

Abstract
P/Q Ca2+ channel activity is inhibited by G protein-coupled receptor activation. Channel inhibition requires a direct Gβγ binding onto the pore-forming subunit, Cav2.1. It is characterized by biophysical changes, including current amplitude reduction, activation kinetic slowing, and an I-V curve shift, which leads to a reluctant mode. Here, we have characterized the contribution of the auxiliary β3-subunit to channel regulation by G proteins. The shift in I-V to a P/Q reluctant mode is exclusively observed in the presence of β3. Along with the observation that Gβγ has no effect on the I-V curve of Cav2.1 alone, we propose that the reluctant mode promoted by Gβγ corresponds to a state in which the β3-subunit has been displaced from its channel-binding site. We validate this hypothesis with a β3-I-II2.1 loop chimera construct. Gβγ binding onto the I-II2.1 loop portion of the chimera releases the β3-binding domain and makes it available for binding onto the I-II loop of Cav1.2, a G protein-insensitive channel. This finding is extended to the full-length Cav2.1 channel by using fluorescence resonance energy transfer. Gβγ injection into Xenopus oocytes displaces a Cy3-labeled β3-subunit from a GFP-tagged Cav2.1 channel. We conclude that β-subunit dissociation from the channel complex constitutes a key step in P/Q calcium channel regulation by G proteins that underlies the reluctant state and is an important process for modulating neurotransmission through G protein-coupled receptors.
P/Q voltage-gated Ca2+ channels, which are localized on the presynaptic site of central and peripheral neurons, control neurotransmitter release (1). They are heteromultimeric proteins composed of the ion pore-forming domain Cav2.1, and three auxiliary subunits, β, α2δ, and γ. The auxiliary β-subunit plays a key role in defining ion channel properties (2). Besides being required for a normal P/Q expression at the plasma membrane (3), it also greatly facilitates channel activation by moderate membrane depolarization (4). All of the modifications in P/Q-channel properties require the binding of the β-subunit onto a discrete cytoplasmic domain (AID), which is present on the I-II loop of the Cav2.1-subunit (5).
P/Q Ca2+ channels are among the effectors of down-regulation of neurotransmitter release by G protein-coupled receptors (6, 7). After activation of the G protein heterotrimer, Gβγ is released and directly binds onto Cav2.1, which is a key step for this membrane-delimited inhibition (8, 9). Channel inhibition is defined by key modifications that include: (i) current amplitude reduction (6), (ii) activation kinetic slowing (6), and (iii) depolarizing shift of the activation curve (10). Changes in steady-state inactivation (10) and inactivation kinetics (11) have also been described. The three first biophysical manifestations contribute greatly to the reduction of Ca2+ entry by G proteins and are considered as essential hallmarks for the identification of a direct channel regulation. As the Ca2+ channel β-subunit, Gβγ also binds onto the I-II loop of Cav2.1 (12), but onto multiple domains. Besides, other Gβγ-binding sites have been identified, such as the N and C termini, although onto different channel types (13–15). Despite these findings, a precise dissection of the channel molecular determinants responsible for each of three main effects of Gβγ is crucially needed. The current reduction, the slowing of channel activation, and the depolarizing shift of the activation curve by G proteins, may not all necessarily occur in response to changes of similar channel structural determinants. For instance, it was found that G protein-coupled receptor activation may trigger Ca2+ current reduction without the usually associated slowing of activation kinetics (16). The depolarizing shift of the activation curve by G proteins, responsible for promoting the reluctant (R) state of the channel, according to Bean (10), is considered as the most critical factor in current reduction by G proteins and deserves an independent study. In this manuscript, we analyzed which channel determinants, and how activated G protein, triggers the R state of P/Q Ca2+ channels. We demonstrate that this particular effect is due to a physical displacement of the β-subunit from its binding site on Cav2.1.
Materials and Methods
Materials. Purified bovine brain Gβγ was from Calbiochem and was used as described (9). Collagenase IA was from Sigma. The GST-AID1.2 fusion protein and the GFP-Cav2.1 fusion construct have been described (5, 17). For GFP-Cav2.1, the N terminus of Cav2.1 was fused to the C terminus of a modified version of GFP (mutations S65T for increased brightness and optimized spectral properties; and V163A, I167T, S175G for better thermosensitivity).
Fluorescence Resonance Energy Transfer (FRET) Measurements. Albino oocytes were analyzed with a ×10 objective by confocal microscopy using the XYλ mode of fluorescence collection of the tcs-sp2 software (Leica, Heidelberg) 4–7 days after injection. The fluorescence was measured through 14 pass-band filters (10 nm width) to reconstruct the emission spectrum. For each measurement, the entire region of the plasma membrane at the equatorial level was considered for the fluorescence intensity. This intensity was normalized to the area of surface analyzed. The FRET levels were estimated as the ratio (575/515) of the fluorescence at 575 nm (emission peak of the acceptor: Cy3), versus the fluorescence at 515 nm (emission peak of the donor: GFP). After injection of Gβγ into oocytes, fluorescence scans were rapidly monitored at various times. The depression, observed ≈545 nm in the spectra of the XY-λ experiments, is due to the use of a triple dichroic mirror (488, 545, and 633 nm). This depression can be avoided by using a neutral 30/70 filter instead of a dichroic mirror. However, the poor reflection of the 488-nm excitation wavelength resulting from the use of this filter gives rise to spectra of much lower dynamics. Altogether, the use of the dichroic mirror provides higher dynamics to our spectra and does not perturb FRET estimation at a 575/515 fluorescence ratio, thereby increasing measurement reliability. Background spectra from control noninjected oocytes were subtracted from spectra from injected cells.
