

Abstract
Excitatory amino acid transporters (EAATs) located on neurons and glia are responsible for limiting extracellular glutamate concentrations, but specific contributions made by neuronal and glial EAATs have not been determined. At climbing fiber to Purkinje cell (PC) synapses in cerebellum, a fraction of released glutamate is rapidly bound and inactivated by neuronal EAATs located on postsynaptic PCs. Because transport involves a stoichiometric movement of ions and is electrogenic, postsynaptic currents mediated by EAATs should permit precise calculation of the amount of postsynaptic glutamate uptake. However, this is possible only if a stoichiometric EAAT current can be isolated from all other contaminating signals. We used synaptic stimulation and photolysis of caged glutamate to characterize the current in PCs that is resistant to high concentrations of glutamate receptor antagonists. Some of this response is inhibited by the high-affinity EAAT antagonist TBOA (dl-threo-β-benzyloxyaspartic acid), whereas the remaining current shows properties inconsistent with glutamate transport. By subtracting this residual non-EAAT current from the response recorded in glutamate receptor antagonists, we have obtained an estimate of postsynaptic uptake near physiological temperature. Analysis of such synaptic EAAT currents suggests that, on average, postsynaptic EAATs take up ≈1,300,000 glutamate molecules in response to a single climbing fiber action potential.
Extracellular neurotransmitter concentrations in the central nervous system are regulated by highly specific systems of reuptake that rapidly bind and quickly move neurotransmitters across membranes. Neurotransmitter reuptake facilitates chemical synaptic transmission by limiting receptor activation and by recycling released neurotransmitter molecules. In the case of glutamate, the primary excitatory neurotransmitter of the central nervous system, uniquely localized excitatory amino acid transporters (EAATs), influence both ionotropic (1–5) and metabotropic glutamate receptor (GluR) (6–9) activation. EAATs on glial (10–12) and neuronal (12–14) processes accomplish this task by relying on the Na+ and K+ electrochemical gradients to move glutamate against its own large concentration gradient.
A single cycle of transport is estimated to take tens of milliseconds (15–20) and is accompanied by the cotransport of 3 Na+, 1 H+ and 1 glutamate- and the countertransport of 1 K+ across the membrane (21, 22). Such stoichiometry results in a net inward movement of two charges per cycle, meaning that glutamate transport is electrogenic. In addition to these stoichiometric fluxes, EAATs become permeable to anions at specific points during the transport cycle (16–19, 23, 24). Under conditions in which anion flux through EAATs is minimized, stoichiometric currents can be used to estimate glutamate flux by taking advantage of the fact that the number of glutamate molecules transported is equal to half the net movement of elementary charges.
In an effort to determine the amount of neuronal glutamate uptake, previous studies have recorded EAAT currents at the only place in the brain where such measurements can be readily made in single neurons, at the climbing fiber (CF)–Purkinje cell (PC) synapse in the cerebellum (25, 26). However, these measurements have either been indirect in that they relied on the EAAT anion current (26), or were compromised by incomplete isolation of the stoichiometric EAAT current (25). To make a more precise estimate of neuronal glutamate uptake, we have carefully analyzed synaptic and photolysis-evoked currents recorded in PCs under various conditions. Our experiments confirm that it is possible to record the stoichiometric EAAT current in PCs. In addition, they identify a glutamate-mediated current that is resistant to standard GluR and EAAT antagonists. We show that this latter current is elicited by release of glutamate from CFs but is not an EAAT current. Isolation of the synaptic EAAT current from this contaminating current suggests that, near physiological temperatures, PC EAATs bind 1.32 ± 0.10 × 106 glutamate molecules after CF activity. Given assumptions about glutamate release at this synapse, we estimate that 16.9 ± 1.3% of the total glutamate released by CFs is removed by postsynaptic EAATs.
Methods
Brain Slice Preparation. Experiments were performed on PCs in 300-μm parasagittal slices of cerebellum from 13- to 17-day-old Sprague–Dawley rats according to institutional guidelines. Animals were anesthetized with halothane and decapitated. The cerebellum was removed and glued to an agar support fixed onto the specimen holder of a vibrating blade microtome (VT1000S, Leica Microsystems). Sections of the vermis were made in ice-cold artificial cerebrospinal fluid (ACSF) containing 119 mM NaCl, 26 mM NaHCO3, 11 mM d-glucose, 2.5 mM KCl, 2.5 mM CaCl2, 1.3 mM MgCl2,and 1 mM NaH2PO4. Slices were first incubated at 35 ± 1°C for 30 min and subsequently stored at 23 ± 1°C in ACSF saturated with 95% O2 and 5% CO2.
