

The neurodegenerative diseases underlying primary progressive aphasia have preferred but not invariant clinical associations. Mesulam et al. reveal that tauopathies most frequently lead to agrammatism, Alzheimer pathology to impaired word retrieval, and TDP type C pathology to impaired word comprehension. The common denominator is greater degeneration of the language-dominant hemisphere.
Keywords: Alzheimers disease, aphasia, ApoE e4, frontotemporal lobar degeneration, hemispheric lateralization
Abstract
Fifty-eight autopsies of patients with primary progressive aphasia are reported. Twenty-three of these were previously described (Mesulam et al., 2008) but had their neuropathological diagnoses updated to fit current criteria. Thirty-five of the cases are new. Their clinical classification was guided as closely as possible by the 2011 consensus guidelines (Gorno-Tempini et al., 2011). Tissue diagnoses included Alzheimer’s disease in 45% and frontotemporal lobar degeneration (FTLD) in the others, with an approximately equal split between TAR DNA binding protein 43 proteinopathies and tauopathies. The most common and distinctive feature for all pathologies associated with primary progressive aphasia was the asymmetric prominence of atrophy, neuronal loss, and disease-specific proteinopathy in the language-dominant (mostly left) hemisphere. The Alzheimer’s disease pathology in primary progressive aphasia displayed multiple atypical features. Males tended to predominate, the neurofibrillary pathology was more intense in the language-dominant hemisphere, the Braak pattern of hippocampo-entorhinal prominence was tilted in favour of the neocortex, and the APOE e4 allele was not a risk factor. Mean onset age was under 65 in the FTLD as well as Alzheimer’s disease groups. The FTLD-TAR DNA binding protein 43 group had the youngest onset and fastest progression whereas the Alzheimer’s disease and FTLD-tau groups did not differ from each other in either onset age or progression rate. Each cellular pathology type had a preferred but not invariant clinical presentation. The most common aphasic manifestation was of the logopenic type for Alzheimer pathology and of the agrammatic type for FTLD-tau. The progressive supranuclear palsy subtype of FTLD-tau consistently caused prominent speech abnormality together with agrammatism whereas FTLD-TAR DNA binding protein 43 of type C consistently led to semantic primary progressive aphasia. The presence of agrammatism made Alzheimer’s disease pathology very unlikely whereas the presence of a logopenic aphasia or word comprehension impairment made FTLD-tau unlikely. The association of logopenic primary progressive aphasia with Alzheimer’s disease pathology was much more modest than has been implied by results of in vivo amyloid imaging studies. Individual features of the aphasia, such as agrammatism and comprehension impairment, were as informative of underlying pathology as more laborious subtype diagnoses. At the single patient level, no clinical pattern was pathognomonic of a specific neuropathology type, highlighting the critical role of biomarkers for diagnosing the underlying disease. During clinical subtyping, some patients were unclassifiable by the 2011 guidelines whereas others simultaneously fit two subtypes. Revisions of criteria for logopenic primary progressive aphasia are proposed to address these challenges.
Introduction
Within the first decade of its delineation as a neurodegenerative syndrome, 63 new patients with primary progressive aphasia (PPA) had been reported in the world literature (Mesulam and Weintraub, 1992). Tissue information was available on 14 and revealed Alzheimer’s disease in some, Pick’s disease in others, and non-specific forms of focal atrophy in the majority. Since then, numerous accounts have illustrated the diversity of the neurodegenerative diseases underlying PPA and their complex relationships to the equally diverse patterns of language impairment. The probabilistic nature of these relationships, together with recent advances in the classification of both PPA (Gorno-Tempini et al., 2011) and frontotemporal lobar degenerations (FTLD) (Mackenzie et al., 2010, 2011), highlight the need to update the evolving clinicopathological correlations of this syndrome.
During the initial characterization of the PPA syndrome, the descriptive term ‘logopenic’ was introduced to designate a type of language impairment that seemed peculiar to PPA but no formal diagnostic criteria were proposed (Mesulam, 1982; Mesulam and Weintraub, 1992). The subsequent publication of the Neary consensus criteria had important implications for nomenclature in this field (Neary et al., 1998). Although the Neary criteria aimed to capture the clinical spectrum of frontotemporal lobar degenerations rather than the phenomenology of PPA, they triggered two major developments in the classification of progressive language disorders. First, they assigned the progressive non-fluent aphasia designation to all cases with progressive loss in the fluency of verbal expression. Second, the Neary et al. (1998) criteria defined semantic dementia as a syndrome with both word comprehension and object recognition impairments, without specifying whether the aphasic or agnosic component needed to be the leading feature.
Although these criteria were not designed to characterize PPA as a whole, their use for that purpose created inadvertent complications. First, the logopenic pattern of aphasia was not recognized as a distinct entity. Second, the semantic dementia designation also subsumed patients whose predominant problem was an associative agnosia rather than an aphasia and who could therefore not receive the PPA diagnosis. Thirdly, PPA patients with a neuropathology other than FTLD appeared implicitly excluded. All three of these problems were addressed by the 2011 international consensus guidelines (Gorno-Tempini et al., 2011): a logopenic variant was identified, inclusion into the semantic subgroup required prior fulfilment of the root PPA criteria, and no assumption was made about the nature of the underlying pathology. Investigations using this approach have reported successful implementation of these guidelines but with limitations in the form of unclassifiable patients and patients who simultaneously fulfil criteria for more than one subtype (Mesulam et al., 2012; Sajjadi et al., 2012; Harris et al., 2013; Mesulam and Weintraub, 2014; Wicklund et al., 2014). The Gorno-Tempini et al. (2011) guidelines also added impaired repetition as a core feature of the logopenic variant, a feature that was not part of the original description of logopenia (Mesulam, 1982), setting the stage for at least two different usages of the term. Nonetheless, these classification guidelines are being used and cited extensively.
The recent reclassification of FTLD has also had a major impact on clinicopathological correlations. In the first 14 PPA cases with autopsy or biopsy information, a non-Alzheimer’s disease ‘focal atrophy’ was the single most common finding (Mesulam and Weintraub, 1992). This type of pathology, also known as ‘dementia lacking distinctive histopathology’ (Knopman et al., 1990), has now been subdivided into numerous species of FTLD, each characterized by specific molecular and morphological patterns of proteinopathy. The two major classes of FTLD, and the ones most relevant to PPA, have been designated FTLD-tau and FTLD-TDP (Mackenzie et al., 2010). The former is characterized by non-Alzheimer tauopathies, the latter by abnormal precipitates of the 43 kD transactive response DNA binding protein TDP-43 (now known as TARDBP). Major FTLD-tau species include Pick’s disease, tauopathy of the corticobasal degeneration-type and tauopathy of the progressive supranuclear palsy (PSP) type, each identified according to the molecular forms and morphology of the hyperphosphorylated tau precipitates. FTLD-TDP is further subdivided into types A, B and C, and D depending on the distribution of the abnormal TDP-43 precipitates.
Much of the existing autopsy information in PPA is derived from isolated case studies. Only a few publications contain sizable series of consecutively autopsied PPA patients; fewer offer neurospychological detail; and still fewer include information on the asymmetry of pathology (Hodges et al., 2004; Kertesz et al., 2005; Forman et al., 2006; Knibb et al., 2006; Alladi et al., 2007; Grossman et al., 2008; Mesulam et al., 2008; Deramecourt et al., 2010; Grossman, 2012; Rohrer et al., 2012; Harris et al., 2013). This relative lack of comprehensive information and the ongoing changes in the classification of PPA and FTLD justify the current report of 58 consecutive autopsies of patients with PPA. They represent a combination of 35 new and 23 previously described but neuropathologically updated cases, the vast majority of which had tissue from both hemispheres so that the asymmetry of neuropathology could be investigated.
Materials and methods
All 58 cases had information on major language domains (word-finding, grammar, comprehension, naming) but only the 35 new cases had information on the additional domains required by the Gorno-Tempini et al. (2011) classification guidelines. Neuropathological associations of subtypes defined by these guidelines were therefore investigated only in the group of the 35 new cases. All other clinical, neuropathological, genetic and demographic analyses combined information from the full set of 58 cases. Whenever appropriate, statistical analyses were done through t-tests and Fisher’s Exact Test.