Further information on Western blotting, chimera constructions, in vitro translation and GST pulldown assays, His-β3 purification and Cy3 labeling, and Xenopus oocyte injection and electrophysiological recording can be found in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results
β-Subunit Expression Is Required for Some Important Biophysical Effects of Gbγ. We first confirmed by immunocytochemistry that the Xenopus oocyte β3-subunit, which has high homology with the rat sequence (18), was undetectable at the protein level; a condition necessary for the correct interpretation of our data (Fig. 1a). We next investigated the biophysical effects of Gβγ on Cav2.1 in the absence and presence of β3 of cRNA origin, to determine the contribution of β3 to Gβγ regulation. Out of β3 and β4, two β-subunits known to bind the P/Q channel, β3 was used because it is known to interact only with AID (19). Injection of 5 ng of Gβγ in Xenopus oocytes expressing Cav2.1 alone produces a significant inhibition of current amplitude at 0 mV (50 ± 3%, n = 5), illustrating the β-independent effects (Fig. 1b). When β3 is coexpressed, Gβγ produces an even greater inhibition of Cav2.1 current amplitude (up to 81 ± 9% at 0 mV, n = 8). An average pre-pulse facilitation of 8% was observed, probably reflecting a partial dissociation of Gβγ from this rabbit Cav2.1 isoform. In both conditions, injection of heat-denatured Gβγ was without effect on current amplitude (Fig. 1c). Current kinetics (activation and inactivation) after the effect of Gβγ is indistinguishable whether or not β3 is expressed (Fig. 1d). Average times to peak of the currents after Gβγ effect were 28.8 ± 8.1 ms (-β3, n = 8) and 28.7 ± 4.1 ms (+β3, n = 10). As interpreted by others (20), these results could suggest that β3 promotes G protein regulation. Alternatively, the following points suggest that this finding results from a displacement of bound β3 from Cav2.1: (i) in the absence of β3,Gβγ has no effect on either the I-V curve (Fig. 2a) or the steady-state inactivation properties (Fig. 2b) of Cav2.1; (ii) β3-subunit is known to displace, toward hyperpolarizing potentials, the I-V curve (-19.5 mV, n = 13) and the steady-state inactivation properties of the channel (-28.9 mV, n = 12; Fig. 2 c and d); and (iii) acute injection of Gβγ reverses the shifting effects of β3 on both the I-V curve (+19.2 mV, n = 18) and the steady-state inactivation (+25.3 mV, n = 10; Fig. 2 c and d). Thus, this reversal in the shift of the I-V curve translates in an apparent extra inhibition (apparent promoting effect of β3) when a 0 mV pulse is used to activate the channel (Fig. 1b).
Fig. 1.

Lack of expression of the endogenous β3-subunit and slow kinetic effect of Gβγ injection. (a) Western blot illustrating the expression of rat β3 from cRNA origin or the integrity of purified His-β3 protein 4 days after injection. Note the lack of expression of endogenous β3 protein.(b) Representative current traces at 0 mV, before (-1 min) and after (3, 7, 14, and 25 min) injection of 5 ng of purified Gβγ, are shown for oocytes expressing either Cav2.1 alone (dark gray circle) or Cav2.1 and β3 (light gray circle). Note the increased current inhibition in the presence of the β3-subunit. (c) Kinetics and β-dependence of the inhibition of Cav2.1 current amplitude by Gβγ. Representative examples are shown. Arrow: injection of 5 ng of Gβγ or HI-Gβγ. Test potential is 0 mV elicited from a holding potential of -90 mV. Filled circles, Cav2.1 alone plus HI-Gβγ; open circles, Cav2.1 plus β3 plus HI-Gβγ; dark gray circles, Cav2.1 plus Gβγ; light gray circles, Cav2.1 plus β3 plus Gβγ. (d) Super-imposed current traces at 0 mV for Cav2.1 plus Gβγ and Cav2.1 plus β3 plus Gβγ.
Fig. 2.

Gβγ reverses the electrophysiological modifications induced by β3-subunit. (a) Current-voltage relationship for Cav2.1 alone (filled circles) and Cav2.1 plus Gβγ (dark gray circles). Essential fitting parameters are V1/2 of 12.3 ± 0.9 mV (-Gβγ) and 14.4 ± 0.6 mV (+Gβγ), and k of 6.0 ± 0.6 mV (-Gβγ) and 5.6 ± 0.4 mV (+Gβγ). (Inset) Box plot representation of the half-activation potentials (V1/2 act) illustrating the lack of any Gβγ-induced shift. (b) Steady-state inactivation curves for Cav2.1 alone (filled circles) and Cav2.1 plus Gβγ (dark gray circles). Fitting parameters are V1/2 of -22.6 ± 2.0 mV (-Gβγ) and -22.6 ± 1.7 mV (+Gβγ), and k of 10.8 ± 1.4 mV (-Gβγ) and 10.8 ± 1.4 mV (+Gβγ). (Inset) Box plot representation of half-inactivation potentials (V1/2 inact) also illustrating the lack of shift by Gβγ.(c) Current-voltage relationship for Cav2.1 alone (filled circles), Cav2.1 plus β3 (open circles), and Cav2.1 plus β3 plus Gβγ (light gray circles). Fits provide the following values: V1/2 of -5.1 ± 0.9 mV (+β3) and 10.9 ± 0.8 mV (+β3 plus Gβγ), and k of 4.6 ± 0.6 mV (+β3) and 6.0 ± 0.6 mV (+β3 plus Gβγ). (Inset) Box plot representation of V1/2 act illustrating the shift by β3 and its reversal by Gβγ.(d) Steady-state inactivation curves. Same conditions and symbols as in c. Fits provide: V1/2 of -46.3 ± 1.0 mV (+β3) and -19.0 ± 1.7 mV (+β3 plus Gβγ), and k of 11.8 ± 0.7 mV (+β3) and 9.8 ± 1.4 mV (+β3 plus Gβγ). (Inset) V1/2 inact also illustrating the shift by β3 and its reversal by Gβγ.In a–d,Gβγ was injected 30 min before electrophysiological recording. IV and inactivation curves are all from different cells but from the same batch.
