

Abstract
Endometriosis, a steroid hormone–dependent disease characterized by aberrant activation of estrogen receptor signaling and progesterone resistance, remains intractable because of the complexity of the pathways underlying its manifestation. We previously showed that eutopic endometria of women with endometriosis exhibit lower expression of Krüppel-like factor 9 (KLF9), a progesterone receptor coregulator in the uterus, relative to that of women without disease. Here we examined whether loss of endometrial KLF9 expression causes ectopic lesion establishment using syngeneic wild-type (WT) mice as recipients of endometrial fragments from WT and Klf9 null donors. We found significantly higher incidence of ectopic lesions with Klf9 null than WT endometria 8 weeks after tissue injection into the intraperitoneal cavity. The increased incidence of lesion establishment with Klf9 null endometria was associated with a higher expression ratio of estrogen receptor 2 isoform relative to that of estrogen receptor 1 and attenuated progesterone receptor levels in endometriotic stromal cells. PCR array analyses of Notch and Hedgehog signaling components in ectopic lesions demonstrated up-regulated expression of select genes (Jag 2, Shh, Gli1, and Stil 1) in Klf9 null lesions relative to that in WT lesions. Immunohistochemical analyses showed increased levels of Notch intracellular domain and Sonic Hedgehog proteins in Klf9 null lesions relative to that in WT lesions, confirming pathway activation. WT recipients with Klf9 null lesions displayed lower systemic levels of TNFα and IL-6 and higher soluble TNF receptor 1 than corresponding recipients with WT lesions. Our results suggest that endometrial KLF9 deficiency promotes endometriotic lesion establishment by the coincident deregulation of Notch-, Hedgehog-, and steroid receptor–regulated pathways.
Endometriosis is a benign gynecologic disorder defined as the presence of functional endometrial stroma and glands (ectopic endometrium) outside of the uterus (eutopic endometrium) (1). The disease affects 1 in 10 reproductive age women, 50% of whom are correspondingly diagnosed with infertility (2). Patients with endometriosis have poor success rates after in vitro fertilization, show higher rates of pregnancy loss and pregnancy-associated complications, and exhibit prevalent comorbidities of ovarian cancer, melanoma, and vaginal infections (3–5). Current treatments for endometriosis are relatively ineffective (6), and recurrence is seen in up to 50% of patients (7), requiring additional surgery within 5 years of the initial procedure (8). Although the etiology of the disease remains unclear, the most accepted is Sampson's transplantation theory of retrograde menstruation (9). The latter does not fully explain the condition, however, because ∼90% of women experience retrograde menstruation, yet only ∼10% develop the disease. Thus, there remains a pressing need for a better understanding of its pathogenesis to enable prevention and provide an effective cure.
Abnormalities in steroid hormone receptor signaling characterize the endometria of women with endometriosis (10). The mitogenic actions of estradiol (E2), which are mediated through 2 estrogen receptor (ESR) isoforms, ESR1 and ESR2 (11), are critical for ectopic lesion establishment, growth, and maintenance (10, 12). Ectopic lesions display altered ESR expression, with ESR2 levels significantly higher than those of ESR1, when compared with corresponding eutopic endometria (12). Progesterone (P4) resistance is similarly associated with endometriosis because lesions fail to regress in a significant number of patients treated with progestins (10, 13). Resistance to P4 can occur at the levels of the progesterone receptor (PGR), coregulators, and downstream effectors (11, 14). Eutopic endometria of women with endometriosis display lower expression of PGR-A and PGR-B isoforms than that of women without the disease (15–17). Further, the ratio of PGR isoform expression (15, 18) and the expression of numerous PGR coregulators (19–21) are altered in ectopic implants relative to those in the eutopic endometrium.
The transcription factor Krüppel-like factor 9 (KLF9), which belongs to the Sp/KLF family consisting of 17 current members (22), has emerged as a potential major player in numerous reproductive dysfunctions associated with aberrant ESR and/or PGR signaling. These include subfertility, uterine hypoplasia, leiomyoma, endometrial adenocarcinoma, endometriosis, and defects in the timing of parturition (17, 23–30). The mechanistic association between these pathologic conditions and KLF9 loss of expression may stem partly from the role of KLF9 as a PGR coregulator (31, 32) and as a context-dependent negative (33) or positive (34) regulator of ESR1 signaling. Consistent with this, we found that the attenuated expression of both KLF9 and PGR (achieved by small interfering RNA targeting) in human endometrial stromal cells in vitro resulted in transcriptional signatures indicative of compromised Wnt, cytokine, and IGF signaling (17), all of which are PGR- and ESR1-regulated pathways. Importantly, eutopic endometria of women with endometriosis and endometria of infertile women display overlap with these dysregulated transcription profiles (16, 17, 35).
Despite observations linking endometrial KLF9 loss of expression in human endometriosis, a causal role for KLF9 in ectopic lesion establishment and underlying mechanism(s) remains poorly understood. In a recent study, a KLF9 family member, KLF11, was shown to arrest progression of endometriosis using Klf11 null mice with surgically induced Klf11 null endometriotic lesions (36). In parallel studies using Klf9 null endometriotic lesions generated in Klf9 null mice, KLF9 loss also led to formation of ectopic lesions, which were comparable to those for WT (control) endometria. However, because the disease model is autologous (recipient and donor are of the same genotype), the effect of endometrial Klf9 null genotype, independent of the systemic environment, on disease establishment and progression could not be readily discerned. In the present study, we examined the pathologic involvement of KLF9 loss in ectopic lesion establishment in a syngeneic immunocompetent mouse model using donor endometria from Klf9 null (knockout) and corresponding Klf9 wild-type (WT) mice. We analyzed generated ectopic endometriotic lesions for proliferation, apoptosis, and steroid receptor expression and assessed Notch and Hedgehog (Hh) signaling activation. Our collective results provide evidence to indicate that a defective endometrium characterized by loss of KLF9 expression promotes establishment of endometriosis in part via the coincident deregulation of Notch, Hh, and steroid receptor-regulated pathways.