Neuropathological diagnoses
Macroscopic atrophy was determined through an inspection of the external surface of the hemispheres and of coronally cut slabs. Atrophy was rated as absent (0), mild (+), moderate (++), or severe (+++) (Bigio, 2013). Samples for histology were taken from homologous cortical areas of both hemispheres. They were processed with standard histological methods, the Gallyas stain, thioflavin-S, and immunohistochemistry with antibodies to phospho-tau, amyloid-β, TDP-43, p62, and alpha-synuclein. The severity of neuronal loss, the density of neurofibrillary tangles, neuritic plaques, TDP-43 precipitates and abnormal tauopathy (Pick bodies, astrocytic plaques, tufted astrocytes, etc.) was rated as absent, mild, moderate or severe (0, +, ++ or +++, respectively). Consensus criteria were used for the diagnoses of Alzheimer’s disease, diffuse Lewy body disease, FTLD-TDP (types A, B, C and D) and FTLD-tau (Pick’s disease-type, PSP-type, and corticobasal degeneration-type) (Mackenzie et al., 2010, 2011; Hyman et al., 2012; Montine et al., 2012; Bigio, 2013). The Alzheimer’s disease diagnosis included the Braak staging for neurofibrillary tangles and the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) scale for neuritic plaques. In addition to the 35 new cases, slides from the 2008 cohort were re-examined and classified according to the current criteria and nomenclature.
Clinical diagnoses in the new cohort
The root diagnosis of PPA was made on the basis of two features (Mesulam, 2001). First, the patient should have had the insidious onset and gradual progression of a language impairment (i.e. aphasia) manifested by deficits in word finding, word usage, word comprehension, or sentence construction. Secondly, the aphasia should have initially arisen as the most salient (i.e. primary) impairment and as the principal factor underlying the disruption of daily living activities. Evidence for this exclusionary component was provided by history and examination. Reliable informants were questioned about the presence of consequential forgetfulness, aberrant behaviours, visuospatial disorientation or object misuse. A structured survey of activities of daily living completed by the informant indicated impairment confined to areas dependent on language skills (Johnson et al., 2004). More quantitative data came from standardized assessments of executive function (Visual-Verbal Test, Tower of London Task, Go-NoGo Test, Trail Making Test), memory (Three Words-Three Shapes Test, WMS-III Faces, Rivermead Behavioural Memory Test) and visuospatial skills (Random Target Cancellation Test, Facial Recognition and Judgement of Line Orientation Tests) (Weintraub et al., 1990, 2012; Wicklund et al., 2004). Given the retrospective nature of chart review in a post-mortem series, not all patients had the same tests, but only those who had both historical and neuropsychological documentation for the relative preservation of non-language domains were included.
The subsequent subtyping of PPA in these 35 cases was guided, wherever possible, by the classification system of Gorno-Tempini et al. (2011). To fulfil the core and ancillary criteria of their classification system, charts were reviewed for information related to the status of speech, fluency of verbal output, grammar, repetition, naming, paraphasias, word comprehension, sentence comprehension, reading, spelling and object knowledge. As the 35 patients in this report were seen over a period of 15 years during which preferred methods of neuropsychological and clinical assessment were evolving, performance levels on different tasks assessing the same domain were translated into a common scale as described below so that each domain could be marked as ‘relatively preserved (0)’, ‘mildly abnormal (+)’ or ‘severely abnormal (++)’. The mixed usage of clinical and neuropsychological data may have introduced uneven implementation of the classification guidelines but this was unavoidable in a retrospective sample. In many of the cases, language function had been tested at several time points. In such instances, two evaluations are included to illustrate changes in the nature of the aphasia over time.
Speech
Dysarthria, laboured articulation, voice distortions and manifestations of speech apraxia such as errors of syllabic stress and duration were considered indicators of speech impairment (Josephs et al., 2006). Assessment of severity was qualitative.
Fluency
Assessment of this domain was based on the fluidity of speech as determined by the rate of word output. It reflected word finding (lexical retrieval) rather than speech (motor programming) impairments. A patient who appeared fluent when engaged in small talk and generalities but who displayed frequent word-finding hesitations when attempting to access infrequently used words was rated as having mildly impaired fluency. Output with consistent rather than intermittent word-finding pauses was rated as showing severe impairment of fluency. In some patients the level of severity was assessed qualitatively based on clinical notes. In others it was based on the quantification of words per minute during a taped narrative of the Cinderella story (Thompson et al., 1995, 2012; Mesulam et al., 2012).
Grammar
Aberrant sentence construction, as manifested by abnormal word order (syntax), distorted use of word endings, misuse of pronouns, and a paucity of small grammatical words (e.g. articles and prepositions) were considered indicative of impairment in this domain. Quotations of statements during the interview, or analysis of writing samples and emails contributed to the assessment of this domain. In some patients, the assessment was also based on the quantitation of grammatical sentences in the taped narrative of the Cinderella story or performance on the Northwestern Anagram Test (Weintraub et al., 2009). Patients who had occasional agrammatism in speech, those who had errors of grammar in writing but not in speech, and those whose Northwestern Anagram Test score or percentage of grammatical sentences were in the 80–60% correct range, were considered to have mild impairments of this domain. Those with more frequent and conspicuous errors (e.g. a patient whose description of the Cookie Theft included the statement ‘falling boy off stool’) or those with scores on the Northwestern Anagram Test <60% were rated as having severe impairments of this domain.
Repetition
Repetition was assessed clinically by asking the patient to repeat single words, meaningful multi-word sentences (e.g. ‘the little girl jumped over the fence’) or a string of grammatical function words (e.g. ‘no ifs ands or buts’). In some patients more quantitative evaluations were based on the Boston Diagnostic Aphasia Examination (BDAE) (Goodglass et al., 2001) or the Western Aphasia Battery—Revised (WAB-R) (Kertesz, 2006). Patients who could repeat simple meaningful sentences but not the string of function words, those who showed somewhat abnormal performance (80–60%) only on the low probability items of the BDAE and those whose performance on the six most difficult items in the repetition subtest of the WAB-R fell in the 80–60% range were classified as having a mild impairment of repetition. Those with deficits in repeating the meaningful multi-word sentence, or with repetition scores <60% on the WAB-R or BDAE low probability items were classified as having a severe impairment.
Naming
In the vast majority of patients this domain was quantified with the Boston Naming Test (Kaplan et al., 1983). Scores of 80–60% were considered indicative of mild impairment, and lower scores as indicative of severe impairment.
Paraphasic errors
These were qualitatively classified as mild or severe based on the frequency of occurrence and described as ‘semantic’ or ‘phonemic’ when the records contained sufficient information.
Single word comprehension errors
This domain was assessed qualitatively by asking the patient to define a word, point to an object denoted by a noun, or more quantitatively with the Peabody Picture Vocabulary Test (Dunn and Dunn, 2006). A Peabody Picture Vocabulary Test performance of 80–60% was classified as mildly abnormal whereas a lower score as severely abnormal.
Sentence comprehension errors
Some patients who had intact word comprehension performed poorly in the comprehension of sentences that were complex either because of length or because of non-canonical structure (If a tiger is eaten by a lion, which animal stays alive?). These abnormalities were classified as mild or severe based on clinical evaluations, occasionally supplemented by performance scores on the WAB-R and Boston Diagnostic Aphasia Examination sentence comprehension items.
Object knowledge
Object knowledge is one of the features that influence the Gorno-Tempini et al. (2011) classification algorithm. This domain was assessed qualitatively by asking the patient to describe the nature of objects they were asked to name, or more quantitatively with the three pictures form of the Pyramids and Palm Trees Test (Howard and Patterson, 1992). Additional information was obtained by asking informants for evidence of object misuse in daily activities. Only one patient (Patient P23) had an impairment of this domain as indicated by performance distinctly <80% on the Pyramids and Palm Trees Test.
Resolving classification problems
The Gorno-Tempini et al. (2011) classification guidelines make it possible for the same patient to fulfil guidelines for both logopenic and agrammatic PPA. For example, an agrammatic patient with spared word and object knowledge would fulfil the agrammatic PPA criteria. The same patient could also fit the logopenic PPA criteria by additionally displaying the two core criteria of word-finding and repetition impairments, and the three ancillary criteria of spared word and object knowledge, spared motor speech, and phonemic paraphasias. In such cases (return visit of Patient P14, initial visit of Patient P15, return visit of Patient P20, initial visit of Patient P22, return visit of Patient P29), we classified the patient as having agrammatic PPA, with the assumption that the agrammatism was the defining feature of the aphasia. Two additional patterns were unclassifiable by the 2011 guidelines. In one type the patient had equally prominent agrammatism and single word comprehension impairments. We classified such patients as having a mixed form of PPA as previously described (Mesulam et al., 2012). In the second and more frequent type of circumstance, the patient was clinically logopenic but lacked the repetition impairment, a pattern that is unclassifiable by the 2011 guidelines. These patients were designated PPA-L* and set apart from patients who also had the impaired repetition required by the 2011 guidelines and who were designated PPA-L. The PPA-L* designation in this report therefore indicates a patient who is descriptively ‘logopenic’ according to the way the term was defined when it was first introduced, but who remains unclassifiable by the Gorno-Tempini et al. (2011) criteria.