Electrophysiology. Recording solutions. Extracellular solution consisted of artificial cerebrospinal fluid plus 80 nM NBQX (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide) and 100 μM picrotoxin. External solutions were saturated with 95% O2 and 5% CO2 and maintained at 33 ± 1°C (unless otherwise indicated) with an in-line solution heater (Warner Instruments) for synaptic stimulation experiments or at 23 ± 1°C for photolysis experiments. For photolysis experiments, 0.5 μM tetrodotoxin was also added to the extracellular solution and, where indicated, 109 mM NaNO3 was substituted for NaCl (10 mM NaCl remained to minimize electrode drift). To inhibit GluRs in experiments using Cs+- and Na+-based pipette solutions, the external solution also contained 20 μM NBQX, 25 μM GYKI 52466 [1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride], 100–500 μM LY 367385 (S-+-α-amino-4-carboxy-2-methylbenzeneacetic acid), and 5 μM CPP [RS-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid]; 100 μM TBOA (dl-threo-β-benzyloxyaspartic acid) was used to block EAATs. To inhibit GluRs in experiments using the K+-based pipette solution, the external solution contained 100 μM NBQX, 100 μM LY 367385, 100 μM RS-α-cyclopropyl-4-phosphonophenylglycine (CPPG) and 5 μM CPP; 200 μM TBOA was used to block EAATs in these experiments. Pipette solutions used in synaptic experiments contained 130 mM CsCH3SO3 or 130 mM KCH3SO3, 10 mM Hepes, 10 mM EGTA, 4 mM NaCl, 1 mM CaCl2, and 0–10 mM N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide (QX-314), adjusted to pH 7.3. Pipette solutions used in photolysis experiments contained 140 mM CsCH3SO3 or 130 mM NaCH3SO3, 10 mM Hepes, 0.2–10 mM EGTA, 1–4 mM MgCl2, 4 mM ATP, and 0.4 mM GTP, adjusted to pH 7.3. Chemicals were obtained from Sigma, Tocris-Cookson, and A.G. Scientific.
PC recordings. Cerebellar slices were visualized at ×40 through a water immersion objective using an upright microscope equipped with infrared differential interference contrast microscopy enhancement (Axioskop 2FS Plus, Carl Zeiss). Whole-cell recordings of PCs held at -70 mV, unless otherwise indicated, were made by using an Axopatch 1D amplifier (Axon Instruments). Recording pipettes were 1.2–1.6 MΩ and series resistances were typically <10 MΩ. All recordings were filtered at 2 kHz and digitized at 10 kHz (synaptic stimulation) or 5 kHz (photolysis experiments). For synaptic experiments, all-or-none responses exhibiting paired pulse depression were activated with an ISO-Flex stimulator (A.M.P.I., Jerusalem) at 0.05 Hz by applying constant current or voltage steps (10–100 μA or 5–30 V/40–100 μs) with a bipolar theta pipette placed in the granule cell layer near the recorded neuron. CF stimulation was adjusted to maximize synaptic delay and minimize stimulus artifact. Photolysis-evoked responses were elicited at 0.0167 Hz by uncaging of 500 μM γ-(CNB-caged)-l-glutamic acid (Molecular Probes) dissolved in 5 ml of recirculating bath solution. Transient (50-ms) UV illumination from a 100 W mercury arc lamp (Carl Zeiss) was provided via the epifluorescence pathway. Timing of the light stimulus was controlled by an electronic shutter (Uniblitz).
Data Acquisition and Analysis. Data were acquired and analyzed by using pclamp 8.2 (Axon Instruments) and igor pro 4.0 (Wavemetrics). Traces were filtered at 1 kHz for display. Single exponentials were fit by using clampfit 9.0 (Axon Instruments); all fits were constrained to decay to a final value of 0. Statistical significance was determined with one-way ANOVA. Reported results represent mean ± SEM.