Materials and Methods
Animals and induction of endometriosis
All animal experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, following protocols approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee. Initial experiments to establish endometriotic lesions used C57BL/6J (Klf9 WT) female mice as recipients (9–10 weeks of age) and same-aged C57BL/6-Tg(UBC-GFP)30Scha/J female mice (designated ubiquitin C (UBC)-green fluorescent protein (GFP); The Jackson Laboratory) as donors. Recipients injected with GFP-expressing endometrium were followed for lesion development for 4 weeks. Subsequent experiments used the same strain of WT mice (8 weeks of age; in estrus) as recipients and either WT or Klf9 null mice (23) of the same age and estrous cycle stage as donors. The induction of endometriosis followed protocols previously reported by Hirata et al (37) and Nowak et al (38). In brief, intact donor uterus was subjected to a full-length midline incision, and the myometrium was scraped from the endometrium by light upward and lateral traction. The intact endometrium was collected, weighed, and minced using closely apposed scalpel blades oriented 180° apart. Minced tissue (40 μg) was reconstituted in 400 μL of PBS and injected into the peritoneal cavity through the abdominal wall on the ventral midline just below the umbilicus, using a syringe with a 20-gauge needle (Fisher Scientific). Sham controls were WT mice injected ip with 400 μL of PBS. Recipient mice were followed for lesion development for 8 weeks. At sacrifice, total body, ovarian, and uterine weights for each mouse were determined.
Imaging and collection of ectopic lesions
In WT recipient mice with UBC-GFP mice as endometrial donors, the presence of lesions in the abdominal cavity was evaluated using a fluorescence stereomicroscope (Carl Zeiss AG) equipped with an Attoarc variable intensity light source controller. For WT recipients with WT and Klf9−/− mice as donors, the presence of lesions was determined using a SteREO Discovery V8 stereomicroscope (Carl Zeiss) equipped with a Canon EOS 1000D camera (Canon, Inc). All lesions were photographed, and their sizes were measured using Axiovision software (Carl Zeiss). Lesion volume was calculated as described in Laschke et al (39). Collected lesions were snap-frozen in liquid nitrogen or placed in 10% neutral-buffered formalin for further processing (below).
RNA isolation, quantitative RT-PCR (QPCR), and focused QPCR arrays
Total RNA was extracted from endometriotic lesions (n = 8–9, each representing an individual mouse/experimental group) with TRIzol reagent (Invitrogen) following the manufacturer's protocol. RNA concentrations were determined using an ND-1000 spectrophotometer (NanoDrop). cDNA was synthesized from 500 ng of total RNA using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories) and analyzed by QPCR using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories) and the Bio-Rad CFX96 Real Time System module and c1000 Touch thermal cycler. Intron-flanking primers were designed to eliminate the amplification of genomic DNA using Primer Express software (Applied Biosystems) and were obtained from Integrated DNA Technologies, Inc. The primer sequences (sense and antisense, respectively) and the corresponding PCR product sizes are shown in Supplemental Table 1 (published on The Endocrine Society's Journals Online web site at http://end.endojournals.org). A standard curve was generated by serially diluting pooled cDNAs from all animals beginning with the most concentrated cDNA pool designated as 10 000 arbitrary units. Target mRNA abundance in endometriotic lesions was normalized to a factor derived from the geometric mean of expression values for cyclophilin A (Ppia) and TATA box binding protein (Tbp), calculated using the GeNorm program (40). Focused PCR Profiler array (Mouse Notch Signaling Pathway PCR Array; SABiosciences) analyses followed protocols described by the manufacturer, using cDNAs prepared from RNAs pooled in equal amounts from ectopic endometrial lesions of 4 individual donor WT and Klf9−/− mice, respectively.
Lesion morphometry and immunohistochemistry
Endometriotic lesions were fixed in 10% neutral-buffered formalin after previously described protocols (24). Sections (5 μm) were mounted on poly(lysine)-coated slides (Fisher Scientific) and processed for morphometric and immunohistochemical analyses (24). For detection and quantification of immunostained cells, sections were treated with 3% hydrogen peroxide for 30 minutes at room temperature and then with Citra-Plus (Biogenex) to unmask antigen (24). Sections were incubated in a blocking solution (VectaStain ABC kits; Vector Laboratories) for 30 minutes to reduce nonspecific staining before incubation for 24 hours with primary antibodies in a humidity chamber at 4°C. Antibodies used at the indicated dilutions (Supplemental Table 2) were obtained from the following sources: (1) rabbit anti-mouse ESR1 (sc-542; Santa Cruz Biotechnology); (2) rabbit anti-mouse ESR2 (Upstate Biotechnology); (3) rabbit anti-mouse PGR (sc-7208; Santa Cruz Biotechnology); (4) rabbit anti-mouse Notch1 (cleaved N-terminal domain, 07-1232; Millipore Corp); (5) rabbit anti-mouse Ki-67 (ab16667; Abcam); and (6) rabbit anti-mouse Sonic Hedgehog (SHH) (sc-9024; Santa Cruz Biotechnology). Incubation with biotinylated, anti-rabbit secondary antibody (1:200 dilution, VectaStain ABC kits; Vector Laboratories) was carried out for 30 minutes at room temperature. Sections were stained with 3,3′-diaminobenzidine (DAB Chromogen; Dako), counterstained with hematoxylin, dehydrated, cleared, and coverslipped for examination under a microscope. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed using an ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore Corp), following the manufacturer's instructions. Control sections were processed similarly, but with the omission of the primary antibody. For nuclear stains (ESR1, ESR2, PGR, Notch intracellular domain [NICD], and TUNEL), approximately 1000 stromal and 100 glandular epithelial cells were counted from 4 to 5 randomly chosen fields (×400 magnification) per slide using an Axiovert 200M microscope equipped with an Axiocam HRc camera and Axiovision software. A total of 3 to 4 slides, with each slide representing an endometriotic lesion from an individual donor mouse per genotype, were evaluated. Results are expressed as the percentage of nuclear immunopositive cells [(number of nuclear positively staining cells/number of total cells counted) × 100]. For cytoplasmic SHH staining (n = 5 lesions/genotype), images were acquired, and staining intensity was determined by computerized quantification using ImageScope (Aperio Technologies Inc). After each image was subjected to color deconvolution to separate brown and blue pigments, an intensity threshold was determined for the 3,3′-diaminobenzidine–positive stain (brown) by scanning, followed by averaging of the intensity range of 5 randomly selected sections; these values were used to determine the relative amount of highest intensity staining for concurrent slides. A total of 5 areas from at least 2 sections per lesion were evaluated. Data are expressed as a percentage of the highest positive staining cells per unit area.