Results
Multiple neuropathological entities were encountered in the total set of the 58 cases, which included the current (Patients P1–35) and the 2008 (Patients X1–23) cohorts (Tables 1–3). When the two cohorts are considered collectively (but with the exclusion of Patients P15 and P16 who had mixed pathologies), 45% of the 56 patients with a single primary pathology had Alzheimer’s disease and 55% non-Alzheimer’s disease pathology. In the non-Alzheimer’s disease group, FTLD-TDP (n = 14) and FTLD-tau (n = 17) were approximately equally represented. The most frequent TDP pathology was of the A type (7 of 15) and the most frequent tau pathology of the corticobasal degeneration type (8 of 17).
Table 1.
Clinical features of Patients P1–P22
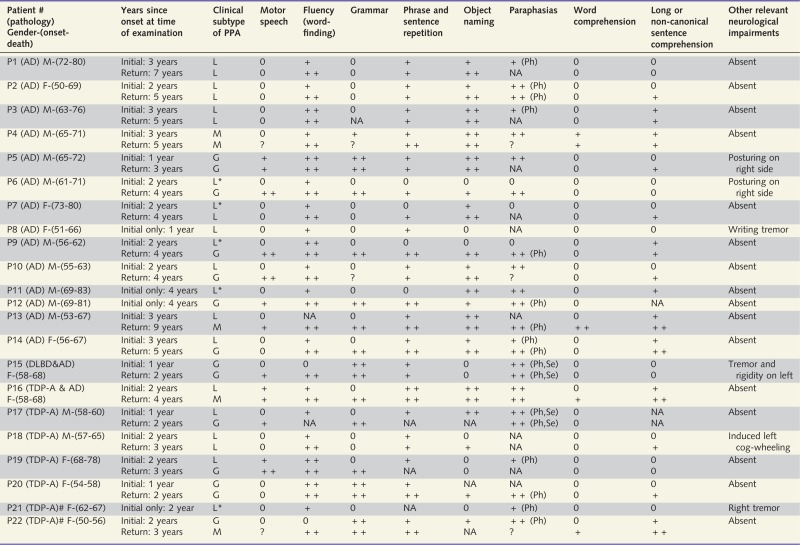 |
The level of impairment in domains is denoted as follows: 0, preserved domain; +, mildly impaired domain; ++, severely impaired domain. #, Patient with GRN mutation.
AD = Alzheimer pathology; DLBD = diffuse Lewy body disease; G = agrammatic PPA; L = logopenic PPA by the 2011 classification; L* = logopenic PPA without repetition impairment and therefore unclassifiable by the 2011 guidelines; M = mixed PPA characterized by combined impairment of grammar and comprehension; NA = information not available; Ph = phonemic paraphasia; Se = semantic paraphasia; TDP-A = frontotemporal lobar degeneration with transactive response DNA-binding protein 43 precipitates of type A; ? = the patient was not able to produce sufficient verbal or written language to judge that domain.
Table 2.
Clinical features of Patients P23–P35
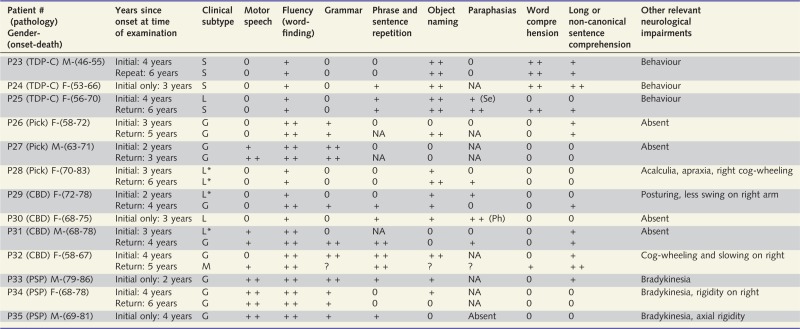 |
The level of impairment in domains is denoted as follows: 0, preserved domain; +, mildly impaired domain; ++, severely impaired domain. Although the results are not shown in Table 2, Patients P23–25 fulfilled the core and ancillary criteria for the diagnosis of semantic PPA through additional tests of object knowledge and surface dysgraphia/dyslexia.
CBD = frontotemporal lobar degeneration of the corticobasal degeneration type; G = agrammatic PPA; L = logopenic PPA by the 2011 classification; L* = logopenic PPA without repetition impairment and therefore unclassifiable by the 2011 guidelines; M = mixed PPA characterized by combined impairment of grammar and comprehension; NA = information not available; Pick = tauopathy of the Pick type; PSP = tauopathy of the progressive supranuclear palsy type; Ph = phonemic paraphasia; S = semantic PPA; Se = semantic paraphasia; TDP-C = frontotemporal lobar degeneration with transactive response DNA binding protein 43 precipitates of type C; ? = the patient was not able to produce sufficient verbal or written language to judge that domain.
Table 3.
Characteristics of patients in the Mesulam et al. (2008) cohort with updated neuropathological classification
| AD | TAU | TAU- CBD | TAU- PiD | TDP- A | TDP-B | |
|---|---|---|---|---|---|---|
| PPA-L/L* (n = 11) | 7 | 1 | 0 | 0 | 2 | 1 |
| PPA-G (n = 6) | 0 | 1 | 3 | 1 | 1 | 0 |
| PPA-S (n = 1) | 1 | 0 | 0 | 0 | 0 | 0 |
| PPA-M (n = 5) | 3 | 0 | 1 | 0 | 0 | 1 |
AD = Alzheimer pathology; PPA-G = agrammatic PPA; PPA-L/L* = patients who were classified as logopenic in a descriptive sense, regardless of the status of repetition, representing a mixture of PPA-L and PPA-L*; PPA-M = mixed PPA characterized by combined impairment of grammar and comprehension; PPA-S = semantic PPA; TAU = frontotemporal lobar degeneration with otherwise unspecified tauopathy; TAU-CBD = frontotemporal lobar degeneration with tauopathy of the corticobasal degeneration-type; TAU-PiD = frontotemporal lobar degeneration with tauopathy of the Pick-type; TDP-A, B = types A and B of frontotemporal lobar degeneration with transactive response DNA-binding protein 43 precipitates.
This cohort of 23 patients did not contain cases with TDP-C or TDP-D.
Gender, age of onset and duration in the combined cohorts
In the combined set of 56 patients with a single primary pathology, the frequency of males was higher in the Alzheimer’s disease (64%) than in the TDP (35%) or tau (47%) groups but the differences did not reach statistical significance (Table 4). Mean age of onset, disease duration and age at death were lower in the TDP group. The TDP versus tau comparison for age of onset (P = 0.027), the TDP versus Alzheimer’s disease comparison for disease duration (P = 0.009), and the TDP versus Alzheimer’s disease and tau comparisons for age at death (P ≤ 0.001) were all significantly different. There were no significant differences in age of onset, duration, or age at death between the Alzheimer’s disease and tau groups. In all three groups, mean age of onset was <65 years (Table 4). Gender did not influence age of onset, age at death or duration of illness.
Table 4.
Gender, onset, duration and ApoE4 frequencies in the new and Mesulam et al. (2008) cohorts combined
| Gender | Onset age | Duration | ApoE4 | |
|---|---|---|---|---|
| AD (n = 25) | 64% M, 36% F | 61.5 ± 9.0 | 11.0 ± 4.9 | 30% |
| FTLD-TDP (n = 14) | 35% M, 65% F | 57.1 ± 6.0 | 7.4 ± 3.4 | 25% |
| FTLD-tau (n = 17) | 47% M, 53% F | 63.8 ± 8.3 | 9.9 ± 3.0 | 20% |
| Combined non-AD (n = 31) | 39% M, 61% F | 60.7 ± 8.0 | 8.7 ± 3.4 | 22% |
The ApoE4 percentages indicate the proportion of patients in a given group with at least one ApoE4 allele. Patients P15 and P16 are excluded because of combined pathologies.
AD = Alzheimer’s disease.