There is some concern that blocking glutamate uptake may significantly slow residual non-EAAT excitatory postsynaptic currents (EPSCs) and thereby compromise the digital subtractions presented in Fig. 5. To determine whether such distortion occurs and to set an appropriate time window for integration, we applied the following procedures. Each subtracted trace (e.g., Fig. 5 C and F) was integrated for different time periods (30 ms, 60 ms, 90 ms, and 120 ms) starting at a time point (<1 ms) after the stimulus artifact had settled. For each trace we compared ratios of these integrals in the form Q60ms/Q30ms, Q90ms/Q60ms, Q120ms/Q90ms to assess the optimal integration window required to recover all charge. Average ratios from this procedure from synaptic experiments using the Cs+-based pipette solution at 23 ± 1°C were: Q60ms/Q30ms = 1.10 ± 0.05 (n = 5); Q90ms/Q60ms = 1.03 ± 0.03 (n = 5); Q120ms/Q90ms = 1.10 ± 0.09 (n = 5). Ratios from synaptic experiments using the Cs+-based pipette solution at 33 ± 1°C were: Q60ms/Q30ms = 1.21 ± 0.07 (n = 15); Q90ms/Q60ms = 1.14 ± 0.05 (n = 15); Q120ms/Q90ms = 1.02 ± 0.03 (n = 15). Ratios from synaptic experiments using the K+-based pipette solution at 33 ± 1°C were: Q60ms/Q30ms = 1.19 ± 0.02 (n = 6); Q90ms/Q60ms = 1.11 ± 0.02 (n = 6); Q120ms/Q90ms = 1.05 ± 0.01 (n = 6). If an outward current caused by the slowed decay of residual EPSCs systematically appeared in the later phase of subtracted traces, the ratio would drop below a value of 1. The finding that these values do not fall below 1.0 in any of the intervals examined confirms that charge was not systematically lost at late times in the decay phase of subtracted responses. On the basis of these results, we chose an integration window of 90 ms for all data presented in Fig. 5 and Table 1.
Fig. 5.

Calculation of glutamate uptake into single PCs. (A) To estimate transport into a single PC recorded by using a Cs+-based internal solution, the CF input is identified in 80 nM NBQX. (B) High concentrations of GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 100 μM LY 367385, 5 μM CPP) leave a small CF-evoked EPSC (thick trace) that is partially inhibited by 100 μM TBOA to leave the residual EPSC (thin trace). (C) Subtracting the residual EPSC (thin trace in B) from the response in GluR antagonists (thick trace in B) yields the TBOA sensitive component that represents a stoichiometric EAAT current under these recording conditions. (D) To estimate transport into a single PC recorded by using a K+-based internal solution, the CF input is identified in 80 nM NBQX. (E) Higher concentrations of GluR antagonists (100 μM NBQX, 100μM LY 367385, 100 μM CPPG, 5 μM CPP) leave a small CF-evoked EPSC (thick trace) that is partially inhibited by 200 μM TBOA to leave the residual EPSC (thin trace). (F) Subtracting the residual EPSC (thin trace in E) from the response in GluR antagonists (thick trace in E) yields the TBOA-sensitive component that represents a stoichiometric EAAT current under these recording conditions. Glutamate uptake can then be estimated under the two conditions tested by integrating the subtracted trace during a 90-ms time interval commencing at the vertical dashed line in C and F and then calculating the corresponding number of glutamates transported based on two elementary charges per cycle of transport.
Table 1. Summary of results from synaptic recordings made under different experimental conditions.
| Cs+-based pipette solution
|
K+-based pipette solution
|
||
|---|---|---|---|
| 23 ± 1°C, n = 5 | 33 ± 1°C, n = 15 | 33 ± 1°C, n = 6 | |
| GluR antagonists, μM | 20 NBQX, 25 GYKI 52466, 100 LY 367385, 5 CPP | 20 NBQX, 25 GYKI 52466, 100 LY 367385, 5 CPP | 100 NBQX, 100 LY 367385, 100 CPPG, 5 CPP |
| Peak current in GluR antagonists, pA | —39.8 ± 2.4 | —57.8 ± 5.1 | —42.2 ± 4.8 |
| TBOA concentration, μM | 100 | 100 | 200 |
| Peak current in GluR antagonists and TBOA, pA | —22.2 ± 2.7 | —31.0 ± 3.5 | —12.3 ± 1.6† |
| Percent TBOA block | 43.9 ± 6.8 | 47.2 ± 3.4 | 70.8 ± 2.4*† |
| EAAT current peak amplitude, pA | —20.7 ± 3.3 | —30.2 ± 3.2 | —32.7 ± 4.5 |
| EAAT current rise time, 20—80% of peak amplitude, ms | 3.2 ± 0.7 | 0.9 ± 0.2* | 1.2 ± 0.3* |
| EAAT current τdecay, ms | 11.6 ± 1.8 | 6.7 ± 0.8* | 8.7 ± 1.4 |
| EAAT current cumulative charge, fC | —317.4 ± 70.8 | —336.7 ± 47.2 | —423.4 ± 32.9 |
Statistical significance was determined by ANOVA and a Tukey—Kramer multiple comparisons post test.