Working Hypothesis. Assuming that Gβγ displaces β3 from its binding site, then the final inhibited state of Cav2.1 should be the same, independent of the presence of β3 (see Fig. 6, which is published as supporting information on the PNAS web site). For the sake of clarity, let us conceptually dissociate the dual effects of the binding of Gβγ on Cav/β into two concomitant effects: (i) the displacement of β from Cav (the focus of this study), and (ii) the inhibition of Cav alone by Gβγ. If our molecular hypothesis is correct, then the following equation (see Supporting Materials and Methods for equation derivation) should allow a theoretical estimation of the apparent promoting effect of β3 on the inhibition of Cav2.1 current induced by Gβγ at a given test potential.
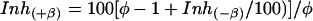 |
where φ represents the change in normalized conductance of the channel produced by β3-subunit dissociation. At 0 mV, the theoretical value of Gβγ inhibition in the presence of a virtual β3-subunit (mean 87 ± 1%, n = 5) is in close agreement with the experimental mean value for Cav2.1/β3 channels (81 ± 9%, n = 8; Fig. 6 d and e). In contrast, the “β-promoting effect” is clearly lost at 30 mV, as verified both experimentally and after modeling. Experimentally, the equal inhibition levels observed at 30 mV for the Cav2.1 and Cav2.1/β3 channels, is further indicative that β3-subunit dissociation has probably no significant effect on Cav2.1 channel properties, other than the depolarizing shift in the I-V curve. The apparent extra inhibition produced by Gβγ at 0 mV, which is observed in the presence of β3, may well result from a displacement of this auxiliary subunit from the channel. We thus propose that binding of Gβγ complex onto Cav2.1 produces two distinct functional effects: (i) a β-independent current amplitude inhibition, and (ii) a β-dependent shift in the I-V curve and steady-state inactivation voltage-dependencies of the channel. In the following experiments, we shall focus our study on the mechanisms of the β-dependent effects of Gβγ regulation.
Structural Rationale. It is now widely accepted that the AID motif, present on the I-II loop, constitutes the main binding site for β on members of the Cav1.X and Cav2.X channel families (ref. 5 and Fig. 3a). This β binding relies exclusively on three important AID residues (Tyr-392, Trp-395, and Ile-396) in the case of Cav2.1 (21). The full-length I-II loop of the Cav2.X family also contains multiple binding sites for Gβγ (refs. 9 and 12 and Fig. 3a). One portion of this binding site is overlapping with the β3 binding site, whereas the others are dispersed over the length of the I-II loop. Owing to this vicinity in Gβγ- and β-subunit-binding sites on the Cav2.1 channel, we hypothesize that the β3-dependent effects of Gβγ are due to a mere displacement of β3 from its unique AID Cav2.1-binding site. To validate this concept, we followed two experimental strategies: (i) a biochemical analysis of the effect of Gβγ on the conformation of a chimeric β3-I-II2.1 protein, and (ii) a dynamic FRET observation of Gβγ effect on β/Cav2.1 interaction in living cells.
Fig. 3.

The Gβγ complex disrupts the AID2.1/BID interaction. (a) Schematic representation of the binding domains for the Gβγ complex and the β-subunit onto the I-II loop of Cav2.1. The regions involved in Gβγ binding are represented as dashed boxes and the β-binding domain (AID) is gray. Note that a complete binding of Gβγ requires the full-length I-II loop. (b) Pull-down demonstration that in vitro-translated [35S]β3 (1–3 pM) specifically binds onto GST-AID1.2 fusion protein (1 μM). T, starting in vitro-translated material; GST, control GST pulldown. Note that AID1.2 is derived from Cav1.2 (α1C, a non-Gβγ-binding channel). (c) A chimera was constructed by direct fusion of the I-II2.1 loop of Cav2.1 to β3 (chimera β3-I-II2.1). In vitro-synthesized [35S]β3-I-II2.1 chimera is unable to bind to GST-AID1.2 fusion protein due to the intramolecular interaction between its endogenous AID and BID domains.(d) Deletion of AID from the β3-I-II2.1 chimera (β3-I-II2.1 ΔAID) restores its association to GST-AID1.2.(e) The preincubation of the [35S]β3-I-II2.1 chimera with increasing concentrations of purified Gβγ complex promotes the binding of the [35S]β3-I-II2.1/Gβγ complex onto GST-AID1.2. Data were fitted with an hyperbolic function y = ax/EC50 + x where a = 0.79 ± 0.11 is the maximal estimated fraction of [35S]β3-I-II2.1 chimera bound to GST-AID1.2 and EC50 = 121 ± 56 nM is the concentration of Gβγ that produces a half-maximal effect. Experiments shown in a–d were successfully repeated three times.