Apolipoprotein E genotypes in the combined cohorts
Apolipoprotein E (ApoE) genotyping was available in 90% of the cases. In the 56 cases with a single primary pathology included for analysis (as noted above, Patients P15 and 16 were excluded because of multiple pathologies), the frequency of an ApoE4 allele was 30% for the Alzheimer’s disease group, 25% for the FTLD-TDP group and 20% for the FTLD-tau group. At the Northwestern Alzheimer’s Disease Brain Bank, the frequency of cases with at least one E4 allele was 59% in a set of 75 patients with the typical amnestic dementia of confirmed Alzheimer’s disease, and 26% in a set of 190 neurologically intact subjects. None of the PPA groups was significantly different from control or from one another and all three were significantly lower in E4 frequency than the amnestic Alzheimer’s disease group. These results confirm, as we have suggested in the past, that E4 is not a risk factor in PPA even when it is caused by Alzheimer’s disease pathology (Rogalski et al., 2011; Gefen et al., 2012).
From neuropathology to clinical phenotype: preferred clinical expressions of pathology types in the new cohort
Information on all parameters required for the subtyping of PPA by the Gorno-Tempini et al. (2011) guidelines was available in the new cohort of 35 patients. Initial clinical evaluation occurred within 4 years of reported onset in all of these patients, and within 2 years in 18 of them. Twenty-seven of the 35 patients had at least two evaluations separated by 1 year or more (Tables 1 and 2).
Alzheimer’s disease
In the group of 14 patients with Alzheimer’s disease as the only primary pathology (Patients P1–14), 78% had the PPA-L (n=7) or PPA-L* (n=4) pattern at the initial examination. This favoured logopenic pattern of clinical expression indicates that the type of Alzheimer pathology that causes PPA tends to spare areas critical for grammar and word comprehension at the initial stages of the disease. However, two patients with Alzheimer pathology did have the agrammatic PPA pattern at the initial examination (at 1 and 4 years after onset) and a third had the combination of agrammatism and comprehension impairment of the mixed PPA pattern at the initial examination (3 years into the disease). Seven of the 11 patients with an initial PPA-L or PPA-L* diagnosis had a follow-up evaluation and four of these (two in each logopenic group) progressed to agrammatic PPA at the second visit. Motor aspects of speech and single word comprehension were almost always preserved at the initial examination. Word-finding or naming impairments were universally present. Ancillary neurological impairments were rare and consisted of induced right upper extremity posturing in two patients and writing tremor in one.
Frontotemporal lobar degeneration-TDP
The TDP-A group (Patients P17–22) had a clinicopathological correspondence pattern similar to that of the Alzheimer’s disease group. The presenting clinical profile was logopenic PPA or logopenic PPA without repetition impairment in four of six cases and agrammatic PPA in the others. In two of five cases with follow-up evaluations, the initial logopenic pattern progressed to agrammatic PPA. In the one left-handed patient with known right hemisphere language dominance (Patient P18), cog-wheeling was noted in the left upper extremity. Patient P21 (right handed) had a tremor in the right upper extremity. One of the two patients with GRN mutations (Patients P21 and P22) presented with logopenic PPA without repetition impairment and the other with severe agrammatism characteristic of the agrammatic PPA type.
The three patients in the TDP-C group (Patients P23–25) were the only patients with severe single word comprehension impairments on a background of relatively preserved speech and grammar, either at the initial encounter or at follow-up. Two had the profile of semantic PPA at the initial visit. The third (Patient P25) had a logopenic PPA pattern with an unusually severe anomia at the initial visit. Such a prodromal ‘anomic’ stage of semantic PPA has been described previously (Mesulam et al., 2012). Severe anomia, out of proportion to the severity of other aphasic impairments was seen in all three cases of TDP-C. No ancillary motor findings were noted but all three patients displayed new compulsive and disinhibited behaviours as the disease progressed.
No TDP-B or TDP-D pathologies were encountered in the new cohort of 35 cases. In the 2008 cohort, two cases had TDP-B pathology. One of these patients presented with the mixed PPA pattern and dysarthria and eventually developed signs of motor neuron disease. The second had the logopenic PPA without repetition impairment pattern 3 years after symptom onset and then progressed to an agrammatic PPA pattern but without signs or symptoms of motor neuron disease.
Frontotemporal lobar degeneration-tau
The overall pattern in the FTLD-tau group (Patients P26–35) was quite different and was dominated by the agrammatic PPA subtype. In 6 of 10 cases the initial aphasia type was agrammatic PPA. In the remaining four cases, PPA-L or PPA-L* was the initial type but progressed to agrammatic PPA in two. The one patient with the persistent PPA-L* pattern and Pick’s disease at autopsy (Patient P28) had an unusual clinical picture characterized by severe acalculia and dysgraphia to the point where she was initially suspected of having a left parietal stroke. She eventually developed severe apraxia and right-sided extrapyramidal impairments reminiscent of the corticobasal syndrome. Because of this clinical picture, Pick’s disease was never suspected. The three PSP-type FTLD-tau cases stood out with a pattern where the speech abnormality, including components of speech apraxia, was nearly as prominent as the aphasic impairment. Only two of four corticobasal degeneration-type FTLD-tau cases, both right-handed, had mild right-sided motor signs. Motor findings were more prominent in the PSP group but without ophthalmoplegia. The three patients with Pick-type FTLD-tau also displayed mild obsessive-compulsive behaviours but no disinhibited behaviours of the type seen in patients with TDP-C.
Mixed pathologies
The one patient with diffuse Lewy body disease and Alzheimer’s disease (Patient P15) had the agrammatic PPA pattern at presentation and follow-up. She was left-handed and developed tremor and rigidity on the left side. The patient with the neuropathology of both Alzheimer’s disease and TDP-A presented with logopenic PPA and rapidly progressed to mixed PPA with no additional motor impairments.
How useful are subtypes and symptoms for inferring underlying pathology types?
In the subset of 35 new cases where sufficient information was available for classification according to the Gorno-Tempini et al. (2011) guidelines, an initial diagnosis of agrammatic PPA was more predictive of FTLD-tau than either of the other two pathologies, with a sensitivity of 60% and specificity of 80%. An initial diagnosis of the logopenic PPA subtype according to these criteria was more predictive of Alzheimer’s disease than of either FTLD-TDP or FTLD-tau. However, the sensitivity of this clinical diagnosis for detecting Alzheimer’s disease was only 50% and its specificity for differentiating Alzheimer’s disease versus non-Alzheimer’s disease pathology was 71%. A persistent logopenic PPA pattern detected both at presentation and at follow-up 5–7 years later was seen in three cases (Patients P1–3), all of who had the pathology of Alzheimer’s disease.
Given the laborious nature of subtype classification, the question was asked whether a simpler process based on the status of two orthogonal symptoms, word comprehension and grammar, might be as informative. This approach had been used previously to subdivide the language impairments of PPA into agrammatic, logopenic, semantic, and mixed types, each with a distinctive pattern of peak atrophy sites (Mesulam et al., 2009, 2012). This procedure allowed us to make use of the 2008 cohort as well, as the grammar and word comprehension abilities of the patients were known. The resultant template incorporated all 58 patients (Fig. 1). The ‘agrammatic’ and ‘semantic’ quadrants overlapped completely with the agrammatic PPA and semantic PPA groups identified by the more elaborate Gorno-Tempini et al. (2011) guidelines, the ‘mixed’ PPA quadrant included patients who would have remained unclassifiable by these guidelines, and the ‘logopenic’ quadrant included patients not only with repetition impairment (as required by these guidelines) but also without repetition impairment (who would have remained unclassifiable). All patients in the ‘logopenic’ quadrant of Fig. 1 were aphasic as manifested by prominent word retrieval impairments. This quadrant also contained the largest number of patients whose clinical pattern changed over time and who evolved from a logopenic pattern into agrammatic, semantic and mixed patterns of aphasia.
Figure 1.

Clinicopathological correlations. A template for classifying all 58 autopsy cases on the basis of single word comprehension and grammaticality of verbal output. All patients had fulfilled the criteria for the root diagnosis of PPA. Classification is based on the single available clinical evaluation for the 23 cases of the Mesulam et al. (2008) cohort and the initial evaluation for the new cohort of 35 cases. The ‘logopenic’ group includes aphasias characterized by word retrieval and naming impairments (regardless of repetition status) but without grammar or word comprehension abnormalities. It therefore incorporates both logopenic PPA and logopenic PPA without repetition impairment. The ‘agrammatic’ group is essentially identical to the agrammatic PPA designation according to the Gorno-Tempini et al. (2011) guidelines. The percentages indicate the distribution of pathology types. AD = Alzheimer pathology; DLBD = diffuse Lewy body disease.