Significance in comparison to column 1 (P < 0.05)
Significance in comparison to column 2 (P < 0.05)
Results
A CF-Evoked Synaptic Current Persists in the Presence of GluR and EAAT Antagonists. CF-evoked synaptic currents are dominated by a large AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor-mediated current (27), although much smaller components can be attributed to group I metabotropic GluRs (mGluRs) (28) and EAATs (4, 28–30). To record CF-evoked stoichiometric EAAT currents, we chose to examine synaptic responses in PCs filled with a pipette solution and recorded at a holding potential expected to minimize the EAAT anion current (see Methods and ref. 19). Experiments commenced in extracellular solution containing a saturating concentration (100 μM) of the γ-aminobutyric acid type A (GABAA), receptor antagonist picrotoxin and a low concentration (80 nM) of the non-N-methyl-d-aspartic acid (NMDA) receptor antagonist NBQX (Fig. 1A). After identifying a CF input, GluRs were inhibited by a mixture of GluR antagonists consisting of 20 μM NBQX, 25 μM GYKI 52466, 100 μM LY 367385, and 5 μM CPP (Fig. 1 A). In the presence of these high concentrations of GluR antagonists, small (-39.8 ± 2.4 pA at 23 ± 1°C, n = 5; -57.8 ± 5.1 pA at 33 ± 1°C, n = 15), all-or-none EPSCs were elicited by CF stimulation (Fig. 1B, thick trace). Similar to previous results (25, 28), addition of the high-affinity nontransported EAAT antagonist TBOA (100 μM) caused incomplete inhibition of the response by 43.9 ± 6.8% at 23 ± 1°C (n = 5) and by 47.2 ± 3.4% at 33 ± 1°C (n = 15) to an average of –22.2 ± 2.7 pA at 23 ± 1°C (n = 5) and –31.0 ± 3.5 pA at 33 ± 1°C (n = 15) (Fig. 1B, thin trace). These data are summarized in Table 1. The remaining current could arise from an incomplete blockade of EAATs by TBOA. However, such GluR and EAAT antagonist-resistant currents (hereafter referred to as “residual EPSCs”) were significantly outward at +35 mV (Fig. 1C) and exhibited linear I–V relationships with reversal potentials near 0 mV (Fig. 1D), properties inconsistent with EAAT currents.
Fig. 1.

A GluR and EAAT antagonist-insensitive residual current is present at CF-PC synapses. (A) CF activation in 80 nM NBQX generates a large inward current that is strongly inhibited by external solution containing GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 100 μM LY 367385, 5 μM CPP). (B) The response recorded in GluR antagonists (thick trace) is incompletely inhibited by the EAAT antagonist TBOA (100 μM), leaving a residual EPSC (thin trace). (C) The residual EPSC remaining in GluR and EAAT antagonists reverses near 0 mV (n = 6). (D) I-V relation for the peak residual EPSC (n = 4).
Photolysis of Caged Glutamate also Elicits a Residual Current in PCs. The data presented above cannot rule out the possibility that residual EPSCs arise from the release by the CF of a neurotransmitter other than glutamate. This issue was examined by characterizing currents elicited upon photolysis of caged glutamate. As previously described, photolysis of caged glutamate generated inward currents in PCs that were mediated in part by AMPA receptors (Fig. 2A) and by mGluRs (31). As was the case for CF EPSCs, a mixture of GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP) incompletely inhibited photolysis-evoked currents (Fig. 2 A). The inward current that persisted in GluR antagonists (Fig. 2B, thick trace) was only partially blocked by 100 μM TBOA (Fig. 2B, thin trace). These residual currents reversed near 0 mV and were prominently outward at +35 mV (Fig. 2C), exhibiting I–V relationships (Fig. 2D) similar to those observed for residual EPSCs. These results are consistent with the hypothesis that residual EPSCs and residual photolysis-evoked currents are caused by a glutamate-activated conductance with nonselective permeability to cations.
Fig. 2.

Photolysis of caged glutamate also elicits a residual current in PCs. (A) Exogenous application of glutamate via photolysis elicits a large inward response in 80 nM NBQX that is inhibited by external solution containing GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP). Horizontal bars above traces indicate shutter open time (50 ms). (B) Similar to synaptic experiments, the photolysis-evoked response in GluR antagonists (thick trace) is only partially blocked by the EAAT antagonist TBOA (100 μM), leaving a residual current (thin trace). (C) The residual current in GluR and EAAT antagonists reverses near 0 mV. (D) I–V relation for the peak residual current (n = 7).