Effect of Gβγ on the Conformation of a Chimeric β3-I-II2.1 Protein. We used a molecular in vitro strategy to test the nature of the interactions between Cav2.1, β3, and Gβγ. First, we designed a chimera molecule between β3 and the I-II loop of Cav2.1 (β3-I-II2.1) with the expectation that the AID domain from I-II2.1 recognizes its binding site on β3 [BID site (22)]. Such a strategy simplifies the number of molecular partners used in Gβγ competition experiments and forces a stoichiometry of one β3 molecule for one I-II loop sequence. Also, the limited amount of translated chimera molecule is easy to saturate by Gβγ binding onto the I-II2.1 part. Fig. 3 b–e summarizes the experiments that lead us to conclude that Gβγ association on the I-II2.1 part of the molecule results in the dissociation of β3 from AID. A GST protein fused to an enlarged AID sequence of Cav1.2 (GST-AID1.2) (5) binds in vitro-translated [35S]β3-subunit (Fig. 3b). On the other hand, fusion of β3 to the I-II loop of Cav2.1 prevents its interaction with GST-AID1.2 (Fig. 3c). To demonstrate that this result is due to an intramolecular interaction between BID and AID2.1 of [35S]β3-I-II2.1, we deleted the 18-aa AID2.1 residues. This deletion restores the ability of the chimera to bind onto GST-AID1.2 (Fig. 3d). Evaluating the binding of β3-I-II2.1 onto GST-AID1.2 represents a good test to identify inhibitors of the internal AID2.1/BID interaction. A molecule will induce the binding of the chimera protein only if it displaces the internal AID2.1/BID interaction without affecting the subsequent BID/AID1.2 interaction. As shown in Fig. 3e, Gβγ is precisely this type of inhibitor, because preincubation of increasing concentrations of purified Gβγ with [35S]β3-I-II2.1 (30 min at 20°C) induces its binding to GST-AID1.2. The use of GST-AID1.2 here appears to be beneficial because, contrary to Cav2.1, Cav1.2, a member of the L-type family, is unable to bind Gβγ (12). The displacement of AID2.1 from its BID-binding site occurs with an EC50 of 121 nM for Gβγ. This value is in close agreement with the reported dissociation constant of Gβγ for the I-II2.1 loop (12) and compares well with the effective concentrations observed in other physiological systems (23). These observations were confirmed in an ex vivo context by evaluating the increase of Cav1.2 current density induced by the unfolding of β3-I-II2.1 by Gβγ (see Supporting Materials and Methods, and Fig. 7, which is published as supporting information on the PNAS web site).
Dynamic Observation of a Gβγ-Induced β Displacement by FRET in Single Living Cells. To follow the dynamic of the interaction between the full-length Cav2.1- and β3-subunits in a cellular context, we designed a FRET strategy. In our case, we favored the combination of GFP-Cav2.1 and chemically labeled Cy3-β3, which constitutes a favorable Förster donor/acceptor pair [Förster radius of 60 Å (24)]. We demonstrated that (i) β3 can be labeled with Cy3-maleimide, and (ii) remains functional after injection in Xenopus oocytes despite the labeling (see Supporting Materials and Methods, and Fig. 8, which is published as supporting information on the PNAS web site). Of note, oocytes have optical properties that make fluorescence studies on living cells valid, only in the vicinity of the plasma membrane. Indeed, the filter effect of the cytoplasm of oocytes drastically decreases fluorescence signals originating from this compartment. This effect can be demonstrated here by the apparent fluorescence distribution to the plasma membrane of mercaptoethanol blocked Cy3-maleimide, a dye that should distribute freely into the cytoplasm (Fig. 4a). Although living oocytes should not be used to study subcellular protein distribution with a confocal microscope, their use remains perfectly valid for fluorescence emission studies in the vicinity of the plasma membrane. We first showed that noninjected albino cells emitted very low levels of fluorescence in our experimental conditions (Fig. 4a). Injection of either Cy3-β3 protein or GFP-Cav2.1 cDNA, alone or in combination, results in strong fluorescence signals in the vicinity of the plasma membrane (Fig. 4a). Fluorescence of GFP-Cav2.1 alone is indicative of the expression of the channel at the plasma membrane in the absence of Cy3-β3, which is in agreement with biophysical data (Figs. 1b and 2 a and b). The more diffuse distribution of Cy3-β3 most likely reflects the cytoplasmic distribution of the protein. We also noted that the apparent distribution of GFP-Cav2.1 fluorescence in the presence of unlabeled β3, or Cy3-β3 fluorescence in the presence of unlabeled Cav2.1, was not modified (not shown). According to our experimental conditions, Cy3-β3 was in molar excess compared with GFP-Cav2.1.
Fig. 4.