Based on this symptomatic approach, the template in Fig. 1 shows that the preservation of both comprehension and grammar (which captures the combined set of logopenic PPA and logopenic PPA without repetition impairment) was most predictive of Alzheimer’s disease pathology (with sensitivity of 56% and specificity of 58%) whereas the presence of agrammatism on a background of preserved comprehension (which captures all agrammatic PPA) was most predictive of FTLD-tau (with a sensitivity of 65% and specificity of 85%). The template in Fig. 1 also showed that Alzheimer’s disease is the most likely pathology associated with mixed PPA and that TDP-C is the most likely pathology associated with semantic PPA. The presence of agrammatism made Alzheimer’s disease pathology unlikely, whereas the presence of a logopenic aphasia or word comprehension impairment made FTLD-tau unlikely. The classification based on this template is therefore as informative of underlying neuropathology as the classification according to the Gorno-Tempini et al. (2011) guidelines.
The status of grammar separated the 49 patients with preserved comprehension into two populations that had significantly different frequencies of underlying neuropathology (Fisher’s exact test, P = 0.001). When grammar was impaired, FTLD-tau was more than twice as common as Alzheimer’s disease or FTLD-TDP pathology. When grammar was preserved, Alzheimer’s disease was more than twice as common as FTLD-tau or FTLD-TDP. The vast majority of the 49 patients with preserved comprehension (top two quadrants of Fig. 1) would have fit the progressive non-fluent aphasia designation of the Neary et al. (1998) criteria. The significantly different distribution of underlying pathologies in the two populations provides additional justification for subdividing progressive non-fluent aphasia into agrammatic and logopenic variants.
Asymmetry of neuropathology
Tissue was available for an analysis of asymmetry in 31 of 35 new cases (Table 5). Twenty-eight of these (90%) had consistently greater atrophy, more neuronal loss or more abnormal protein precipitates (neurofibrillary tangles, neuritic plaques, TDP-43 or tau-positive inclusions) in the language-dominant hemisphere (left hemisphere in 26 right-handed subjects and right hemisphere in two left-handed subjects). In one of the left-handed subjects (Patient P18) with known right hemisphere dominance for language (Mesulam et al., 2005) and FTLD-TDP at autopsy, the superficial atrophy and neuronal loss was distinctly greater in the language-dominant right hemisphere although the TDP precipitates did not show consistent asymmetry. In some of the cases with Alzheimer’s disease, the neurofibrillary tangle distribution was not only skewed to the left but also deviated from the Braak pattern of hippocampo-entorhinal predominance (Figs 2 and 3). In Patient P9 quantitative MRI had been obtained 7 months before death and revealed a close correspondence between neurofibrillary tangle numbers and sites of peak atrophy in the left hemisphere (Fig. 3) (Gefen et al., 2012). Asymmetry in the distribution of neurodegenerative markers was also seen in cases of FTLD-TDP and FTLD-tau (Fig. 4).
Table 5.
Patterns of asymmetry
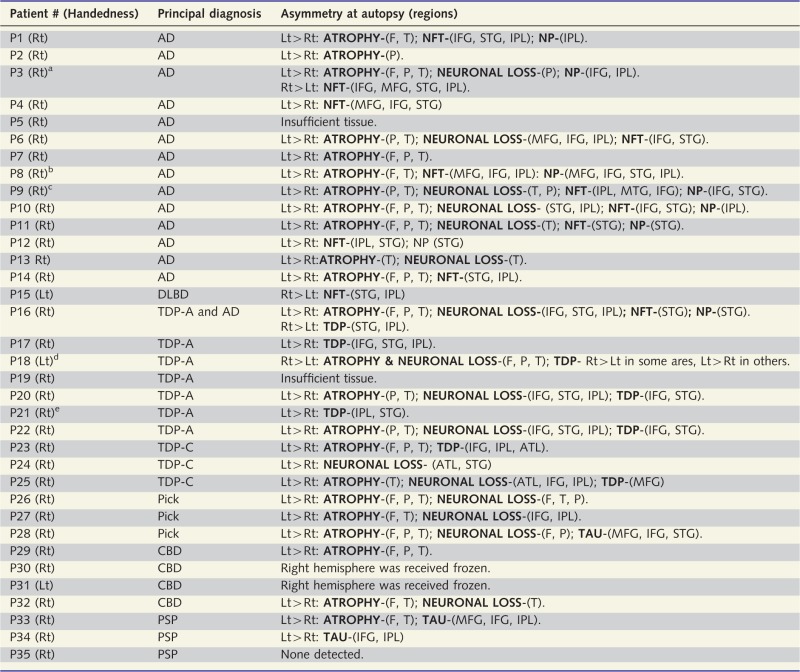 |
The one common denominator that cuts across all pathology types is the frequently asymmetric degeneration of the language-dominant hemisphere.
AD = Alzheimer pathology; ATL = anterior temporal lobe; DLBD = diffuse Lewy body disease; CBD = frontotemporal degeneration with tauopathy of the corticobasal degeneration type; F = frontal lobe; IFG = inferior frontal gyrus; IPL = inferior parietal lobule; Lt = left; MFG = middle frontal gyrus; NFT = neurofibrillary tangles of the Alzheimer-type; NP = neuritic amyloid plaques; TAU = markers of tauopathy; TDP = abnormal TDP-43 precipitates; P = parietal lobe; PSP = frontotemporal lobar degeneration with tauopathy of the progressive supranuclear palsy type; PiD = frontotemporal lobar degeneration with tauopathy of the Pick type; Rt = right; STG = superior temporal gyrus; T = temporal lobe; TDP-A, B, C = frontotemporal degeneration with transactive response DNA binding protein 43 precipitates of types A, B or C.
aNeurofibrillary tangle and neuritic plaque counts reported as Patient P3 in Gefen et al. (2012).
bNeurofibrillary tangle and neuritic plaque counts reported as Patient P2 in Gefen et al. (2012).
cNeurofibrillary tangle and neuritic plaque counts reported as Patient P7 in Gefen et al. (2012).
dEvidence for right hemisphere language dominance in this patient was reported in Mesulam et al. (2005).
eTDP counts reported in Gliebus et al. (2010).
Figure 2.

Atypical distribution of Alzheimer pathology in Patient P6. The photomicrographs show neurofibrillary tangles and neuritic plaques in thioflavin-S stained tissue. Magnification is ×100 except in the entorhinal area where it is ×40. Lesions are much denser in the language-dominant left superior temporal gyrus (STG). Furthermore, the principles of Braak staging do not apply in any strict fashion as neocortex contains more lesions than entorhinal cortex and the CA1 region of the hippocampus.
Figure 3.

Atypical distribution of Alzheimer pathology in Patient P9. Top: Quantitative imaging within 7 months before death shows focal peak atrophy sites in the left temporoparietal junction (TPJ). Bottom: The number of neurofibrillary tangles per cubic millimetre is greater in language-related neocortical areas than in entorhinal cortex (ENTO) and more in the language-dominant left hemisphere than in the right. Data taken from Gefen et al. (2012). PPA-L* = logopenic PPA with intact repetition at the initial evaluation 2 years after onset; STG = superior temporal gyrus.
Figure 4.

Asymmetry of proteinopathy in frontotemporal lobar degenerations causing PPA. (A) Number of abnormal TDP-43 precipitates in Patient P21 in posterior inferoparietal cortex (PIPL), anterior inferoparietal cortex (AIPL), superior temporal gyrus (STG), inferior temporal gyrus (ITG) and entorhinal cortex (EC). Data taken from Gliebus et al. (2010). (B) Asymmetry of tauopathy shown by immunohistochemistry in the inferior frontal gyrus (IFG) of Patient P28 with FTLD-tau (Pick-type). (C) Asymmetry of tauopathy shown by immunohistochemistry in the inferior frontal gyrus of Patient P29 with FTLD-tau (corticobasal degeneration-type). (D) Tau-positive astrocytic plaque characteristic of corticobasal degeneration (CBD) pathology in Patient P29.
Focal and prominent asymmetrical atrophy of dorsal frontoparietal areas in the language-dominant hemisphere was frequently seen in Alzheimer’s disease, TDP-A, corticobasal degeneration and Pick pathologies without distinguishing features that differentiated one disease type from another (Fig. 5). In some cases the atrophy was so focal and severe that it raised the suspicion of a cerebrovascular lesion at the time of brain removal. TDP-C had a distinctive pattern of asymmetrical anterior temporal lobe atrophy. Surface atrophy appeared relatively mild in PSP.
Figure 5.

Similar appearance of asymmetry in PPA caused by Alzheimer’s disease and FTLD-tau. Arrows point to areas of prominent atrophy in the left inferior frontal gyrus of Patient P7 who had Alzheimer pathology (A) and Patient P29 who had frontotemporal lobar degeneration with tauopathy of the corticobasal degeneration-type (B). The asterisks mark the relatively spared contralateral inferior frontal gyrus of the right hemisphere. AD = Alzheimer’s disease; CBD = corticobasal degeneration.
