX genes citZ (citrate synthase II), citC (isocitrate dehydrogenase), and citH (malate dehydrogenase). These three genes are organized in an analogous cluster in BACSU but only citC is PHX. Clusters of PHX TCA cycle genes occur in many proteobacterial genomes but not most
genomes do not possess succinate dehydrogenase homologs. The second TCA cluster in BACHA contains the PHX genes citZ (citrate synthase II), citC (isocitrate dehydrogenase), and citH (malate dehydrogenase). These three genes are organized in an analogous cluster in BACSU but only citC is PHX. Clusters of PHX TCA cycle genes occur in many proteobacterial genomes but not most 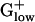 genomes.
genomes.
PHX Genes in Fermentation Pathways. The pyruvate-formate lyase (pfl) gene, which is essential to a mixed acid fermentation pathway, shows very high E(g) values in LACLA, STRPY, STAAU, STRPN, LISIN, LISMO, and CLOPE. Actually, many genes from the mixed acid fermentation pathways are PHX in these genomes (Table 5). Intriguingly, the pyruvate-formate lyase gene is absent from the two Bacillus genomes, BACSU and BACHA. Actually, BACSU, “more” aerobic than most 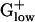 organisms, apparently uses pyruvate dehydrogenase rather than pyruvate-formate lyase during fermentative growth (ref. 21 and Table 5). The enzymes alcohol/acetaldehyde dehydrogenase and l-lactate dehydrogenase both function in mixed acid fermentation (as well as other fermentations) and are potently PHX in the majority of the
organisms, apparently uses pyruvate dehydrogenase rather than pyruvate-formate lyase during fermentative growth (ref. 21 and Table 5). The enzymes alcohol/acetaldehyde dehydrogenase and l-lactate dehydrogenase both function in mixed acid fermentation (as well as other fermentations) and are potently PHX in the majority of the 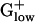 genomes.
genomes.
Multifunctional Proteins and PHX Levels. Multifunctional proteins often attain very high predicted expression levels. For example, polynucleotide phosphorylase is fundamental in RNA processing and mRNA degradation, and the gene reaches the highest E(g) value, 2.66, among all of the Escherichia coli genes. Polynucleotide phosphorylase is also a component of the mRNA degradosome, which involves RNase E, DnaK, RhlB helicase, and enolase (22). RNase E is PHX in E. coli, with an E(g) value of 1.22, but is generally missing from 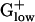 genomes. RNase P, which in the
genomes. RNase P, which in the 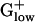 plays similar roles in maturation of ribosomal RNA, is not PHX in
plays similar roles in maturation of ribosomal RNA, is not PHX in 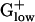 genomes. In
genomes. In 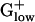 genomes, polynucleotide phosphorylase has very high E(g) values in LISMO (1.76) and LISIN (1.70). Enolase attains high E(g) values in all
genomes, polynucleotide phosphorylase has very high E(g) values in LISMO (1.76) and LISIN (1.70). Enolase attains high E(g) values in all 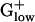 genomes. We have conjectured that a protein that performs multiple functions, which require abundant concentration of the protein, tend to attain higher E(g) values than the average PHX gene (10, 12).
genomes. We have conjectured that a protein that performs multiple functions, which require abundant concentration of the protein, tend to attain higher E(g) values than the average PHX gene (10, 12).
The enzyme aconitase interconverts citrate and isocitrate in the TCA cycle. Aconitase also serves as a sensor, detecting changes in the redox state and assaying iron content within the cell (23, 24). This protein can further function as a transcriptional activator that specifically regulates gene expression for the transferrin receptor and controls levels of ferritin in the cell (24). At its iron sulfur center, aconitase can be inactivated by oxidative stress or iron deprivation. Aconitase of BACSU binds RNA (25). Aconitase has the highest E(g) value, 2.56, in Deinococcus radiodurans (26) and its gene is PHX in virtually all aerobic genomes but generally not in the 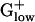 genomes. This finding is consistent with the anaerobic propensity of most
genomes. This finding is consistent with the anaerobic propensity of most 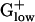 bacteria with lesser need for TCA cycle enzymes or means for response to oxidative stress.
bacteria with lesser need for TCA cycle enzymes or means for response to oxidative stress.