Experimental conditions to observe FRET in oocytes. (a) Optical (T, transmission light) and fluorescence images of Xenopus oocytes in various experimental conditions: NI (noninjected), Cy3 (injected with Cy3-maleimide only), Cy3-β3 (injected at 1.75 ng), GFP-Cav2.1, and the combination GFP-Cav2.1 plus Cy3-β3. Fluorescence is artificially colored to reflect color codes for the emission spectra in b. Similar fluorescence distributions were observed for unlabeled Cav2.1 plus Cy3-β3 (compared with Cy3-β3), and GFP-Cav2.1 plus unlabeled β3 (compared with GFP-Cav2.1). (b) Average emission spectra for various experimental conditions. Noninjected, filled circles and dotted line (n = 4). Green refers to GFP-Cav2.1 plus unlabeled β3 (n = 5), dark red to unlabeled Cav2.1 plus Cy3-β3 (n = 4), and red to GFP-Cav2.1 plus Cy3-β3 (n = 6). Arrows indicate the amount of crosstalks at 575 nm that were used to correct for FRETN calculations. (c) Average emission spectra. Red line and symbols, as in b. Blue line and symbols, sum of emission spectra for GFP-Cav2.1 plus unlabeled β3, and for unlabeled Cav2.1 plus Cy3-β3 (n = 9) after subtraction of background emission (from noninjected oocytes).
Fig. 4b represents average emission spectra from cells in four experimental conditions (noninjected, injected with GFP-Cav2.1 plus unlabeled β3, injected with unlabeled Cav2.1 plus Cy3-β3, and injected with GFP-Cav2.1 plus Cy3-β3). The fluorochromes were excited at 488 nm and the emission spectra recorded from 500 to 640 nm with 14 successive 10-nm-wide band paths. The settings (laser power and photomultiplier gain) of the confocal were kept constant during all of the measurements. The fluorescence spectrum of the noninjected oocytes (dotted line) is very low, compared with injected oocytes, and was subtracted from the data shown in Fig. 5 and for FRET calculations. Under the same conditions, oocytes expressing GFP-Cav2.1 plus unlabeled β3 give a stronger fluorescence signal with a maximal emission at λmax of 515 nm (Fig. 4b, green line). Although the excitation wavelength at 488 nm limits the excitation of Cy3-β3 (λex at 543 nm), we observed a fluorescence emission spectra with a maximal emission at 575 nm for oocytes injected with 1.75 ng of Cy3-β3 and expressing unlabeled Cav2.1 (Fig. 4b, dark red line). The fluorescence intensity level of oocytes injected with Cy3-β3 demonstrated a good reproducibility. Also, the absence of unlabeled Cav2.1 did not alter the observed emission spectra of Cy3-β3. When both GFP-Cav2.1 and Cy3-β3 are present in the oocyte (Fig. 4b, red line), the emission signal strongly decreases at 515 nm, whereas it increases significantly at 575 nm, suggesting that FRET might occur between these two molecules. This effect is illustrated by the comparison of average emission spectra of oocytes expressing GFP-Cav2.1 and Cy3-β3 with the sum of the average emission spectra of cells expressing GFP-Cav2.1 plus unlabeled β3 and of cells expressing unlabeled Cav2.1 plus Cy3-β3 (Fig. 5c, blue line). This summed spectrum should be representative of the theoretical spectrum for oocytes containing both GFP-Cav2.1 and Cy3-β3 if there was no FRET. Comparing the two spectra in Fig. 5c, the FRET occurring between GFP-Cav2.1 and Cy3-β3 can be evidenced by the decreased emission at 515 nm, and the increased emission at 575 nm.
Fig. 5.

Analysis of Gβγ-induced P/Q calcium channel disassembly. (a) Average emission spectra and effect of the injection of 5 ng of Gβγ for oocytes in various experimental conditions: (Top) Unlabeled Cav2.1 plus Cy3-β3. (Middle) GFP-Cav2.1 plus unlabeled β3.(Bottom) GFP-Cav2.1 plus Cy3-β3. •, noninjected; ○, Gβγ-injected. (b) Box plot representation of FRETN values for noninjected Gβγ oocytes (-), Gβγ-injected (5 ng), and injection of 5 ng of HI-Gβγ. (c) Kinetics of FRET decrease induced by 5 ng of Gβγ injection. Illustration of two representative cells. (d) Model of calcium channel regulation by the Gβγ complex. W, β-bound W state; R, Gβγ-bound R state; R′,Gβγ-unbound R state.