Intracellular Na+ Substitution Does Not Inhibit the Photolysis-Evoked Residual Current. EAATs derive energy from the Na+ and K+ electrochemical gradients through the cotransport of 3 Na+ and countertransport of 1 K+ (21, 22). For this reason, disrupting the Na+ gradient impairs glutamate transport (32) and alters EAAT currents (17–19). We took advantage of this Na+ dependence to further confirm that the photolysis-evoked residual current is not an EAAT current. PCs were dialyzed with a Na+-based internal solution, and photolysis-evoked currents were recorded in the presence of GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP) and 100 μM TBOA. Analysis was confined to times >20 min after breakthrough into whole-cell mode to allow for maximal Na+ dialysis. Residual responses persisted even under these conditions (Fig. 3A). Moreover, their I-V relationship (Fig. 3B) was indistinguishable from that of residual currents recorded with a Cs+-based internal solution (Fig. 2D). Thus, even in the face of manipulations expected to severely impair EAAT function, namely pharmacological blockade of EAATs and removal of the electrochemical driving force for transport, a residual current remained. These features strongly imply that this current is not an EAAT current.
Fig. 3.

Intracellular Na+ substitution does not inhibit the photolysis-evoked residual current. (A) Dialysis of a PC with a Na+-based internal solution for >20 min after breakthrough should prevent glutamate transport, but this manipulation does not affect residual currents recorded in GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP) and TBOA (100 μM). Horizontal bar above traces indicates shutter open time (50 ms). (B) Average I–V relationships for residual currents recorded in Na+-filled cells (open circles, n = 7) and under control conditions (filled circles, same data as in Fig. 2D).
The Photolysis-Evoked Residual Current Shows Minimal Anion Permeability. All EAATs are known to generate uncoupled anion fluxes that can be quite large when highly permeable anions such as 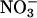 are present on either side of the membrane (16–19, 23, 25, 26, 33). With this common feature of EAATs in mind, photolysis-evoked responses were examined in a NaNO3-based external solution, a condition expected to enhance the EAAT anion current and to shift the reversal potential of the EAAT current in the negative direction. As expected in the absence of EAAT antagonists, a prominent anion current component characteristic of glutamate transport was evident upon glutamate photolysis in GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP) (Fig. 4A). Inhibiting EAATs with 100 μM TBOA caused a large positive shift in the reversal potential of the photolysisevoked response in NaNO3 (Fig. 4D), revealing a current (Fig. 4 B and C) indistinguishable from the residual current described previously (Fig. 2C). These results confirm that the large EAAT-associated anion current detected in PCs is blocked by TBOA, leaving the residual response. The apparent lack of anion permeability of the residual current is further evidence that these responses do not arise from the activity of EAATs.
are present on either side of the membrane (16–19, 23, 25, 26, 33). With this common feature of EAATs in mind, photolysis-evoked responses were examined in a NaNO3-based external solution, a condition expected to enhance the EAAT anion current and to shift the reversal potential of the EAAT current in the negative direction. As expected in the absence of EAAT antagonists, a prominent anion current component characteristic of glutamate transport was evident upon glutamate photolysis in GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP) (Fig. 4A). Inhibiting EAATs with 100 μM TBOA caused a large positive shift in the reversal potential of the photolysisevoked response in NaNO3 (Fig. 4D), revealing a current (Fig. 4 B and C) indistinguishable from the residual current described previously (Fig. 2C). These results confirm that the large EAAT-associated anion current detected in PCs is blocked by TBOA, leaving the residual response. The apparent lack of anion permeability of the residual current is further evidence that these responses do not arise from the activity of EAATs.
Fig. 4.

Unlike EAAT currents, photolysis-evoked residual currents have no detectable anion permeability. (A) Photolysis of caged glutamate in a NaNO3-based external solution containing GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 500 μM LY 367385, 5 μM CPP), but no EAAT antagonists, elicits a prominent EAAT anion current that reverses near -60 mV. Horizontal bars above traces indicate shutter open time (50 ms). (B) Inhibiting EAATs with TBOA (100 μM) dramatically shifts the reversal potential and reveals the residual current at -35 mV. (C) The residual current recorded in a NaNO3-based external solution reverses near 0 mV. (D) Average I–V relationships for the photolysis-evoked currents in a NaNO3-based external solution containing GluR antagonists before (open triangles, n = 8) and after (filled triangles, n = 5) TBOA application.