Apart from structural roles in ribosome formation, several RPs act in multifunctional capacities (27). For example, the S9 protein is an accessory protein in DNA repair. The RP L25 (93 amino acids in E. coli) is homologous to the general stress protein (Ctc) of ≈200 amino acids in length. Ctc is present in the ribosome complex of BACSU (28). However, in almost all genomes, the Ctc and L25 protein genes are mutually exclusive. The L25 protein is PHX in proteobacteria but the longer Ctc generally does not qualify as PHX in the 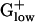 genomes (the exceptions are STAAU and LISMO).
genomes (the exceptions are STAAU and LISMO).
PA Genes Among 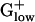 Genomes. The percentage of alien genes in
Genomes. The percentage of alien genes in 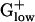 genomes is markedly low, <3.6% (with the exception being BACSU, 4.7%, Table 1) compared with Gram-negative genomes, which contain more PA genes (in the range of 4–10%). Numbers and types of PA genes differ significantly among
genomes is markedly low, <3.6% (with the exception being BACSU, 4.7%, Table 1) compared with Gram-negative genomes, which contain more PA genes (in the range of 4–10%). Numbers and types of PA genes differ significantly among 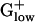 genomes. The differences putatively pertain to different evolutionary histories of the organisms and amount of lateral transfer and genomic flux. Many PA genes are unknown conserved proteins and most are hypothetical ORFs. The second most common PA types consist of variant transposase collections or prophage genes. Generally, PA clusters in all genomes are composed mainly of ORFs, transposases, phage-related proteins, uncharacterized regulatory proteins, methylation/restriction systems, and genes acting in pathogenicity. Other common PA genes include RNA methyltransferase, modification regulators, and two-component response regulators (cf. ref. 14).
genomes. The differences putatively pertain to different evolutionary histories of the organisms and amount of lateral transfer and genomic flux. Many PA genes are unknown conserved proteins and most are hypothetical ORFs. The second most common PA types consist of variant transposase collections or prophage genes. Generally, PA clusters in all genomes are composed mainly of ORFs, transposases, phage-related proteins, uncharacterized regulatory proteins, methylation/restriction systems, and genes acting in pathogenicity. Other common PA genes include RNA methyltransferase, modification regulators, and two-component response regulators (cf. ref. 14).
The BACHA alien genes are replete with transposases. By contrast, BACSU contains few transposase genes and none are PA. One PA cluster of BACHA (positions 3833–3845 kb) features several genes involved in exopolysaccharide biosynthesis. A PA cluster of capsular polysaccharide biosynthesis proteins exists in STRPN. Clusters of PA lipopolysaccharide biosynthesis genes often occur in Gram-negative bacteria (13, 14).
Both LISIN and LISMO contain the same cluster (putative operon) of PA genes, comprising four genes involved in methionine metabolism (homologs of 5-methyltetrahydrofolate-homocysteine methyltransferase, cystathionine β-lyase, cystathionine γ-synthase, and cobalamin-independent methionine synthase). Perhaps the whole methionine biosynthesis operon has been acquired in a relatively recent lateral transfer.
PHX ORFs. The majority of PHX genes are generally annotated, based on strong sequence similarity or experimental evidence. However, in most genomes, a small fraction of ORFs or poorly characterized hypothetical genes is also found among the PHX. Although some ORFs may be erroneously characterized as PHX due to limitations of our method, a PHX status of homologous ORFs in multiple genomes provides strong support for significant roles of these ORFs in the 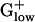 bacteria. The most notable examples of such ORFs (those PHX in three or more genomes) are listed in Table 6, which is published as supporting information on the PNAS web site). We propose that these genes may code for proteins which are highly relevant for these bacteria and are suitable targets for experimental manipulations.
bacteria. The most notable examples of such ORFs (those PHX in three or more genomes) are listed in Table 6, which is published as supporting information on the PNAS web site). We propose that these genes may code for proteins which are highly relevant for these bacteria and are suitable targets for experimental manipulations.