Next, we analyzed the effect of injecting 5 ng of Gβγ onto the average emission spectrum of oocytes containing both GFP-Cav2.1 and Cy3-β3. As shown in Fig. 5a, injection of Gβγ induces a drastic modification of the emission spectrum. The observed changes (decreased emission at 575 nm, and increased emission at 515 nm) are coherent, with a reduction in FRET efficiency. This observation qualitatively validates the concept that Gβγ is able to dissociate Cy3-β3 from GFP-Cav2.1. A similar Gβγ injection has no effect on the fluorescence emission spectra of oocytes containing GFP-Cav2.1 plus unlabeled β3 or unlabeled Cav2.1 plus Cy3-β3 (Fig. 5a). To quantify precisely the FRET changes between GFP-Cav2.1 and β3-Cy3 and to normalize for fluorescence intensity variability of the fluorophores concentrations in different cells, we followed the method of Gordon et al. (25) for FRET calculation. By using the fluorescence recorded at 515 and 575 nm in our experimental conditions, the equation for normalized FRET (FRETN) calculations is:
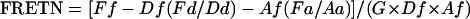 |
where Ff is the fluorescence at 575 nm of GFP-Cav2.1 plus Cy3-β3 (excitation at 488 nm); Df is the fluorescence at 515 nm of GFP-Cav2.1 + Cy3-β3 (excitation at 488 nm); Fd is the fluorescence at 575 nm of GFP-Cav2.1 plus unlabeled β3 (excitation at 488 nm); Dd is the fluorescence at 515 nm of GFP-Cav2.1 alone (excitation at 488 nm); Af is the fluorescence at 575 nm of GFP-Cav2.1 plus Cy3-β3 (excitation at 543 nm); Fa is the fluorescence at 575 nm of unlabeled Cav2.1 plus Cy3-β3 (excitation at 488 nm); Aa is the fluorescence at 575 nm of unlabeled Cav2.1 plus Cy3-β3 (excitation at 543 nm); and G is a constant related to the respective transmissions of the three filter sets used (F, A, and D) and to the quantum yield of the two fluorophores. As suggested (25), G will be set arbitrary to a value of 1. In this equation, the term “- Df(Fd/Dd)” corresponds to the correction of FRET signal for donor fluorescence crosstalk (GFP), whereas the term “- Af(Fa/Aa)” corresponds to the correction of FRET signal for acceptor fluorescence crosstalk (Cy3). These crosstalks can be observed in the individual cases presented in Fig. 4b (see arrows). Using the equation, we came up with an average FRETN(-Gβγ) value of 0.0156 ± 0.0048 (n = 5) before injection of Gβγ (Fig. 5b). This value is decreased to FRETN(+Gβγ) =-0.0012 ± 0.0013 (n = 5) 15 min after injection of 5 ng of Gβγ in the same cells. As a control, injection of 5 ng of heat-inactivated Gβγ (HI-Gβγ) produced no decrease of FRETN [FRETN(HI-Gβγ) = 0.0224 ± 0.0095 (n = 3)]. In theory, a FRET decrease can be due to either a change of the relative orientation of the two fluorophores, or to an increase in the interfluorochrome distance. Because the orientation of the flexible Cy3-maleimide fluorochromes is randomly changing, a possible reorientation of GFP, induced by Gβγ interaction with Cav2.1 (maybe including its N terminus), should statistically not impact the FRET value very much. The presence of three different Cy3-maleimide fluorophores, located at distant sites on β3, all outside the interaction site with Cav2.1, further minimizes the chances that GFP reorientation should affect the FRET value to the extent we observe. Assuming a random relative orientation, the large Förster radius between GFP and Cy3 (60 Å) allows the observation of FRET for distances up to 100 Å between GFP and any one of the three Cy3 present on the β3-subunit. Therefore, the most likely explanation for the decrease in FRET, induced by Gβγ, should be a large increase in interfluorochrome distance, compatible with a dissociation of the GFP-Cav2.1/Cy3-β3 complex. Assuming that FRET decrease induced by Gβγ is truly due to the dissociation of Cy3-β3 from GFP-Cav2.1, then, as noticed earlier (25), FRETN is directly proportional to the concentration ratio [GFP-Cav2.1-Cy3-β3 complex]/([total GFP-Cav2.1] · [total Cy3-β3]). Due to the large molar excess of Cy3-β3 in oocytes, all of the GFP-Cav2.1 present at the plasma membrane can be considered to be associated with Cy3-β3 before injection of Gβγ. This assumption is validated by the biophysical studies (Figs. 2 and 8) and earlier studies (3). Furthermore, Gβγ has no effect on the total concentrations of Cy3-β3 and GFP-Cav2.1 present in oocytes. Therefore, the FRETN value that decreases to 0 after Gβγ injection is indicative that Gβγ displaces all of the Cy3-β3 from GFP-Cav2.1 channels.
Our approach on living oocytes provided the possibility to monitor FRET changes at various times after Gβγ injection (Fig. 5c). FRETN decrease reached completion 3 min after Gβγ injection.
Discussion
Activation of G proteins results in the binding of Gβγ onto the I-II loop of Cav (9, 12) and mediates inhibition of presynaptic Ca2+ currents (7). The contribution of the β-subunit to the regulation of channel activity by Gβγ has never been clearly elucidated (11). Initially, it was thought that β antagonized the effect of G proteins (26). Recent studies (20) show, on the contrary, that β is required for the willing mode of the channel and promotes G protein regulation. Some of the discrepancies reported on the mechanisms of regulation by G proteins may simply stem from the fact that data based exclusively on biophysical approaches have been interpreted directly in molecular terms. As shown in this manuscript, some of these modulations are independent of the presence of the β-subunit. We purposely developed molecular approaches in parallel with biophysical studies to exclusively address the molecular mechanisms underlying the β-dependent Gβγ modulation.