Peritoneal fluid collection and ELISA
Peritoneal fluids were collected by injecting 1 mL of cold PBS-containing protease inhibitors (Halt Protease Inhibitor Cocktail; Thermo Scientific) into the peritoneum of donor mice immediately before lesion isolation using a 27-gauge needle (Fisher Scientific). Fluid was then retrieved using the same syringe after gentle massaging of the peritoneum. Fluid was centrifuged at 1500 rpm for 10 minutes at 4°C to remove peritoneal cells, and supernatant was stored at −80°C. Peritoneal fluid cytokines were quantified for TNFα and soluble TNF receptor 1 (sTNFR1) levels (n = 8–9 individual mouse/donor lesion group) using mouse TNFα and mouse sTNFR1 Quantikine ELISAs (R&D Systems).
Serum RIA and ELISA
Approximately 500 μL of whole blood was collected by closed cardiac puncture from recipient mice. Serum was separated by centrifugation of whole blood at 4600 × g for 1 hour and stored at −20°C before analysis. Serum E2, P4, and IL-6 levels were measured using an Ultrasensitive Estradiol Kit (Beckman Coulter), a Progesterone EIA Kit (Cayman Chemical), and a mouse IL-6 Quantikine ELISA Kit (R&D Systems), respectively.
Statistical analysis
Statistical analysis was performed using SigmaStat software (version 3.5; Systat Software). For comparisons of lesion volumes and numbers, RNA transcript levels, percentage of immunostaining cells, serum levels of steroid hormones and IL-6, and serum and peritoneal TNFα and sTNFR1 levels as a function of donor genotype, data (expressed as means ± SEM) were evaluated for statistical differences by the Student t test. Lesion incidence between the 2 groups was analyzed by the Fisher exact test. Differences were considered to be statistically significant at P < .05. Tendencies for differences were considered for .05 < P < .10.
Results
Validation of endometriosis induction in immunocompetent mice
We first established a mouse model of experimental endometriosis integrating procedures from studies described previously (37, 38) and confirmed that endometriosis-like lesions developed in immunocompetent animals. Intraperitoneal injection of UBC-GFP–expressing endometrial fragments into WT recipients resulted in endometriotic lesions that morphologically resembled those found in other rodent models of endometriosis (27, 36, 37, 38, 41) (Figure 1, A and B). Hematoxylin and eosin (H&E) staining of lesion sections demonstrated the presence of well-organized uterine stromal and glandular architecture, confirming their endometrial origin (Figure 1C).
Figure 1.
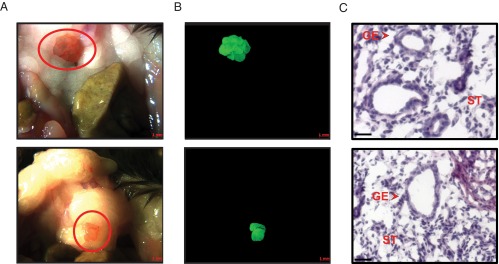
Macroscopic and histological validation of endometriotic lesions in immunocompetent mice. The formation of endometriotic-like lesions in the peritoneal cavity of recipient mice injected ip with UBC-GFP–expressing endometrial fragments was evaluated by brightfield (A) and fluorescence (B) microscopy. Scale bar corresponds to 1 mm. C, H&E staining of lesion sections was visualized at ×400 magnification. Scale bar corresponds to 20 μm. GE, glandular epithelium; ST, stroma.
Loss of endometrial KLF9 results in higher lesion incidence
To test the hypothesis that a defective endometrium due to loss of KLF9 transcriptional control promotes endometriosis, we used the above model with recipient WT mice and donor endometria derived from either WT or Klf9−/− mice of the same age and estrous cycle stage (Figure 2A). Representative brightfield macroscopic images and H&E-stained microscopic images of lesions are shown in Figure 2, B and C. Lesions were readily distinguishable from surrounding tissues based on gross appearance and resembled those that developed in a comparable mouse model (37, 41). WT and Klf9−/− lesions were found primarily adhering to the peritoneal wall, in the abdominal fat pad, and in the intestinal mesentery, with no preference to any one location. Lesions developing in these locations are consistent with other reports using comparable methods of endometriosis induction in mice (37, 41). Endometriotic lesions were evaluated for incidence, number, and volume in recipient mice at sacrifice. There was a significant increase in the incidence of lesions in those injected with Klf9 null (100% incidence) than with WT (65% incidence) endometrial fragments (Table 1). However, the numbers and volumes of established lesions were not affected by loss of Klf9 expression in donor endometria. Sham controls did not manifest any lesions (data not shown). Serum E2 and P4 levels did not differ in recipient mice with or without lesions and irrespective of donor genotype (Supplemental Figure 1). These results suggest that an endometrium defective in Klf9 expression promotes ectopic lesion establishment.
Figure 2.

Experimental design and characterization of endometrial-like lesions in a mouse model of endometriosis. A, Schematic representation of endometrial ectopic lesion generation and their analyses. Klf9 WT mice were used as recipients of endometrial fragments from WT and Klf9−/− null mice (donors) following previously published procedures (38, 39). N is the number of mice used for each experimental paradigm.. B, Representative images of ectopic lesions isolated from WT recipients 8 weeks after ip administration of WT or Klf9 null endometrium. Scale bar corresponds to 1 mm. C, H&E-stained sections of representative WT or Klf9−/− lesions at ×400 magnification. Scale bar corresponds to 20 μm. GE, glandular epithelium; ST, stroma. D, Representative pictures of immunostained stromal cells of WT and Klf9−/− ectopic lesions for proliferation marker Ki67. E, Representative pictures of immunostained stromal cells of WT and Klf9−/− ectopic lesions for apoptotic status (TUNEL). For D and E, the percentage of staining cells (expressed as means ± SEM) was determined by counting the number of immunopositive nuclei over the total number of stromal cells per field. Red arrows demonstrate Ki67-positive (D) and TUNEL-staining (E) stromal cells. Three to four lesions per genotype were analyzed. Scale bar corresponds to 20 μm. *, P < .05 by the Student t test.
Table 1.
Parameters of Lesions Derived from WT and Klf9 Null Endometrium
| Lesion Parameters | Donor Genotype |
|
|---|---|---|
| WT | Klf9−/− | |
| No. (per mouse) | 2.1 ± 0.37 | 1.8 ± 0.31 |
| Volume (per mouse), mm3 | 27.82 ± 10.53 | 37.8 ± 11.30 |
| Incidence, % (n) | 64.7 (11/17)a | 100% (18/18)a |
Lesion parameters were collected at 8 weeks postinduction.
P ≤ .01, Fisher exact test.