Measuring Neuronal Glutamate Uptake. The findings described above identify at least two components of glutamate-evoked current that persist in high concentrations of GluR antagonists. The first is an EAAT current that is blocked by TBOA. The second has several properties that are inconsistent with glutamate transport: it persists in TBOA, persists in a symmetrical Na+ gradient, and shows very low anion permeability. Indeed, recent evidence suggests that this current is a glutamate-gated, mixed-cation current (30). Because of the small size of the stoichiometric EAAT current compared to this residual cation current, integration of CF EPSCs recorded in high concentrations of GluR antagonists will significantly overestimate postsynaptic uptake. Therefore, under various experimental conditions discussed below, we used a subtractive procedure to first isolate and then integrate only the TBOA-sensitive component, procedures outlined in Fig. 5 and summarized in Table 1.
The first sets of experiments were made with Cs+-based pipette solutions. CF inputs were identified under conditions where non-N-methyl-d-aspartic acid (NMDA) receptors were partially inhibited (Fig. 5A). Average responses were then recorded, first after applying high concentrations of GluR antagonists (20 μM NBQX, 25 μM GYKI 52466, 100 μM LY 367385, 5 μM CPP) (Fig. 5B, thick trace), and second after addition of 100 μM TBOA (Fig. 5B, thin trace). Digital subtraction of the latter current trace from the response recorded before TBOA application yields a trace representing the stoichiometric EAAT current (Fig. 5C). This response was integrated within a 90-ms window immediately after current onset (see Methods). Applied to 5 CF inputs at room temperature and 15 CF inputs at near physiological temperature, this strategy uncovered an average TBOA-sensitive charge of -317.4 ± 70.8 fC at 23 ± 1°C (n = 5) and -336.7 ± 47.2 fC at 33 ± 1°C (n = 15). As expected, EAAT currents recorded near physiological temperature were significantly faster to rise and decay compared to measurements made at room temperature (Table 1), reflecting the more rapid turnover of transporters at elevated temperature.
Results published while this paper was being reviewed reported that the residual EPSC is generated by GluRs with a low affinity for NBQX (30). This work also suggests that TBOA may depress glutamate release from CFs because of accumulated glutamate that activates presynaptic mGluRs. To address these concerns and also obtain a more physiologically accurate measure of glutamate transport, we used the subtractive strategy described above but instead recorded CF responses at 33 ± 1°C with a K+-based pipette solution in the presence of a group II/III mGluR antagonist (100 μM CPPG) and higher concentrations of AMPA/kainate receptor antagonists (100 μM NBQX). A higher concentration of TBOA (200 μM) was also used to isolate the transporter current. Table 1 compares the results obtained under these conditions with those described earlier. These manipulations revealed significantly smaller residual EPSCs (Fig. 5 D–F and Table 1) and a TBOA-sensitive charge of -423.4 ± 32.9 fC (n = 6), which was slightly larger but not significantly different from measurements made by using Cs+-based solutions and lower concentrations of GluR and EAAT antagonists in the absence of CPPG. Assuming an inward flow of +2e per transport cycle (21, 22), the net charge movements measured in this latter condition correspond to 1.32 ± 0.10 × 106 (n = 6) glutamate molecules transported into the postsynaptic PC after CF activation at 33 ± 1°C.
Discussion
In mature rats, each CF provides an exceptionally strong synaptic input onto a few PCs in the cerebellum (34, 35). Glutamate released at this synapse rapidly elicits a large non-N-methyl-d-aspartic acid (NMDA) receptor-mediated EPSC that is strongly inhibited by ionotropic GluR antagonists. Under conditions where the EAAT anion conductance is minimized, GluR antagonists block a fraction (≈50–70%) of this CF-evoked EPSC (28, 29). Despite the incomplete inhibition, the current remaining in GluR antagonists has been interpreted to be a synaptically evoked stoichiometric EAAT current, and this response was used to estimate the amount and time course of postsynaptic glutamate uptake (25).
We have carefully examined currents that persist in GluR antagonists in response to either CF synaptic stimulation or photolysis of caged glutamate and find two components. One is TBOA sensitive and is presumably a stoichiometric EAAT current. The other component, termed “residual current,” has several features inconsistent with glutamate transport. First, we have confirmed that the residual current is not blocked by high concentrations of TBOA, a competitive antagonist for EAAC1 and EAAT4 (36, 37), the two EAATs expressed by PCs (12, 14, 24, 38). Second, I–V analysis of the residual synaptic and photolysis-evoked currents shows that they uniformly reverse polarity at potentials near 0 mV, which would not be expected of stoichiometric EAAT currents or of responses consisting of mixed stoichiometric and anion currents. Third, the residual current is unaffected by imposing a symmetrical Na+ gradient, a condition that would drastically alter the activity of any Na+-dependent transporter (18, 19). Fourth, although the I–V relationship of photolysis-evoked EAAT currents shows a clear shift when the anion gradient is altered, the I–V relationship of the residual current is unchanged. Taken together, these data strongly argue that the TBOA-insensitive residual current is not an EAAT current. Rather, on the basis of its glutamate dependence, kinetics and reversal potential, the residual current is most likely mediated by ionotropic GluRs. Indeed, recently published results suggest that this current arises from a low-affinity kainate receptor comprised of GluR5 subunits (30). Complete characterization of this component of the CF EPSC will depend on pharmacological agents effective at GluRs containing different AMPA or kainate receptor subunits.