Two cases had conflicting patterns. Patient P16 (right-handed) with primary diagnoses of both FTLD-TDP (type A) and Alzheimer’s disease had more atrophy, neuronal loss and Alzheimer’s disease markers (neurofibrillary tangles and neuritic plaques) in the left hemisphere but more TDP-43 precipitates in the right (Fig. 6). In Patient P3 who was also right-handed and had Alzheimer’s disease pathology as the primary diagnosis, atrophy was more pronounced and neuritic plaques were more numerous in the left hemisphere but the neurofibrillary tangles were more pronounced in the right hemisphere. In both of these cases with conflicting patterns in vivo imaging (single-photon emission computed tomography in Patient P3 and MRI in Patient P16) had shown greater hypoperfusion and atrophy in the left.
Figure 6.

Conflicting asymmetry in PPA with TDP-type A and Alzheimer’s disease pathologies in right-handed Patient P16. Top: TDP-43 precipitates show rightward preponderance in the superior temporal gyrus (STG). Bottom: Thioflavin-S positive neurofibrillary tangles and neuritic plaques of Alzheimer pathology show the reverse asymmetry, in a pattern that is more concordant with the aphasic phenotype in a right-handed person. AD = Alzheimer’s disease.
In the case with mixed diffuse Lewy body disease and Alzheimer’s disease pathology (Patient P15, left-handed) there were more neurofibrillary tangles in the right hemisphere, but no asymmetry of Lewy bodies or neurites. It is interesting to note that in both cases of mixed pathology (Patients P15 and P16), the neurofibrillary tangles rather than the proteinopathy of the additional pathological entity showed the most predilection for the language-dominant hemisphere. In Patient P35 neither the external examination of the brain at autopsy nor the histological sections revealed asymmetry, but the MRI had shown greater frontal and temporal atrophy on the left. In the Mesulam et al. (2008) cohort, 12 of 19 cases with sufficient tissue showed similar leftward asymmetries of atrophy and other markers of neuropathology.
Discussion
The post-mortem examination of 58 consecutive PPA autopsies, including 35 new cases and 23 previously reported cases reanalysed to meet the most current neuropathological classification standards, revealed nine distinct neuropathological entities: Alzheimer disease, diffuse Lewy body disease, TDP-A (with and without GRN mutations), TDP-B, TDP-C, and FTLD-tau of the Pick-, corticobasal degeneration- and PSP-types. The diffuse Lewy body disease case and one of the TDP-A cases also had Alzheimer pathology. Each of these neuropathological patterns, including the joint presence of diffuse Lewy body disease and TDP-A with Alzheimer pathology has been reported in conjunction with PPA in previously published case reports and autopsy series (Caselli et al., 2002; Hodges et al., 2004; Knibb et al., 2006; Mesulam et al., 2008; Grossman, 2012; Harris et al., 2013; Perry et al., 2013).
The availability of tissue from both hemispheres in the vast majority of cases allowed us to show that the one unifying common denominator was the greater severity of the atrophy, neuronal loss and disease-specific proteinopathy in the language-dominant hemisphere. It is remarkable that the asymmetry of neurodegeneration persisted into the time of autopsy, many years after the onset of the selective aphasic phenotype. Asymmetry of neurodegeneration is therefore the core feature of PPA not only at disease onset but also as the disease progresses. This asymmetry cannot be attributed to the cellular or molecular nature of the underlying disease as it was observed in all pathology types.
The nature of the putative patient-specific susceptibility factors that underlie the asymmetry of neurodegeneration in PPA remains unknown. One potential clue emerged from the discovery that PPA patients had a higher frequency of personal or family history of learning disability, including dyslexia, when compared to controls or patients with other dementia syndromes (Rogalski et al., 2008; Miller et al., 2013). Patient P1 (Case 4 in Rogalski et al., 2008), for example, was dyslexic and had three dyslexic sons who had difficulty completing high school, but who then proceeded to build successful careers as adults. The association with learning disability and dyslexia led to the speculation that PPA could reflect the tardive manifestation of a developmental or genetic vulnerability of the language network that remains compensated during much of adulthood but that eventually becomes the locus of least resistance for the expression of an independently arising neurodegenerative process. The same neurodegenerative process would presumably display different anatomical distributions, and therefore different phenotypes, in persons with different vulnerability profiles, explaining why identical genetic mutations of GRN or MAPT can display such heterogeneity of clinical expression.
Conceivably, some of the genetic risk factors linked to dyslexia could interact with the primary neurodegenerative process and enhance its impact on the language network (Rogalski et al., 2013). Such inborn risk factors could promote dyslexia as a developmental event in some family members and PPA as a late degenerative event in others. Interestingly, some of the candidate genes for dyslexia do seem to have an influence on the asymmetry of cortical function. For example, healthy subjects bearing the molecular variants of KIAA0319/TTRAP/THEM2 previously identified as enhancing the risk of dyslexia showed a reduced left-hemispheric asymmetry of functional activation in the superior temporal sulcus during a reading task (Pinel et al., 2012). Several genes are known to be differentially expressed in the left and right hemispheres and could presumably also influence the asymmetric vulnerability to neurodegeneration (Sun et al., 2005). Although mutations in the forkhead box P2 gene (FOXP2) have been linked to speech and language impairment, PPA and controls have not shown differences in the frequencies of at least two polymorphisms of this gene (Premi et al., 2012). The identification of factors underlying the asymmetry of atrophy in PPA would have considerable relevance for understanding the general principles that influence selective vulnerability in neurodegenerative diseases.
The peculiarities of Alzheimer pathology in primary progressive aphasia
In ‘typical’ Alzheimer’s disease, the hippocampo-entorhinal region bears the brunt of the neurodegeneration, ApoE4 is a major risk factor, no consistent hemispheric asymmetry is present, symptoms usually emerge after the age of 65, females tend to be overrepresented, and memory loss (amnesia) tends to be the most common salient impairment.
None of these ‘typical’ features could be identified in the group of PPA patients with Alzheimer’s disease at autopsy. Mean onset in this group was under 65 years of age, males were slightly more numerous, ApoE4 was not a risk factor, amnesia was not present during the initial years, and the distribution of neurodegeneration was asymmetrical. In some cases, there were more neurofibrillary tangles in language-related neocortices than in the hippocampo-entorhinal complex, a pattern that does not even fit the principles of Braak staging (Gefen et al., 2012).
The Alzheimer’s disease that causes PPA is therefore biologically, anatomically and clinically distinct from the typical late-onset Alzheimer’s disease. It is becoming increasingly clear that Alzheimer’s disease is not a unitary disease and that it has distinct subtypes, such as the one that causes PPA. Other Alzheimer’s disease ‘subtypes’ include frontal-type dementias and the progressive visuospatial impairments of posterior cortical atrophy. In the former, neurofibrillary tangles can be more numerous in the frontal lobes than in the entorhinal cortex whereas in the latter the neurofibrillary tangles show unusually high concentrations in occipito-parietal cortex and the superior colliculus (Hof et al., 1997; Johnson et al., 1999). It is interesting to note that in all three of these atypical forms, the clinical phenotype more closely reflects the anatomical distributions of the neurofibrillary tangles than of the amyloid plaques. In keeping with these observations, in vivo amyloid imaging in patients with PPA and in those with typical amnestic dementias has shown a poor concordance between clinical features and distributions of amyloid labelling (Lehmann et al., 2013). The genotyping results also lead to the interesting implication that the E4 allele may be a risk factor for only some manifestations of Alzheimer’s disease but not for all (Rogalski et al., 2011).
Challenges in the subtyping of primary progressive aphasia
As the Gorno-Tempini et al. (2011) classification guidelines were being used to subtype the 35 cases in this study, two challenges related to logopenic PPA were encountered. First, strict adherence to these guidelines left as unclassifiable eight patients who had word retrieval impairments on a background of relatively preserved grammar and comprehension, a pattern that fit the original clinical description of a logopenic language impairment (Mesulam and Weintraub, 1992). These patients were not classifiable by the Gorno-Tempini et al. (2011) system because of preserved repetition abilities. A second challenge was encountered in the form of patients who fit criteria for both logopenic PPA and agrammatic PPA. Making impaired repetition an ancillary rather than core feature for logopenic PPA and replacing it with the core requirement that grammar be intact would have circumvented both challenges, at least in our sample, and might be worth considering as a potential revision to the Gorno-Tempini et al. (2011) guidelines (Mesulam and Weintraub, 2014). Partial justification for such a revision comes from a quantitative study where ‘logopenic PPA’ was defined without the requirement of abnormal repetition (Mesulam et al., 2009). The atrophy map in this set of patients was nearly identical to the atrophy map of patients who fit the Gorno-Tempini et al. (2011) requirement of poor repetition in logopenic PPA (Mesulam et al., 2012).