Discussion
The 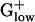 genomes are distinct from those of other bacterial groups in myriad ways: (i) The percent of genes encoded from the leading strand in
genomes are distinct from those of other bacterial groups in myriad ways: (i) The percent of genes encoded from the leading strand in 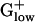 genomes pervasively exceeds 75%. This finding contrasts sharply with γ- and α-proteobacterial genomes, and high G+C Gram-positive genomes (29). (ii) BACSU and almost all
genomes pervasively exceeds 75%. This finding contrasts sharply with γ- and α-proteobacterial genomes, and high G+C Gram-positive genomes (29). (ii) BACSU and almost all 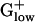 genomes contain PolC, the polymerase responsible for asymmetric synthesis of the leading strand during replication, whereas the DnaE enzyme synthesizes the lagging strand, unlike other genomes in which DnaE replicates both strands (29). (iii) The principal glycolysis genes (gap, pgk, tpi, pgm, and eno) of
genomes contain PolC, the polymerase responsible for asymmetric synthesis of the leading strand during replication, whereas the DnaE enzyme synthesizes the lagging strand, unlike other genomes in which DnaE replicates both strands (29). (iii) The principal glycolysis genes (gap, pgk, tpi, pgm, and eno) of 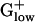 genomes are PHX and generally are encoded in an operon. This organization is not present in E. coli and most Gram-negative genomes. All genes of this operon are invariably PHX in BACSU, LISMO, LISIN, STAAU, CLOAC, and CLOPE with a PHX amino acid antiporter gene positioned between pgm and eno in the CLOPE genome. The same operon is also present in BACHA, but only with gap, pgk, and eno PHX, whereas tpi and pgm are near PHX levels [E(g) = 0.96 and 0.88, respectively]. Excepting tpi, the remaining four genes are transcribed in the same order in which they act in the glycolytic pathway. This glycolysis operon is missing from LACLA, STRPY, and STRPN. On the other hand, LACLA features a PHX cluster combining the glycolysis genes pfk (6-phosphofructokinase) and pyk (pyruvate kinase) with ldh (lactate dehydrogenase), all strongly PHX. In the genomes STRPY and STRPN, pfk and pyk are contiguous, whereas ldh is separated. Perhaps the association of ldh with glycolytic genes in a single operon reflects the importance of lactic acid fermentation in LACLA metabolism. (iv) Hexokinase, which converts glucose 6 to glucose-6-phosphate, is a prominent glycolysis gene functioning in most eukaryotes but is generally absent from prokaryotes. However, the
genomes are PHX and generally are encoded in an operon. This organization is not present in E. coli and most Gram-negative genomes. All genes of this operon are invariably PHX in BACSU, LISMO, LISIN, STAAU, CLOAC, and CLOPE with a PHX amino acid antiporter gene positioned between pgm and eno in the CLOPE genome. The same operon is also present in BACHA, but only with gap, pgk, and eno PHX, whereas tpi and pgm are near PHX levels [E(g) = 0.96 and 0.88, respectively]. Excepting tpi, the remaining four genes are transcribed in the same order in which they act in the glycolytic pathway. This glycolysis operon is missing from LACLA, STRPY, and STRPN. On the other hand, LACLA features a PHX cluster combining the glycolysis genes pfk (6-phosphofructokinase) and pyk (pyruvate kinase) with ldh (lactate dehydrogenase), all strongly PHX. In the genomes STRPY and STRPN, pfk and pyk are contiguous, whereas ldh is separated. Perhaps the association of ldh with glycolytic genes in a single operon reflects the importance of lactic acid fermentation in LACLA metabolism. (iv) Hexokinase, which converts glucose 6 to glucose-6-phosphate, is a prominent glycolysis gene functioning in most eukaryotes but is generally absent from prokaryotes. However, the 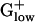 genomes distinguish PTS enzymes, with many PHX, to produce glucose-6-phosphate from other hexoses and from glucose transported into the cell by using a mechanism coupling phosphorylation to transmembrane transport. (v) The S1 giant RP gene is of reduced length, ≈380–410 codons (see Tables 2 and 4), compared with the S1 gene of Gram-negative bacteria whose length generally exceeds 500 amino acids. The S1 RP does not exist in archaea. (vi) Each
genomes distinguish PTS enzymes, with many PHX, to produce glucose-6-phosphate from other hexoses and from glucose transported into the cell by using a mechanism coupling phosphorylation to transmembrane transport. (v) The S1 giant RP gene is of reduced length, ≈380–410 codons (see Tables 2 and 4), compared with the S1 gene of Gram-negative bacteria whose length generally exceeds 500 amino acids. The S1 RP does not exist in archaea. (vi) Each 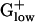 genome encodes only one copy of the translation elongation factor EF-Tu, whereas γ- and α-proteobacteria generally possess two or more gene copies.
genome encodes only one copy of the translation elongation factor EF-Tu, whereas γ- and α-proteobacteria generally possess two or more gene copies.