Evidence That Gβγ Displaces β from Its Cav-Binding Site. Our approaches were developed to study the relationship between Cav2.1, β3, and Gβγ. The biophysical experiments were aimed at solving some of the contradictory reports from the literature. To avoid many drawbacks related to the activation of G protein-coupled receptors, we directly injected controlled amounts of purified Gβγ complex. The biophysical effects of Gβγ are not due to an indirect alteration of the phosphorylation state of Cav2.1 because we have similar results in the presence of 500 ng/ml staurosporin (data not shown). Modeling of the biophysical data suggests that Gβγ may functionally displace β3 from the channel. We aimed to know if this functional displacement was actually due to a physical removal of β3 from its unique binding site on Cav2.1 or an allosteric effect of Gβγ that prevented the β3 regulation. By using a pull-down assay, we first demonstrated that Gβγ disrupts the intramolecular interaction between both the AID2.1 and BID sites of the β3-I-II2.1 chimera. Recently, Herlitze et al. (28) came to an opposite conclusion. However, these authors used a much shorter fragment of the I-II loop that inconveniently misses several Gβγ-binding sites. Although Gβγ has the ability to bind to AID (12), we envision that interaction of Gβγ with these other sites should favor some kind of unzipping of β3 from AID. In addition, these authors omitted Gγ from their studies (28), which is worrisome, because we expect Gγ to play a key function in G protein regulation. Gγ has structural homology with the III-IV loop of Cav2.1 (29). Because this loop interacts with several critical AID residues of the I-II loop (29), we expect that Gγ contributes to β3-subunit displacement. Obviously, the precise submolecular sequence of events responsible for β3 dissociation from the AID site will require further dissection.
To extend these observations to the full-length Cav2.1- and β3-subunit in a cell context, we developed a FRET strategy. When Cy3-labeled β3 protein and GFP-Cav2.1 fusion protein were coinjected in oocytes, we observed a strong FRET signal resulting from their direct association. The dynamic decrease of FRET signal induced by Gβγ in this system gives a real-time demonstration of the displacement of Cy3-β3 from GFP-Cav2.1. Theoretically, a decrease of FRET can be interpreted as either a change in conformation of the GFP-Cav2.1/Cy3-β3 complex or a dissociation of Cy3-β3 from the channel. Assuming a random relative orientation, the large Förster radius of the GFP/Cy3 pair (R0 = 60 Å) allows the observation of FRET for distances up to 100 Å between GFP and any one of the three Cy3. For comparison, the theoretical Stokes radius of globular proteins like the β3-subunit is ≈50 Å. Thus, the three Cy3 labels must contribute statistically to the FRET. The complete suppression of FRET induced by Gβγ (as estimated from FRETN) indicates that β3 should be displaced by a distance greater than its own Stokes radius. Of course, it cannot be completely excluded that this FRET suppression results from a simple reorientation of the respective emission and excitation dipolar moments of the fluorophores. Reorientation of the GFP fluorophore is possible because this rigid tag is attached to the N terminus of Cav2.1, a possible binding site of Gβγ. However, three flexible Cy3 acceptor fluorophores located at different locations on β3 should randomize the FRET signal, no matter what orientation GFP or β3 may take on the channel. Also, it cannot be argued that Gβγ produces a simple swinging of β3 by favoring its attachment onto a different site, because the I-II loop is the only binding domain of this β isoform. Finally, although the allosteric model defended by others (28) cannot be excluded only on the basis of the present FRET experiment, the convergence of these data with those obtained here by the other experimental approaches gives a solid support to the hypothesis of a physical dissociation of the Cav-β heterodimer by Gβγ.
The Shift of the Channel from a W State to an R State Results from the β Displacement. It was initially reported that the neurotransmitter-induced inhibition of neuronal calcium currents results mainly from a change in channel voltage dependence (10). Bean (10) proposed that calcium channels can shift from a W state, characterized by a low-voltage activation potential, to an R state requiring stronger depolarization for activation. This shift can be induced by the binding of Gβγ onto the channel (11). In the absence of β3, we did not observe any effect of Gβγ on the channel's voltage dependence of activation. On the other hand, the same experiment performed with β3 resulted in an important shift of the voltage dependence of activation. This biophysical observation, associated to the molecular data discussed above, demonstrate that the shift of the channel from the W state to the R state is not directly induced by the binding of Gβγ as generally proposed, but rather, by the displacement of β from the channel as a consequence of Gβγ binding. Here also, an allosteric model of regulation in which both β3 and Gβγ would remain associated to the channel is difficult to envision. It would seem as an extraordinary coincidence that the biophysical properties of Cav2.1 with β3 and Gβγ bound onto it would be similar to those of Cav2.1 alone, particularly considering that β3 and Gβγ produce such drastic effects alone. With these results, we generated a model of P/Q channel regulation by G-protein (Fig. 5d). In this model, the channel is in the W state in the absence of Gβγ. Activation of G proteins leads to Gβγ binding onto the channel and the departure of the Ca2+ channel β-subunit, resulting in the R state. It should be emphasized that the R state remains a state in which the β-independent G protein regulation remains effective (reduced current amplitude in our study). It should be differentiated from another R state (R′) in which the channel is neither associated to the Gβγ nor to the β-subunit.
Physiological Importance. Free β-subunit has recently been shown to regulate gene silencing through nuclear relocalization (30). Here, displacement of β from Cav may represent one of the mechanisms whereby G proteins would trigger the relocalization of β in the nucleus. Hence, this unique mechanism of β dissociation could take part in a new pathway of transcription regulation by G protein-coupled receptors.
Supplementary Material
Acknowledgments
We thank K. G. Beam for providing GFP-Cav2.1, and J. Nargeot, C. C. Chen, K. P. Campbell, and M. Gola for critical comments. M.D.W. is supported by an Action Concertée Incitative Grant from the Research Ministry and G.S. is supported by the Fondation pour la Recherche Médicale.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FRET, fluorescence resonance energy transfer; FRETN, normalized FRET; W, willing; R, reluctant; HI, heat-inactivated.