As in all other neurodegenerative diseases, the clinical picture of PPA changes over time, leading to considerable longitudinal shifts in subtype classification. This turned out to be particularly pertinent to the logopenic subtype where 7 of 11 patients with an initial logopenic PPA diagnosis (by the 2011 guidelines) progressed to agrammatic PPA, semantic PPA and mixed PPA by the second visit. Whether clinicopathological correlations should be based on the initial aphasia pattern or on its subsequent trajectory is a question that remains to be resolved.
Relationship of pathology to clinical features of the aphasia
The 35 autopsy cases revealed preferred but not invariant clinicopathological correlations. When FTLD-tau subtypes caused PPA, they most commonly (but not always) led to an aphasia characterized by agrammatism either initially or at follow-up, reflecting the predilection of these tauopathies for posterior frontal cortex where Broca’s area is located (Whitwell et al., 2010a). In fact, the presence of agrammatism in a patient with intact single word comprehension makes it unlikely to find Alzheimer’s disease at autopsy. The additional feature of a prominent motor speech abnormality in a patient with agrammatic PPA was the preferred manifestation of FTLD-tau of the PSP subtype and was only seen with this pathology in our sample. The close relationship of PSP pathology to motor speech abnormalities, especially apraxia of speech, has been described previously and may reflect the subcortical extension of the degeneration into nuclei and fibre pathways that coordinate speech production (Josephs et al., 2006; Ito, 2009). In fact, PSP pathology can give rise to pure apraxia of speech in the absence of aphasia (Josephs et al., 2006). However, PSP pathology in PPA has also been described without major speech abnormality and the combination of prominent apraxia of speech with non-fluent aphasia can also occur as a consequence of corticobasal degeneration pathology (Mochizuki et al., 2003; Josephs et al., 2006; Deramecourt et al., 2010). In our patients, PSP pathology was not associated with the characteristic ophthalmoplegia of the PSP syndrome, underlining the need to distinguish the PSP syndrome from PSP pathology.
All three FTLD-TDP type C cases in the new cohort of 35 patients had the semantic PPA variant at the initial or follow-up examination. The anatomical basis of this association lies in the selective vulnerability of the anterior temporal lobe to the neurodegenerative effects of TDP-C, leading to semantic PPA when the disease is asymmetrically concentrated within the language-dominant hemisphere (Mackenzie et al., 2006; Rohrer et al., 2010; Whitwell et al., 2010b). The semantic PPA subtype has also been described with Alzheimer’s disease pathology (Knibb et al., 2006), as in the case of one of the patients in the Mesulam et al. (2008) cohort (Table 3). So although TDP-C may selectively target the anterior temporal lobe and lead to semantic PPA, the presence of this clinical pattern does not always indicate TDP-C pathology.
When Alzheimer’s disease caused PPA, it most commonly (but not always) emerged as a logopenic language impairment characterized by word-finding and naming impairments in the absence of grammar or comprehension deficits, presumably reflecting the predilection of Alzheimer’s disease pathology, for the temporoparietal components of the language network (Gorno-Tempini et al., 2004). The relationship of logopenic PPA to Alzheimer’s disease pathology and the assumption that this clinical diagnosis could provide a marker for underlying Alzheimer pathology has attracted a great deal of attention. A study based on amyloid imaging as an Alzheimer’s disease biomarker did in fact report positive scans in 92% of the logopenic patients (Leyton et al., 2011). Our results indicate a much more modest relationship between the clinical diagnosis of logopenic PPA by the Gorno-Tempini et al. (2011) guidelines and Alzheimer’s disease. Interestingly, all three patients who had a stable logopenic PPA pattern for 5 years or more (Patients P1–3) had Alzheimer’s disease pathology at post-mortem. A longitudinally stable logopenic PPA pattern may therefore have a particularly high correlation with Alzheimer’s disease pathology.
The usefulness of clinical features for surmising the underlying pathology
The current results reinforce the conclusion that clinical characterization in PPA increases the precision with which the identity of the most probable pathology can be surmised. When implemented according to the 2011 guidelines, such characterization requires the assessment of at least 10 separate domains of language function. A less rigorous method, based on the status of two cardinal features, comprehension and grammar, can be about as informative of the underlying pathology as the subtyping by these guidelines. Sensitivity and specificity are quite modest with either approach, underscoring the need for additional evidence based on reliable biomarkers. At the present time, amyloid imaging with PET and CSF levels of tau and amyloid can help to determine whether or not a patient with PPA has Alzheimer’s disease pathology. In the future, advances in tau imaging are likely to differentiate FTLD-tau from FTLD-TDP in PPA patients with negative Alzheimer’s disease biomarkers.
Conclusion
The multiplicity of cellular pathologies that can cause the same clinical phenotype and the multiplicity of clinical phenotypes that can be caused by the same cellular pathology continue to bewilder attempts at establishing consistent clinicopathological correlations in neurodegenerative diseases. Primary progressive aphasia was one of the first entities to highlight the general principle that clinical manifestations reflect the anatomical distribution rather than the cellular nature of the underlying neurodegenerative disease (Weintraub and Mesulam, 2009). In any given case, the anatomical distribution of neuronal loss is likely to reflect the outcome of complex interactions between patient-specific factors that delineate loci of least resistance and disease-specific factors that constrain the set of possible distributions. This is why PPA can be caused by so many neurodegenerative diseases, and why each of these entities leads to preferred but not invariant aphasia subtypes. The patient-specific factors that cause multiple disease entities to be expressed asymmetrically in the language-dominant hemisphere remain to be identified. Progress in addressing this question may help to clarify the determinants of selective vulnerability in neurodegenerative diseases and perhaps also the molecular roots of hemispheric dominance in the human brain.
Acknowledgements
We thank Anne F. Koronkiewicz and Melanie Peterson, Brain Bank Research Assistants, for their contributions to this work.
Glossary
Abbreviations
- FTLD
frontotemporal lobar degeneration
- PPA
primary progressive aphasia
- PSP
progressive supranuclear palsy
- TDP
TAR DNA binding protein
Funding
Supported by DC008552 from the National Institute on Deafness and Communication Disorders, and AG13854 (Alzheimer Disease Centre) from the National Institute on Aging.