There are differences between E. coli and 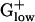 genomes in biosynthetic pathways that use TCA cycle genes. For example, E. coli uses succinyl-CoA in the biosynthesis of lysine and methionine, whereas BACSU uses acetyl-CoA. E. coli possesses isocitrate lyase, the first enzyme of the glyoxylate shunt, which competes with isocitrate dehydrogenase. This enzyme is effective for acquiring a carbon gain in the metabolism of fatty acids but most of the
genomes in biosynthetic pathways that use TCA cycle genes. For example, E. coli uses succinyl-CoA in the biosynthesis of lysine and methionine, whereas BACSU uses acetyl-CoA. E. coli possesses isocitrate lyase, the first enzyme of the glyoxylate shunt, which competes with isocitrate dehydrogenase. This enzyme is effective for acquiring a carbon gain in the metabolism of fatty acids but most of the 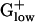 genomes lack isocitrate lyase. Aerobic and anaerobic lifestyles can be best distinguished with anaerobic-specific pathways. For anaerobic respiration, BACSU relies exclusively on nitrate or nitrite as its terminal electron acceptor, whereas E. coli has many alternative acceptors. Two nitroreductase homologs yodC and ydgI are PHX in BACSU but not in other
genomes lack isocitrate lyase. Aerobic and anaerobic lifestyles can be best distinguished with anaerobic-specific pathways. For anaerobic respiration, BACSU relies exclusively on nitrate or nitrite as its terminal electron acceptor, whereas E. coli has many alternative acceptors. Two nitroreductase homologs yodC and ydgI are PHX in BACSU but not in other 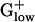 genomes.
genomes.
The predicted expression levels [E(g) values] correlate significantly with 2D gel assessments of protein abundances for organisms when such experimental data are available (12), and in some cases, our predictions have been confirmed by experiments (28, 30). However, the E(g) values have different interpretations than experimental data. The experimental measurements relate to protein or mRNA abundances in a specific environment. By contrast, the E(g) values relate to codon frequencies, which evolve over an extended period, presumably in adaptation to the conditions encountered by the organism. Thus, the E(g) values are not affected by laboratory cultivations and possible discrepancies between the experimental data, and the theoretical predictions may reflect differences in the cell metabolism or physiology under laboratory conditions and in the natural habitat.
Supplementary Material
Acknowledgments
We thank Drs. Bill Burkholder and Allan Campbell for helpful discussions and suggestions regarding the manuscript. This work was supported by National Institutes of Health Grants 5R01GM10452-37 and 5R01HG00335-14.
Abbreviations: PHX, predicted highly expressed;  , the low G+C group of Gram-positive bacteria; PTS, phosphotransferase system; LACLA, Lactococcus lactis; STRPY, Streptococcus pyogenes; STRPN, Streptococcus pneumoniae; LISMO, Listeria monocytogenes; LISIN, Listeria innocua; STAAU, Staphylococcus aureus; BACHA, Bacillus halodurans; BACSU, Bacillus subtilis; CLOAC, Clostridium acetobutylicum; CLOPE, Clostridium perfringens; RP, ribosomal protein, TF, translation/transcription processing factor; CH, chaperone and degradation; PA, putative alien.
, the low G+C group of Gram-positive bacteria; PTS, phosphotransferase system; LACLA, Lactococcus lactis; STRPY, Streptococcus pyogenes; STRPN, Streptococcus pneumoniae; LISMO, Listeria monocytogenes; LISIN, Listeria innocua; STAAU, Staphylococcus aureus; BACHA, Bacillus halodurans; BACSU, Bacillus subtilis; CLOAC, Clostridium acetobutylicum; CLOPE, Clostridium perfringens; RP, ribosomal protein, TF, translation/transcription processing factor; CH, chaperone and degradation; PA, putative alien.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ferretti, J. J., McShan, W. M., Ajdic, D., Savic, D. J., Savic, G., Lyon, K., Primeaux, C., Sezate, S., Suvorov, A. N., Kenton, S., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 4658-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tettelin, H., Nelson, K. E., Paulsen, I. T., Eisen, J. A., Read, T. D., Peterson, S., Heidelberg, J., DeBoy, R. T., Haft, D. H., Dodson, R. J., et al. (2001) Science 293, 498-506. [DOI] [PubMed] [Google Scholar]