Klf9 null lesions display increased proliferation and decreased apoptosis status
Increased proliferation and decreased apoptosis are associated with ectopic lesion progression and maintenance (1). In ectopic lesions formed from Klf9 null endometria, the percentage of stromal cells displaying nuclear Ki67 immunoreactivity was higher than in those formed from WT endometria (Figure 2D). By contrast, Klf9 null endometriotic lesions exhibited significantly less TUNEL-immunopositive cells than WT lesions, indicating attenuated apoptotic status (Figure 2E).
Klf9 null and WT ectopic lesions differ in ESR isoform expression
We previously demonstrated that KLF9 influences context-dependent ESR1 expression in endometrial cells in vivo (26, 34) and in vitro (26, 33). To determine whether differing ESR expression in established lesions as a function of endometrial genotype may contribute to the increased incidence of ectopic lesions, we evaluated ESR1 and ESR2 transcript and protein levels in WT and Klf9−/− endometriotic lesions by QPCR and immunohistochemistry, respectively. The limited amount of ectopic tissues harvested from each mouse precluded performing the more quantitative Western blot analysis for ESR1 and ESR2 proteins. Esr1 mRNA levels were lower, whereas those of Esr2 mRNA were higher in Klf9−/− than WT lesions, resulting in a significantly greater Esr2/Esr1 ratio in Klf9−/− lesions (Figure 3A). Immunohistochemical analyses localized immunoreactive ESR1 and ESR2 predominantly in nuclei of glandular and stromal compartments of WT and Klf9−/− lesions. Interestingly, although no differences in the percentage of nuclear ESR1-positive cells were noted in endometrial glands between lesion types (data not shown), the percentage of nuclear stromal ESR1-positive cells was significantly lower in Klf9−/− than in WT lesions (Figure 3B). Similar to that observed for Esr2 transcript levels, nuclear ESR2 immunoreactivity was higher in stromal cells of Klf9−/− than of WT lesions (Figure 3C). The stromal-specific expression changes for both ESR1 and ESR2 in Klf9−/− relative to those of WT lesions are consistent with the predominant stromal localization of KLF9 (23, 24).
Figure 3.

ESR isoforms are differentially expressed in endometriotic lesions from WT and Klf9 null endometria. A, Transcript levels of Esr1 and Esr2 and their ratios were determined in WT and Klf9−/− lesions by QPCR. Data (means ± SEM) are expressed as fold change and were obtained from n = 8 to 9 lesion RNAs, with each lesion isolated from an individual mouse per experimental group. B and C, Representative pictures of immunostained stromal cells of WT and Klf9−/− ectopic lesions for ESR1 (B) and ESR2 (C). The percentages of immunostained cells (expressed as means ± SEM) were determined by counting the number of immunopositive nuclei over the total number of stromal cells per field. Red arrowheads demonstrate immunopositive stromal cells. Three to four lesions per genotype were analyzed. Scale bar corresponds to 20 μm. *, P < .05 by the Student t test.
Lower PGR expression is associated with Klf9-null ectopic lesions
P4 resistance is considered to underlie the development of endometriosis (1, 10, 44). In previous studies, we showed that KLF9 is a PGR coregulator in endometrial cells (31, 32) and that co-loss of KLF9 and PGR characterized eutopic endometria of women with endometriosis (17). To evaluate whether loss of KLF9 alters PGR expression in endometriotic lesions, which may underlie increased lesion incidence, we compared PGR transcript and protein levels in lesions formed from WT and Klf9−/− endometria by QPCR and immunohistochemistry, respectively. Total Pgr expression was significantly lower in Klf9−/− than in WT lesions; however, there were no corresponding differences in Pgr-b mRNA levels between lesions (Figure 4A). PGR immunoreactivity was largely undetectable in glands of lesions for both genotypes (data not shown). In contrast, nuclear-localized PGR immunoreactivity was highly prominent in stroma of these lesions (Figure 4B), with the percentage of total nuclear PGR-positive cells being significantly lower in Klf9−/− than in WT lesions (Figure 4C).
Figure 4.

PGR expression is attenuated in endometriotic lesions from Klf9 null relative to WT endometria. A, Transcript levels of total Pgr and Pgr-b were quantified in WT and Klf9−/− lesions by QPCR. Data (means ± SEM) are expressed as fold change and were obtained from n = 8 to 9 lesion RNAs, with each lesion isolated from an individual mouse per experimental group. B, Representative pictures of immunostained stromal cells of WT and Klf9−/− ectopic lesions for PGR. C, The percentages of immunostained cells (expressed as means ± SEM) were determined by counting the number of immunopositive nuclei over the total number of stromal cells per field. Red arrowheads demonstrate immunopositive stromal cells. Three to four lesions per genotype were analyzed. Scale bar corresponds to 20 μm. D, Transcript levels of PGR-responsive genes were quantified in WT and Klf9−/− lesions by QPCR. Data (means ± SEM) are expressed as fold change and were obtained from n = 4 to 5 lesion RNAs, with each lesion isolated from an individual mouse per experimental group. *, P < .05 by the Student t test.
To determine whether Klf9 null lesions with lower PGR expression (compared with WT lesions) is associated with altered expression of PGR-regulated target genes, as we previously reported in human endometrial stromal cells with co-knockdown of KLF9 and PGR expression (17), transcript levels for Wnt2, Wnt4, Dickkopf homolog 1 (Dkk1), stanniocalcin 1 (Stc1), early growth response (Egr1), and forkhead box O1a (Foxo1a) were evaluated in Klf9 null and WT ectopic endometrial lesions by QPCR. Consistent with our previous report (17), transcript levels for Wnt2 and Stc1were higher in Klf9 null than in WT lesions, whereas that for Foxo1a did not differ with lesion genotype (Figure 4D). However, in contrast to our previous report (17), transcript levels for Wnt4 and Dkk1 increased, whereas that for Egr1 did not change in Klf9 null relative to WT lesions (Figure 4D).