References
- 1.Turner, T. J., Adams, M. E. & Dunlap, K. (1992) Science 258, 310-313. [DOI] [PubMed] [Google Scholar]
- 2.Isom, L. L., De Jongh, K. S. & Catterall, W. A. (1994) Neuron 12, 1183-1194. [DOI] [PubMed] [Google Scholar]
- 3.Bichet, D., Cornet, V., Geib, S., Carlier, E., Volsen, S., Hoshi, T., Mori, Y. & De Waard, M. (2000) Neuron 25, 177-190. [DOI] [PubMed] [Google Scholar]
- 4.De Waard, M. & Campbell, K. P. (1995) J. Physiol. (London) 485, 619-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pragnell, M., De Waard, M., Mori, Y., Tanabe, T., Snutch, T. P. & Campbell, K. P. (1994) Nature 368, 67-70. [DOI] [PubMed] [Google Scholar]
- 6.Dunlap, K. & Fischbach, G. D. (1981) J. Physiol. (London) 317, 519-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolphin, A. C. (1998) J. Physiol. (London) 506, 3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herlitze, S., Zhong H., Scheuer T. & Catterall, W. A. (1996) Nature 380, 258-262. [DOI] [PubMed] [Google Scholar]
- 9.Zamponi, G. W., Bourinet, E., Nelson, D., Nargeot, J. & Snutch, T. P. (1997) Nature 385, 442-446. [DOI] [PubMed] [Google Scholar]
- 10.Bean, B. P. (1989) Nature 340, 153-156. [DOI] [PubMed] [Google Scholar]
- 11.Zamponi, G. W. (2001) Cell Biochem. Biophys. 34, 79-94. [DOI] [PubMed] [Google Scholar]
- 12.De Waard, M., Liu, H., Walker, D., Scott, V. E. S., Gurnett, C. A. & Campbell, K. P. (1997) Nature 385, 446-450. [DOI] [PubMed] [Google Scholar]
- 13.Zhang, F., Ellinor, P. T., Aldrich, R. W. & Tsien, R. W. (1996) Neuron 17, 991-1003. [DOI] [PubMed] [Google Scholar]
- 14.Qin, N., Platano, D., Olcese, R., Stefani, E. & Birnbaumer, L. (1997) Proc. Natl. Acad. Sci. USA 94, 8866-8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolphin, A. C., Page, K. M., Berrow, N. S., Stephens, G. J. & Canti, C. (1999) Ann. N.Y. Acad. Sci. 868, 160-174. [DOI] [PubMed] [Google Scholar]
- 16.Twitchell, W. A. & Rane, S. G. (1993) Neuron 10, 701-709. [DOI] [PubMed] [Google Scholar]
- 17.Grabner, M., Dirksen, R. T. & Beam, K. G. (1998) Proc. Natl. Acad. Sci. USA 95, 1903-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tareilus, E., Roux, M., Qin, N., Olcese, R., Zhou, J., Stefani, E. & Birnbaumer, L. (1997) Proc. Natl. Acad. Sci. USA 94, 1703-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker, D., Bichet, D., Geib, S., Mori, E., Cornet, V., Snutch, T. P., Mori, Y. & De Waard, M. (1999) J. Biol. Chem. 274, 12383-12390. [DOI] [PubMed] [Google Scholar]
- 20.Meir, A., Bell, D. C., Stephens, G. J., Page, K. M. & Dolphin, A. C. (2000) Biophys. J. 79, 731-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Waard, M., Scott, V. E. S., Pragnell, M. & Campbell, K. P. (1996) FEBS Lett. 380, 272-276. [DOI] [PubMed] [Google Scholar]
- 22.De Waard, M., Pragnell, M. & Campbell, K. P. (1994) Neuron 13, 495-503. [DOI] [PubMed] [Google Scholar]
- 23.Dascal, N. (2001) Trends Endocrinol. Metab. 12, 391-398. [DOI] [PubMed] [Google Scholar]
- 24.Wouters, F. S. & Bastiaens, P. I. H. (1999) Curr. Biol. 9, 1127-1130. [DOI] [PubMed] [Google Scholar]
- 25.Gordon, G. W., Berry, G., Liang, X. H., Levine, B. & Herman, B. (1998) Biophys. J. 74, 2702-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourinet, E., Soong, T. W., Stea, A. & Snutch, T. P. (1996) Proc. Natl. Acad. Sci. USA 93, 1486-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herlitze, S., Zhong, H., Scheuer, T. & Catterall, W. A. (2001) Proc. Natl. Acad. Sci. USA 98, 4699-4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hümmer, A., Delzeith, O., Gomez, S. R., Moreno, R. L., Mark, M. D. & Herlitze, S. (2003) J. Biol. Chem. 278, 49386-49400. [DOI] [PubMed] [Google Scholar]
- 29.Fathallah, M., Sandoz, G., Mabrouk, K., Geib, S., Villaz, M., Sabatier, J. M. & De Waard, M. (2002) Eur. J. Neurosci. 16, 219-228. [DOI] [PubMed] [Google Scholar]
- 30.Hibino, H., Pironkova, R., Onwumere, O., Rousset, M., Charnet, P., Hudspeth, A. J. & Lesage, F. (2003) Proc. Natl. Acad. Sci. USA 100, 307-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.