References
- Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, et al. Focal cortical presentations of Alzheimer's disease. Brain. 2007;130:2636–45. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- Bigio EH. Making the diagnosis of frontotemporal lobar degeneration. Arch Pathol Lab Med. 2013;137:314–25. doi: 10.5858/arpa.2012-0075-RA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli RJ, Beach TG, Sue LI, Connor DJ, Sabbagh MN. Progressive aphasia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14:55–8. doi: 10.1159/000064925. [DOI] [PubMed] [Google Scholar]
- Deramecourt V, Lebert F, Debachy B, Mackowiak-Cardoliani MA, Bombois S, Kerdraon O, et al. Prediction of pathology in primary progressive language and speech disorders. Neurology. 2010;74:42–9. doi: 10.1212/WNL.0b013e3181c7198e. [DOI] [PubMed] [Google Scholar]
- Dunn LA, Dunn LM. Peabody picture vocabulary test-4. Minneapolis: Pearson; 2006. [Google Scholar]
- Forman MS, Jennifer F, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59:952–62. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gefen T, Gasho K, Rademaker A, Lalehzari M, Weintraub S, Rogalski E, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain. 2012;135:1554–65. doi: 10.1093/brain/aws076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliebus G, Bigio E, Gasho K, Mishra M, Caplan D, Mesulam M-M, et al. Asymmetric TDP-43 distribution in primary progressive aphasia with progranulin mutation. Neurology. 2010;74:1607–10. doi: 10.1212/WNL.0b013e3181df0a1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E, Barresi B. Boston diagnostic aphasia examination. 3rd edn. Austin: Pro-Ed; 2001. [Google Scholar]
- Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–46. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis A, Weintraub S, Kertesz A, Mendez MF, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–14. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M. The non-fluent/agrammatic variant of primary progressive aphasia. Lancet Neurol. 2012;11:545–55. doi: 10.1016/S1474-4422(12)70099-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M, Xie SX, Libon DJW, Massimo L, Moore P, Vesely L, et al. Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology. 2008;70:2036–45. doi: 10.1212/01.wnl.0000303816.25065.bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JM, Gall C, Thompson JC, Richardson AMT, Neary D, Du Plessis D, et al. Classification and pathology of primary progressive aphasia. Neurology. 2013;81:1–8. doi: 10.1212/01.wnl.0000436070.28137.7b. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- Hof PR, Vogt BA, Bouras C, Morrison JH. Atypical form of Alzheimer's disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res. 1997;37:3609–25. doi: 10.1016/S0042-6989(96)00240-4. [DOI] [PubMed] [Google Scholar]
- Howard D, Patterson K. Pyramids and palm trees: a test of symantic access from pictures and words. Edmonds, Suffolk, UK: Thames Valley Test Company; 1992. [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio E, Cairns NJ, Carillo MC, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S. Progressive supranuclear palsy and diffusion tensor imaging. Eur Neurol Rev. 2009;4:108–11. [Google Scholar]
- Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999;56:1233–9. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- Johnson N, Barion A, Rademaker A, Rehkemper G, Weintraub S. The activities of daily living questionnaire (ADLQ): a validation study in patients with dementia. Alzheimer Dis Assoc Disord. 2004;18:223–30. [PubMed] [Google Scholar]
- Josephs KA, Duffy RJ, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006;129:1385–98. doi: 10.1093/brain/awl078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S. The Boston naming test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- Kertesz A. Western Aphasia Battery-Revised (WAB-R) Austin: Pro-Ed; 2006. [Google Scholar]
- Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol. 2006;59:156–65. doi: 10.1002/ana.20700. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Mastri AR, Frey WH, II, Sung JH, Rustan T. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–6. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Ghosh PM, Madison C, Lafirce R, Jr, Corbetta-Rastelli C, Weiner MW, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain. 2013;136:844–58. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton CE, Villemange VL, Savage S, Pike KE, Ballard KJ, Piguet O, et al. Subtypes of progressive aphasia: application of the international consensus criteria and validation using β-amyloid imaging. Brain. 2011;134:3030–43. doi: 10.1093/brain/awr216. [DOI] [PubMed] [Google Scholar]
- Mackenzie I, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–49. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio E, Cairns NJ, Alafuzoff I, Krill J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–3. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M, Weintraub S. Is it time to revisit the classification of primary progressive aphasia? Neurology. 2014 doi: 10.1212/WNL.0000000000000272. (Epub ahead of print.) [DOI] [PubMed] [Google Scholar]
- Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11:592–8. doi: 10.1002/ana.410110607. [DOI] [PubMed] [Google Scholar]
- Mesulam M-M. Primary progressive aphasia. Ann Neurol. 2001;49:425–32. [PubMed] [Google Scholar]
- Mesulam M-M, Weintraub S. Spectrum of primary progressive aphasia. In: Rossor MN, editor. Unusual dementias. London: Baillière Tindall; 1992. pp. 583–609. [PubMed] [Google Scholar]
- Mesulam M-M, Weintraub S, Parrish T, Gitelman DR. Primary progressive aphasia: reversed asymmetry of atrophy and right hemisphere language dominance. Neurology. 2005;64:556–7. doi: 10.1212/01.WNL.0000150545.46351.DE. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. 2008;63:709–19. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M, Wieneke C, Rogalski E, Cobia D, Thompson C, Weintraub S. Quantitative template for subtyping primary progressive aphasia. Arch Neurol. 2009;66:1545–51. doi: 10.1001/archneurol.2009.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M-M, Wieneke C, Thompson C, Rogalski E, Weintraub S. Quantitative classification of primary progressive aphasia at early and mild impairment stages. Brain. 2012;135:1537–53. doi: 10.1093/brain/aws080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ZA, Mandelli MA, Rankin KP, Henry ML, Babiak MC, Frazier DT, et al. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain. 2013;136:3461–73. doi: 10.1093/brain/awt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, Shoji S. Progressive supranuclear palsy presenting with primary progressive aphasia-Clinicopathological report of an autopsy case. Acta Neuropathol. 2003;105:610–4. doi: 10.1007/s00401-003-0682-5. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for a neuropathologic assesment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Perry DC, Lehmann M, Yokoyama JS, Karydas AM, Lee JJ, Coppola G, et al. Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol. 2013;70:774–8. doi: 10.1001/2013.jamaneurol.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinel P, Fauchereau F, Moreno A, Barbot A, Lathrop M, Zelenika D, et al. Genetic variants of FOXP2 and KIAA0319/TTRAP/THEM2 locus are associated with altered brain activation in distinct language-related regions. J Neurosci. 2012;32:817–25. doi: 10.1523/JNEUROSCI.5996-10.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premi E, Pilotto A, Alberici A, Papetti A, Archetti S, Seripa D, et al. FOXP2, ApoE, and PRNP: new modulators in primary progressive aphasia. J Alzheimers Dis. 2012;28:941–50. doi: 10.3233/JAD-2011-111541. [DOI] [PubMed] [Google Scholar]
- Rogalski E, Johnson N, Weintraub S, Mesulam M-M. Increased frequency of learning disability in patients with primary progressive aphasia and their first degree relatives. Arch Neurol. 2008;65:244–8. doi: 10.1001/archneurol.2007.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Rademaker A, Helenewski I, Johnson N, Bigio E, Mishra M, et al. APOE e4 is a susceptibility factor in amnestic but not aphasic dementias. Am J Alzheimers Dis Other Demen. 2011;25:159–63. doi: 10.1097/WAD.0b013e318201f249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski E, Weintraub S, Mesulam M-M. Are there susceptibility factors for primary progressive aphasia? Brain Lang. 2013;127:135–8. doi: 10.1016/j.bandl.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Geser F, Zhou J, Gennatas ED, Sidhu M, Trojanowski JQ, et al. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. 2010;75:2204–11. doi: 10.1212/WNL.0b013e318202038c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Rossor MN, Warren J. Alzheimer pathology in primary progressive aphasia. Neurobiol Age. 2012;33:744–52. doi: 10.1016/j.neurobiolaging.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajjadi SA, Patterson K, Arnold RJ, Watson PC, Nestor PJ. Primary progressive aphasia: a tale of two syndromes and the rest. Neurology. 2012;78:1670–7. doi: 10.1212/WNL.0b013e3182574f79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Patoine C, Abu-Khalil A, Visvader J, Sum E, Cherry TJ, et al. Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science. 2005;308:1794–8. doi: 10.1126/science.1110324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CK, Cho S, Hsu C-J, Wieneke C, Rademaker A, Weitner BB, et al. Dissociations between fluency and agrammatism in primary progressive aphasia. Aphasiology. 2012;26:20–43. doi: 10.1080/02687038.2011.584691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CK, Shapiro LP, Tait ME, Jacobs B, Schneider SL, Ballard K. A system for the linguistic analysis of agrammatic language production. Brain Lang. 1995;51:124–9. [Google Scholar]
- Weintraub S, Mesulam M. With or without FUS, it is the anatomy that dictates the dementia phenotype. Brain. 2009;132:2906–8. doi: 10.1093/brain/awp286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub S, Mesulam M-M, Wieneke C, Rademaker A, Rogalski EJ, Thompson CK. The Northwestern Anagram Test: measuring sentence production in primary progressive aphasia. Am J Alzheimers Dis Other Demen. 2009;24:408–16. doi: 10.1177/1533317509343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub S, Rogalski E, Shaw E, Salwani S, Rademaker A, Wieneke C, et al. Verbal and nonverbal memory in primary progressive aphasia: the Three Words-Three Shapes Test. Behav Neurol. 2012;26:67–76. doi: 10.3233/BEN-2012-110239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub S, Rubin NP, Mesulam MM. Primary progressive aphasia. Longitudinal course, neuropsychological profile, and language features. Arch Neurol. 1990;47:1329–35. doi: 10.1001/archneur.1990.00530120075013. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr, Boeve B, Parisi JE, Ahlskog JE, Drubach DA, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010a;75:1879–87. doi: 10.1212/WNL.0b013e3181feb2e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Jack CR, Jr, Parisi JE, Senjem ML, Knopman D, Boeve B, et al. Does TDP-43 type confer a distinct pattern of atrophy in frontotemporal lobar degeneration? Neurology. 2010b;75:2212–20. doi: 10.1212/WNL.0b013e31820203c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklund A, Johnson N, Weintraub N. Preservation of reasoning in primary progressive aphasia: further differentiation from Alzheimer's disease and the behavioral presentation of frontotemporal dementia. J Clin Exper Neuropsychol. 2004;26:347–55. doi: 10.1080/13803390490510077. [DOI] [PubMed] [Google Scholar]
- Wicklund MR, Duffy JR, Strand EA, Machulda MM, Whitwell JL. Quantitative application of the primary progressive aphasia consensus criteria. Neurology. 2014 doi: 10.1212/WNL.0000000000000261. (Epub ahead of print.) [DOI] [PMC free article] [PubMed] [Google Scholar]