Klf9−/− ectopic lesions demonstrate enhanced Notch and Hh signaling component expression
Emerging evidence suggests the stem cell origin of endometriosis (44, 45). Consistent with this, endometriotic lesions in women display higher expression of the adult stem cell (ASC) markers Musashi and SOX-2 than secretory endometrium (46, 47). Notch and Hh signaling pathways are critical for ASC maintenance and proliferation in a number of tissues (48, 49). KLF9 has been reported to inhibit expansion of glioblastoma-initiating (stem) cells via suppression of Notch signaling (50). Based on these cumulative studies, we evaluated whether the higher incidence of ectopic endometriotic lesions generated with Klf9 null relative to that with WT endometria might be due in part to activation of Notch and Hh signaling consequent to loss of endometrial Klf9. We used a Notch signaling focused PCR array to compare the relative expression of genes implicated in Notch and Hh signaling pathways in WT and Klf9−/− endometriotic lesions. From a total of 84 genes included in the focused array, 23 were up-regulated and 3 were down-regulated by at least 1.5-fold in Klf9 null relative to WT lesions (Supplemental Table 3). A subset of up-regulated genes, consistent with Notch and Hh signaling pathway activation, was examined for differential expression by QPCR using cDNAs prepared from RNAs from 8 to 9 independent lesions, each lesion representing an individual mouse. Within the Notch signaling pathway, transcript levels of Notch ligand Jag2 were significantly increased, whereas those of downstream effectors Hes1, Wnt11, and Nr4a2 tended to increase in Klf9−/− relative to WT lesions (Figure 5A). Notch1 gene expression did not differ in lesions with or without Klf9. The transcript levels of the Hh pathway–associated components, ligand Shh, nuclear effector Gli1, and target Stil, were significantly greater with that for Smo tending to increase in Klf9 null relative to WT lesions (Figure 5A).
Figure 5.

Loss of Klf9 expression in endometriotic lesions is associated with activation of Notch and Hh signaling pathways. A, Validation of differential gene expression of select Notch and Hedgehog signaling components from focused PCR microarray analyses of ectopic lesions from WT and Klf9 null endometria by QPCR. Data (means ± SEM) are expressed as fold change and were obtained from n = 8 to 9 lesion RNAs, with each lesion isolated from an individual mouse per experimental group. B, Representative pictures and quantification of immunostained stromal cells of WT and Klf9−/− ectopic lesions for NICD. C, Representative pictures and quantification of immunostained stromal cells of WT and Klf9−/− ectopic lesions for Hh ligand SHH. The bottom panels represent magnification of the boxed areas indicated in the corresponding top panel. For both B and C, the percentages of nuclear (NICD)– or cytoplasmic (SHH)–staining cells (expressed as means ± SEM) were determined as described in Materials and Methods. Red arrowheads demonstrate immunopositive stromal cells. Three to four lesions per genotype were analyzed. Scale bar corresponds to 20 μm. *, P < .05 by the Student t test.
To further confirm Notch signaling activation in ectopic lesions with loss of KLF9, we performed immunohistochemical analysis on WT and Klf9 null lesions with an antibody specific for the activated NICD (48). Klf9−/− ectopic lesions had a significantly greater percentage of stromal cells positive for immunoreactive nuclear NICD than WT lesions (Figure 5B). For Hh signaling activation, immunoreactive SHH was evaluated in cytoplasmic compartments of stromal cells following procedures previously described for the SHH paralog, Indian Hedgehog (51). Relative to WT ectopic lesions, Klf9−/− lesions had higher SHH immunoreactivity in stromal cells (Figure 5C). Control sections evaluated in parallel in the absence of anti-Notch or anti-SHH antibodies did not show any staining (data not shown).
Peritoneal fluid and serum cytokine levels in mice with Klf9−/− vs WT ectopic lesions
Women with endometriosis have altered peritoneal and systemic levels of proinflammatory cytokines, which are believed to contribute to disease progression (52, 53). We previously reported that co-knockdown of KLF9 and PGR in human endometrial stromal cells in vitro led to elevations in numerous inflammatory and cytokine pathway component mRNAs (17). To examine whether mice with Klf9 null lesions relative to those with WT lesions display enhanced peritoneal inflammation, we quantified the levels of TNFα and the related sTNFR1 in peritoneal fluids. Peritoneal fluid levels of TNFα and sTNFR1 did not differ between mice injected with WT or Klf9−/− endometrium (Figure 6A). In contrast, serum levels of TNFα tended to decrease, whereas those for sTNFR1, a natural antagonist of TNFα, tended to increase in mice with Klf9 null lesions (Figure 6B). Mice with Klf9−/− lesions also had significantly less IL-6 in the sera than mice with WT lesions (Figure 6B). Results suggest that the presence/maintenance of ectopic lesions generated by the specific loss of endometrial Klf9 is associated with systemic (sera) rather than local (peritoneum) changes in inflammation status.
Figure 6.
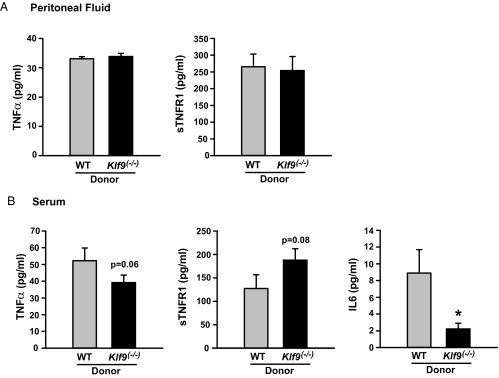
Loss of Klf9 expression in endometriotic lesions alters serum but not peritoneal levels of inflammatory mediators involved in endometriosis. A, Levels of TNFα and sTNFR1 in peritoneal fluids of recipient mice with endometriotic lesions formed from WT or Klf9−/− endometrium. B, Serum levels of TNFα, sTNFR1, and IL6 in recipient mice with endometriotic lesions formed from WT or Klf9−/− endometrium. Data (means ± SEM) are from n = 8 to 9 individual mice per donor lesion group. *, P < .01 by the Student t test.