- 3.Bolotin, A., Wincker, P., Mauger, S., Jaillon, O., Malarme, K., Weissenbach, J., Ehrlich, S. D. & Sorokin, A. (2001) Genome Res. 11, 731-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glaser, P., Frangeul, L., Buchrieser, C., Rusniok, C., Amend, A., Baquero, F., Berche, P., Bloecker, H., Brandt, P., Chakraborty, T., et al. (2001) Science 294, 849-852. [DOI] [PubMed] [Google Scholar]
- 5.Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., Bertero, M. G., Bessieres, P., Bolotin, A., Borchert, S., et al. (1997) Nature 390, 249-256. [DOI] [PubMed] [Google Scholar]
- 6.Takami, H., Nakasone, K., Takaki, Y., Maeno, G., Sasaki, R., Masui, N., Fuji, F., Hirama, C., Nakamura, Y., Ogasawara, N., et al. (2000) Nucleic Acids Res. 28, 4317-4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., et al. (2001) Lancet 357, 1225-1240. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu, T., Ohtani, K., Hirakawa, H., Ohshima, K., Yamashita, A., Shiba, T., Ogasawara, N., Hattori, M., Kuhara, S. & Hayashi, H. (2002) Proc. Natl. Acad. Sci. USA 99, 996-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.N ölling, J., Breton, G., Omelchenko, M. V., Makarova, K. S., Zeng, Q., Gibson, R., Lee, H. M., Dubois, J., Qiu, D., Hitti, J., et al. (2001) J. Bacteriol. 183, 4823-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlin, S. & Mrázek, J. (2000) J. Bacteriol. 182, 5238-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen, R., Bussemaker, H. J. & Gerstein, M. (2003) Nucleic Acids Res. 31, 2242-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karlin, S., Mrázek, J., Campbell, A. & Kaiser, D. (2001) J. Bacteriol. 183, 5025-5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karlin, S. (2001) Trends Microbiol. 9, 335-343. [DOI] [PubMed] [Google Scholar]
- 14.Mrázek, J. & Karlin, S. (1999) Ann. N.Y. Acad. Sci. 870, 314-329. [DOI] [PubMed] [Google Scholar]
- 15.Sengupta, J., Agrawal, R. K. & Frank, J. (2001) Proc. Natl. Acad. Sci. USA 98, 11991-11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teter, S. A., Houry, W. A., Ang, D., Tradler, T., Rockabrand, D., Fischer, G., Blum, P., Georgopoulos, C. & Hartl, F. U. (1999) Cell 97, 755-765. [DOI] [PubMed] [Google Scholar]
- 17.Fink, A. L. (1999) Physiol. Rev. 79, 425-449. [DOI] [PubMed] [Google Scholar]
- 18.Mrázek, J., Bhaya, D., Grossman, A. R. & Karlin, S. (2001) Nucleic Acids Res. 29, 1590-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajdic, D., McShan, W. M., McLaughlin, R. E., Savic, G., Chang, J., Carson, M. B., Primeaux, C., Tian, R., Kenton, S., Jia, H., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 14434-14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida, K. I., Fujita, Y. & Ehrlich, S. D. (2000) J. Bacteriol. 182, 5454-5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano, M. M., Dailly, Y. P., Zuber, P. & Clark, D. P. (1997) J. Bacteriol. 179, 6749-6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpousis, A. J. (2002) Biochem. Soc. Trans. 30, 150-155. [PubMed] [Google Scholar]
- 23.Hentze, M. W. & Kuhn, L. C. (1996) Proc. Natl. Acad. Sci. USA 93, 8175-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rouault, T. A. & Klausner, R. D. (1996) Trends Biochem. Sci. 21, 174-177. [PubMed] [Google Scholar]
- 25.Alen, C. & Sonenshein, A. L. (1999) Proc. Natl. Acad. Sci. USA 96, 10412-10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karlin, S. & Mrázek, J. (2001) Proc. Natl. Acad. Sci. USA 98, 5240-5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wool, I. G. (1996) Trends Biochem. Sci. 21, 164-165. [PubMed] [Google Scholar]
- 28.Schmalisch, M., Langbein, I. & Stulke, J. (2002) J. Mol. Microbiol. Biotechnol. 4, 495-501. [PubMed] [Google Scholar]
- 29.Rocha, E. (2002) Trends Microbiol. 10, 393-395. [DOI] [PubMed] [Google Scholar]
- 30.Karunakaran, K. P., Noguchi, Y., Read, T. D., Cherkasov, A., Kwee, J., Shen, C., Nelson, C. C. & Brunham, R. C. (2003) J. Bacteriol. 185, 1958-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.