Because the residual current is not an EAAT current, integrating the synaptic current that remains in GluR antagonists significantly overestimates PC glutamate uptake and may yield an inaccurate picture of EAAT activation kinetics at synapses. A more accurate measurement of transport can be made if only the TBOA-sensitive synaptic component is integrated. Applying this strategy to responses recorded near physiological temperature suggests that neuronal EAATs in PCs transport 1.32 ± 0.10 × 106 glutamate molecules in response to synaptic stimulation. To calculate the fraction of glutamate transported into the postsynaptic neuron from this value, it is necessary to estimate the amount of glutamate released. CFs are believed to release 2.5–4 vesicles (4) containing 4,700 ± 150 transmitter molecules (39) from 510 ± 50 functional release sites (40). Combining these assumptions (510 sites releasing 3.25 vesicles containing 4,700 glutamate molecules) with our average estimate of uptake recorded with a K+-based pipette solution implies that EAATs on a PC remove ≈16.9 ± 1.3% of the total glutamate released in response to a single CF action potential. To our knowledge, this estimate represents the first measurement of pure stoichiometric transporter currents at a synapse.
Postsynaptic glutamate uptake at the CF synapse could serve several purposes. Strategic clearance could isolate patches of postsynaptic receptors opposed to CF boutons in the face of prominent multivesicular release (4). In addition, this mechanism may limit activation of the mGluRs present at CF synapses (28, 29). Although the absolute magnitude of postsynaptic uptake is small, the privileged localization of these uptake sites makes it difficult to generalize about their influence on glutamate concentrations, especially when considering small spatial scales. Ultimately, simulations of glutamate diffusion that incorporate GluR-binding sites and EAATs into realistic spaces will be required to address these issues.
Acknowledgments
We thank members of the Otis laboratory for helpful discussions. This work was supported by National Institutes of Health Grant NS040499.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: EAAT, excitatory amino acid transporter; GluR, glutamate receptor; CF, climbing fiber; PC, Purkinje cell; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide; GYKI 52466, 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride; LY 367385, S-+-α-amino-4-carboxy-2-methylbenzeneacetic acid; CPP, RS-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid; TBOA, dl-threo-β-benzyloxyaspartic acid; EPSC, excitatory postsynaptic current; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; mGluR, metabotropic GluR; CPPG, RS-α-cyclopropyl-4-phosphophenylglycine.
References
- 1.Barbour, B., Keller, B. U., Llano, I. & Marty, A. (1994) Neuron 12, 1331-1343. [DOI] [PubMed] [Google Scholar]
- 2.Diamond, J. S. & Jahr, C. E. (1997) J. Neurosci. 17, 4672-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Otis, T. S., Wu, Y. C. & Trussell, L. O. (1996) J. Neurosci. 16, 1634-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadiche, J. I. & Jahr, C. E. (2001) Neuron 32, 301-313. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi, M., Sarantis, M. & Attwell, D. (1996) J. Physiol. (London) 497, 523-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heuss, C., Scanziani, M., Gahwiler, B. H. & Gerber, U. (1999) Nat. Neurosci. 2, 1070-1077. [DOI] [PubMed] [Google Scholar]
- 7.Scanziani, M., Salin, P. A., Vogt, K. E., Malenka, R. C. & Nicoll, R. A. (1997) Nature 385, 630-634. [DOI] [PubMed] [Google Scholar]
- 8.Brasnjo, G. & Otis, T. S. (2001) Neuron 31, 607-616. [DOI] [PubMed] [Google Scholar]
- 9.Reichelt, W. & Knopfel, T. (2002) J. Neurophysiol. 87, 1974-1980. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhry, F. A., Lehre, K. P., van Lookeren Campagne, M., Ottersen, O. P., Danbolt, N. C. & Storm-Mathisen, J. (1995) Neuron 15, 711-720. [DOI] [PubMed] [Google Scholar]