Discussion
In the current study, we developed a mouse model of experimental endometriosis to test the hypothesis that a defective endometrium, due to loss of expression of the PGR coregulator KLF9, recapitulating that found in eutopic endometria of women with endometriosis (17), can promote the establishment of endometrial ectopic lesions. This mouse model, in which recipient mice are immunocompetent and reproductive competent, is well suited to address this question because it makes use of donor mouse endometrium that differs in the presence or absence of KLF9 expression and, thus, allows for investigations of immune and systemic pathways affected by the presence of ectopic lesions of the 2 genotypes. Our findings reported here offer several important contributions to the understanding of endometriosis. First, we provide direct evidence to indicate that a deficiency in endometrial KLF9 expression causes ectopic lesion establishment. This result positions KLF9 in the category of several PGR coregulators whose aberrant expression is demonstrated to promote endometriosis (20, 21) and, together with the findings reported for KLF11 (36), suggests a pathologic role for KLF family members in uterine dysfunction. Second, we established a link between KLF9 and the expression of several components of the Notch and Hh signaling pathways in ectopic lesions. Our identification of activation of Notch and Hh signaling as relevant to endometriosis with loss of endometrial KLF9 regulation (either directly or indirectly) is a novel finding and congruent with reports linking aberrant activation of these pathways to stem cell expansion (48, 49) and with the emerging evidence for the stem cell origin of endometriosis (44, 45). Finally, we confirmed that PGR and ESR signaling is tightly coupled to KLF9 from findings reported here that loss of KLF9 expression in ectopic lesions is associated with significant alterations in expression ratios of ESR2 and ESR1 isoforms, substantial loss of PGR expression, and deregulation of expression of a subset of PGR target genes. Based on these findings, we propose a model (Figure 7) that integrates the key components involved in steroid hormone (ESR and PGR), Notch (Jag2 and NICD), and Hh (SHH, Gli, and Stil) signaling pathways with proliferation (Ki67) and apoptotic (TUNEL) status that together may mediate the pathologic steps involved in lesion establishment with loss of KLF9 transcriptional control.
Figure 7.

Proposed model integrating potential pathways dysregulated with loss of endometrial Klf9 leading to ectopic lesion establishment and progression. KLF9 is required for proper regulation of PGR and ESR expression or activity and maintains (directly or indirectly) the appropriate activation of Notch and Hh signaling. In the absence of endometrial stromal KLF9 (depicted as crossed-out KLF9), promotion of proliferation, attenuated apoptotic status, and aberrant immune response consequent to dysregulated ESR, PGR, Notch, and Hh signaling lead to lesion development and survival. The regulation by PGR and ESR of Notch and Hh signaling is a possibility that was not addressed in this study.
In our experimental model of endometriosis, Klf9 null endometria induced the formation in recipient (WT) mice of ectopic lesions at 100% incidence, which is significantly higher (by 35%) than that found in recipient mice injected with Klf9 WT endometria. Whereas ip injection of tissue may cause a “wounding reaction” that permits lesion establishment even with normal (WT) endometria, as shown for a similar mouse model of lesion establishment (41), our data unequivocally demonstrate the specific contribution of endometrial Klf9 null mutation to formation of ectopic lesions. Interestingly, there was no change in lesion numbers, and lesion volume showed only very modest (nonsignificant) differences with donor endometrial genotype at 8 weeks postinjection; these values suggest the possibility of lesion resorption after an extended period postinitiation. Alternatively, the normal physiology of the recipient mice (ie, immunocompetent) may have limited continuous lesion growth. A more detailed time course of lesion development will be necessary to address these possibilities, which would also better define the optimal window for more exhaustive analyses of signaling pathways that are promoted with Klf9 loss for lesion formation.
The present study highlights for the first time a novel role of KLF9 in ESR2 regulation. Normally, ESR2 expression is low to negligible in the uterus; however, this ESR isoform (rather than ESR1) has been reported to primarily mediate estrogenic response in endometriotic lesions (54). Thus, the higher expression ratio of ESR2 to ESR1 in ectopic lesions of Klf9 null endometria is consistent with the loss of KLF9 driving ESR2 dysregulation, thus, allowing for establishment of endometriotic lesions. Given the lack of candidate KLF9/ESR target genes that are known currently, WT and Klf9 KO ectopic lesions were not further evaluated for levels of expression of potential ESR1 or ESR2 target genes.
P4 resistance is a hallmark of endometriosis, with both PGR-A and PGR-B isoform levels diminished in ectopic lesions (relative to eutopic endometria), with one report indicating with PGR-B rather than PGR-A loss occurring preferentially (18, 55). However, others have reported lower levels of PGR-A than PGR-B (compared with eutopic endometria) in other endometriosis models (56). Although we previously demonstrated a positive, albeit modest, regulation by KLF9 of PGR expression in uterine endometrium (17, 24) and myometrium (28), the specific PGR isoform targeted by KLF9 remains unknown. However, the distinct expression levels of PGR target genes between WT and Klf9 null lesions, a number of which (eg, Wnt2 and Stc1) were up-regulated in agreement with our previous findings in human endometrial stromal cells with co-knockdown of KLF9 and PGR (17), support the transcriptional deregulation of PGR signaling with Klf9 loss. Given the complex cross-regulation of PGR expression in uterine cells at the levels of the respective promoter or posttranscriptional events, further studies to dissect the individual control of PGR isoform expression by KLF9 will be required to expand our understanding of the contribution of KLF9 to the disease.
Another novel finding of this study is the implication for aberrant Notch and Hh activation to contribute to endometriotic lesion establishment and survival in the absence of Klf9. Both signaling pathways are critical for the proliferation and maintenance of ASCs (45, 46). Stem cells are thought to contribute to the cyclical regeneration of the endometrium (57, 58) and have been linked to the development of endometriosis (45, 46, 59). Whereas members of the KLF family and, in particular, KLF9 have not been linked specifically to endometrial stem cell biology and pathology, it is tempting to speculate on this possibility, given the concurrent increases in levels of the Notch ligand Jag2 transcript and activated NICD and in multiple components of the Hh pathway, including ligand SHH, intracellular mediator Gli1, and downstream target Stil, in Klf9 null relative to WT lesions. Related to Hh signaling, a number of studies have demonstrated the functional importance of the Hh pathway in maintaining P4 sensitivity in pregnancy and endometrial function by direct PGR regulation of the Hh ligand, Indian Hedgehog (60, 61). Importantly, Indian Hedgehog (62) and KLF9 (17) both display decreased expression in eutopic endometria of women with endometriosis. Thus, a potential link between compromised fertility (23, 24) and increased incidence of endometriosis (this study) associated with KLF9 loss in mouse models is a likelihood that can be further explored in women with endometriosis exhibiting attenuated Klf9 expression (17). Whether KLF9 is a direct regulator of Notch and Hh signaling or functions as an indirect mediator through ESR and PGR also requires further study.
Another pathway considered to be involved in endometriosis development is the complex network of inflammatory factors, which promote recruitment of immune cells that in turn support angiogenesis and adhesion of ectopic tissues for lesion establishment and survival (52, 53). Numerous clinical studies have implicated TNFα, sTNFR1, and IL-6 in the development and/or progression of endometriosis based on their increased levels in sera or peritoneal fluids of patients with endometriosis (53, 63, 64). The lack of discernible differences in peritoneal levels of TNFα and sTNFR1 as a function of donor genotype and the lower TNFα and IL-6 and higher sTNFR1 in sera of recipient WT mice are consistent with studies demonstrating decreased serum TNFα levels in women with relative to those without endometriosis (52, 64) and with progressively lower serum IL-6 levels in women with more severe disease (65, 66). A similar trend for both cytokines has also been shown in adolescent girls with severe endometriosis (67). Although a causative association between Klf9 null ectopic lesions and lower systemic cytokine levels indicative of attenuated immune response was not explored in the present study, these results suggest that the presence of Klf9 null lesions initiates a less immunogenic systemic response, consistent with the persistence (ie, less resorption) of Klf9 null lesions (100% incidence) relative to that of WT lesions. In a rat model of endometriosis, lower levels of serum IL-6 were found after 8 weeks of endometriosis induction compared with that after 4 weeks, suggesting IL-6 may be critical for earlier stages of disease development (68). We posit that although higher inflammation status may be necessary for the survival of WT lesions, Klf9 null lesions may have acquired other mechanisms (ie, increased Notch signaling) to maintain lesion growth. Understanding the precise mechanisms by which KLF9 mediates endometrial cell immunogenicity is a topic for future investigations.
In summary, our studies suggest that a defective endometrium due to loss of KLF9 expression promotes lesion establishment and survival in an immunocompetent mouse model of experimental endometriosis. We suggest that in endometrial stromal cells, KLF9 provides coincident regulation of Notch, Hh, and steroid receptor signaling pathways, which when deregulated, underlie establishment and/or progression of the disease. The studies presented herein lend credence to further studies into the role of KLF9 in ASC biology, the aberrant expansion of which may partly contribute to the pathophysiology of endometriosis. Finally, given the demonstrated context-dependent functions of KLF9 (33, 34, 69), similar to those of other KLF family members (70), explorations into the utility of drugs that promote KLF9 expression may lead to novel approaches for mitigating endometriosis and other pathological conditions associated with KLF9 dysregulation.
Acknowledgments
We thank the members of Dr R. C. M. Simmen's and Dr F. A. Simmen's laboratories for helpful discussions during the course of this work.
This study was supported in part by grants from the National Institutes of Health (HD21961) and the Arkansas Biosciences/Arkansas Children's Hospital Research Institute. C. D. Simmons was a recipient of the National Institutes of Health Research Supplement to Promote Diversity in Health-Related Research Award and of the Southern Regional Education Board State Doctoral Scholars Program.
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 1178
- ASC
- adult stem cell
- E2
- estradiol
- ESR
- estrogen receptor
- GFP
- green fluorescent protein
- H&E
- hematoxylin and eosin
- Hh
- Hedgehog
- KLF9
- Krüppel-like factor 9
- P4
- progesterone
- PGR
- progesterone receptor
- QPCR
- quantitative RT-PCR
- SHH
- Sonic Hedgehog
- sTNFR1
- soluble TNF receptor 1
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
- UBC
- ubiquitin C
- WT
- wild-type.
References
- 1. Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–1799 [DOI] [PubMed] [Google Scholar]
- 2. Eskenazi B, Warner ML. Epidemiology of endometriosis. Obstet Gynecol Clin North Am. 1997;24:235–258 [DOI] [PubMed] [Google Scholar]
- 3. Barnhart K, Dunsmoor-Su R, Coutifaris C. Effect of endometriosis on in vitro fertilization. Fertil Steril. 2002;77:1148–1155 [DOI] [PubMed] [Google Scholar]
- 4. Stephansson O, Kieler H, Granath F, Falconer H. Endometriosis, assisted reproduction technology, and risk of adverse pregnancy outcome. Hum Reprod. 2009;24:2341–2347 [DOI] [PubMed] [Google Scholar]
- 5. Gemmill JA, Stratton P, Cleary SD, Ballweg ML, Sinaii N. Cancers, infections, and endocrine diseases in women with endometriosis. Fertil Steril. 2010;94:1627–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schenken RS. Pathogenesis, clinical features, and diagnosis of endometriosis. In: UpToDate, Barbieri RL, Barss VA. (eds). Waltham, MA: UpToDate; 2009 [Google Scholar]
- 7. Guo SW. Recurrence of endometriosis and its control. Hum Reprod Update. 2009;15:441–461 [DOI] [PubMed] [Google Scholar]
- 8. Abbott JA, Hawe J, Clayton RD, Garry R. The effects and effectiveness of laparoscopic excision of endometriosis: a prospective study with 2–5 year follow-up. Hum Reprod. 2003;18:1922–1927 [DOI] [PubMed] [Google Scholar]
- 9. Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissues into the peritoneal cavity. Am J Obstet Gynecol. 1927;14:422–469 [Google Scholar]
- 10. Bulun SE, Cheng YH, Pavone ME, et al. Estrogen receptor-β, estrogen receptor-α, and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28:36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486 [DOI] [PubMed] [Google Scholar]
- 12. Kitawaki J, Kado N, Ishihara H, Koshiba H, Kitaoka Y, Honjo H. Endometriosis: the pathophysiology as an estrogen-dependent disease. J Steroid Biochem Mol Biol. 2002;83:149–155 [DOI] [PubMed] [Google Scholar]
- 13. Lessey BA. Medical management of endometriosis and infertility. Fertil Steril. 2000;73:1089–1096 [DOI] [PubMed] [Google Scholar]
- 14. Lonard DM, O'Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125:411–414 [DOI] [PubMed] [Google Scholar]
- 15. Igarashi TM, Bruner-Tran KL, Yeaman GR, et al. Reduced expression of progesterone receptor-B in the endometrium of women with endometriosis and in cocultures of endometrial cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fertil Steril. 2005;84:67–74 [DOI] [PubMed] [Google Scholar]
- 16. Burney RO, Talbi S, Hamilton AE, et al. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology. 2007;148:3814–3826 [DOI] [PubMed] [Google Scholar]
- 17. Pabona JM, Simmen FA, Nikiforov MA, et al. Krüppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2012;97:E376–E392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab. 2000;85:2897–2902 [DOI] [PubMed] [Google Scholar]