- 11.Lehre, K. P., Levy, L. M., Ottersen, O. P., Storm-Mathisen, J. & Danbolt, N. C. (1995) J. Neurosci. 15, 1835-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothstein, J. D., Martin, L., Levey, A. I., Dykes-Hoberg, M., Jin, L., Wu, D., Nash, N. & Kuncl, R. W. (1994) Neuron 13, 713-725. [DOI] [PubMed] [Google Scholar]
- 13.Furuta, A., Rothstein, J. D. & Martin, L. J. (1997) J. Neurosci. 17, 8363-8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dehnes, Y., Chaudhry, F. A., Ullensvang, K., Lehre, K. P., Storm-Mathisen, J. & Danbolt, N. C. (1998) J. Neurosci. 18, 3606-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wadiche, J. I., Arriza, J. L., Amara, S. G. & Kavanaugh, M. P. (1995) Neuron 14, 1019-1027. [DOI] [PubMed] [Google Scholar]
- 16.Wadiche, J. I. & Kavanaugh, M. P. (1998) J. Neurosci. 18, 7650-7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otis, T. S. & Jahr, C. E. (1998) J. Neurosci. 18, 7099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otis, T. S. & Kavanaugh, M. P. (2000) J. Neurosci. 20, 2749-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergles, D. E., Tzingounis, A. V. & Jahr, C. E. (2002) J. Neurosci. 22, 10153-10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergles, D. E. & Jahr, C. E. (1998) J. Neurosci. 18, 7709-7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy, L. M., Warr, O. & Attwell, D. (1998) J. Neurosci. 18, 9620-9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zerangue, N. & Kavanaugh, M. P. (1996) Nature 383, 634-637. [DOI] [PubMed] [Google Scholar]
- 23.Wadiche, J. I., Amara, S. G. & Kavanaugh, M. P. (1995) Neuron 15, 721-728. [DOI] [PubMed] [Google Scholar]
- 24.Fairman, W. A., Vandenberg, R. J., Arriza, J. L., Kavanaugh, M. P. & Amara, S. G. (1995) Nature 375, 599-603. [DOI] [PubMed] [Google Scholar]
- 25.Auger, C. & Attwell, D. (2000) Neuron 28, 547-558. [DOI] [PubMed] [Google Scholar]
- 26.Otis, T. S., Kavanaugh, M. P. & Jahr, C. E. (1997) Science 277, 1515-1518. [DOI] [PubMed] [Google Scholar]
- 27.Konnerth, A., Llano, I. & Armstrong, C. M. (1990) Proc. Natl. Acad. Sci. USA 87, 2662-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dzubay, J. A. & Otis, T. S. (2002) Neuron 36, 1159-1167. [DOI] [PubMed] [Google Scholar]
- 29.Auger, C. & Marty, A. (1997) Neuron 19, 139-150. [DOI] [PubMed] [Google Scholar]
- 30.Huang, Y. H., Dykes-Hoberg, M., Tanaka, K., Rothstein, J. D. & Bergles, D. E. (2004) J. Neurosci. 24, 103-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canepari, M., Papageorgiou, G., Corrie, J. E., Watkins, C. & Ogden, D. (2001) J. Physiol. (London) 533, 765-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanner, B. I. & Sharon, I. (1978) Biochemistry 17, 3949-3953. [DOI] [PubMed] [Google Scholar]
- 33.Clark, B. A. & Barbour, B. (1997) J Physiol (London) 502, 335-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palay, S. L. & Chan-Palay, V. (1974) Cerebellar Cortex: Cytology and Organization (Springer, New York).
- 35.Sugihara, I., Wu, H. S. & Shinoda, Y. (2001) J. Neurosci. 21, 7715-7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimamoto, K., Lebrun, B., Yasuda-Kamatani, Y., Sakaitani, M., Shigeri, Y., Yumoto, N. & Nakajima, T. (1998) Mol. Pharmacol. 53, 195-201. [DOI] [PubMed] [Google Scholar]
- 37.Shigeri, Y., Shimamoto, K., Yasuda-Kamatani, Y., Seal, R. P., Yumoto, N., Nakajima, T. & Amara, S. G. (2001) J. Neurochem. 79, 297-302. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka, J., Ichikawa, R., Watanabe, M., Tanaka, K. & Inoue, Y. (1997) NeuroReport 8, 2461-2464. [DOI] [PubMed] [Google Scholar]
- 39.Bruns, D. & Jahn, R. (1995) Nature 377, 62-65. [DOI] [PubMed] [Google Scholar]
- 40.Silver, R. A., Momiyama, A. & Cull-Candy, S. G. (1998) J. Physiol. (London) 510, 881-902. [DOI] [PMC free article] [PubMed] [Google Scholar]