- 19. Aghajanova L, Velarde MC, Giudice LC. The progesterone receptor coactivator Hic-5 is involved in the pathophysiology of endometriosis. Endocrinology. 2009;150:3863–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han SJ, Hawkins SM, Begum K, et al. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat Med. 2012;18:1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirota Y, Tranguch S, Daikoku T, et al. Deficiency of immunophilin FKBP52 promotes endometriosis. Am J Pathol. 2008;173:1747–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005;85:551–556 [DOI] [PubMed] [Google Scholar]
- 23. Simmen RC, Eason RR, McQuown JR, et al. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Krüppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem. 2004;279: 29286–29294 [DOI] [PubMed] [Google Scholar]
- 24. Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Kruppel-like factor9/basic transcription element binding protein-1 alters peri-implantation uterine development in mice. Biol Reprod. 2005;73:472–481 [DOI] [PubMed] [Google Scholar]
- 25. Rackow BW, Taylor HS. Submucosal uterine leiomyomas have a global effect on molecular determinants of endometrial receptivity. Fertil Steril. 2010;93:2027–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simmons CD, Pabona JM, Heard ME, et al. Krüppel-like factor 9 loss-of-expression in human endometrial carcinoma links altered expression of growth-regulatory genes with aberrant proliferative response to estrogen. Biol Reprod. 2011;85:378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee B, Du H, Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod. 2009;80:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zeng Z, Velarde MC, Simmen FA, Simmen RC. Delayed parturition and altered myometrial progesterone receptor isoform A expression in mice null for Krüppel-like factor 9. Biol Reprod. 2008;78:1029–1037 [DOI] [PubMed] [Google Scholar]
- 29. Simmen FA, Su Y, Xiao R, Zeng Z, Simmen RC. The Krüppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod Biol Endocrinol. 2008;6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Panda H, Pelakh L, Chuang TD, Luo X, Bukulmez O, Chegini N. Endometrial miR-200c is altered during transformation into cancerous states and targets the expression of ZEBs, VEGFA, FLT1, IKKβ, KLF9, and FBLN5. Reprod Sci. 2012;19:786–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang D, Zhang XL, Michel FJ, Blum JL, Simmen FA, Simmen RC. Direct interaction of the Krüppel-like family (KLF) member, BTEB1, and PR mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology. 2002;143:62–73 [DOI] [PubMed] [Google Scholar]
- 32. Velarde MC, Iruthayanathan M, Eason RR, Zhang D, Simmen FA, Simmen RC. Progesterone receptor transactivation of the secretory leukocyte protease inhibitor gene in Ishikawa endometrial epithelial cells involves recruitment of Krüppel-like factor 9/basic transcription element binding protein-1. Endocrinology. 2006;147:1969–1978 [DOI] [PubMed] [Google Scholar]
- 33. Velarde MC, Zeng Z, McQuown JR, Simmen FA, Simmen RC. Krüppel-like factor 9 is a negative regulator of ligand-dependent estrogen receptor α signaling in Ishikawa endometrial adenocarcinoma cells. Mol Endocrinol. 2007;21:2988–3001 [DOI] [PubMed] [Google Scholar]
- 34. Pabona JM, Velarde MC, Zeng Z, Simmen FA, Simmen RC. Nuclear receptor co-regulator Krüppel-like factor 9 and prohibitin-2 expression in estrogen-induced epithelial cell proliferation in the mouse uterus, J Endocrinol. 2009;200:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kao LC, Germeyer A, Tulac S, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144:2870–2881 [DOI] [PubMed] [Google Scholar]
- 36. Daftary GS, Zheng Y, Tabaa ZM, et al. A novel role of the Sp/KLF transcription factor KLF11 in arresting progression of endometriosis. PLoS One. 2013;8:e60165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirata T, Osuga Y, Yoshino O, et al. Development of an experimental model of endometriosis using mice that ubiquitously express green fluorescent protein. Hum Reprod. 2005;20:2092–2096 [DOI] [PubMed] [Google Scholar]
- 38. Nowak NM, Fischer OM, Gust TC, Fuhrmann U, Habenicht UF, Schmidt A. Intraperitoneal inflammation decreases endometriosis in a mouse model. Hum Reprod. 2008;23:2466–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laschke MW, Körbel C, Rudzitis-Auth J, et al. High-resolution ultrasound imaging: a novel technique for the noninvasive in vivo analysis of endometriotic lesion and cyst formation in small animal models. Am J Pathol. 2010;176:585–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burns KA, Rodriguez KF, Hewitt SC, Janardhan KS, Young SL, Korach KS. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology. 2012;153:3960–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Simmons CD, Pabona JM, Zeng Z, et al. Response of adult mouse uterus to early disruption of estrogen receptor-α signaling is influenced by Krüppel-like factor 9. J Endocrinol. 2010;205:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Osteen KG, Bruner-Tran KL, Eisenberg E. Reduced progesterone action during endometrial maturation: a potential risk factor for the development of endometriosis. Fertil Steril. 2005;83:529–537 [DOI] [PubMed] [Google Scholar]
- 44. Gargett BE, Chan RW. Endometrial stem/progenitor cells and proliferative disorders of the endometrium. Minerva Ginecol. 2006;58:511–526 [PubMed] [Google Scholar]
- 45. Sasson IE, Taylor HS. Stem cells and the pathogenesis of endometriosis. Ann NY Acad Sci. 2008;1127:106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Götte M, Wolf M, Staebler A, et al. Increased expression of the adult stem cell marker Musashi-1 in endometriosis and endometrial carcinoma. J Pathol. 2008;215:317–329 [DOI] [PubMed] [Google Scholar]
- 47. Götte M, Wolf M, Staebler A, Buchweitz O, Kiesel L, Schuring AN. Aberrant expression of the pluripotency marker SOX-2 in endometriosis. Fertil Steril. 2011;95:338–341 [DOI] [PubMed] [Google Scholar]
- 48. Aster JC, Blacklow SC. Targeting the notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol. 2012;30:2418–2420 [DOI] [PubMed] [Google Scholar]
- 49. McMillan R, Matsui W. Molecular pathways: the Hedgehog signaling pathway in cancer. Clin Cancer Res. 2012;18:4883–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ying M, Sang Y, Li Y, et al. Krüppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29:20–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei Q, Levens ED, Stefansson L, Nieman LK. Indian Hedgehog and its targets in human endometrium: menstrual cycle expression and response to CDB-2914. J Clin Endocrinol Metab. 2010;95:5330–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lebovic DI, Mueller MD, Taylor RN. Immunobiology of endometriosis. Fertil Steril. 2001;75:1–10 [DOI] [PubMed] [Google Scholar]
- 53. Mihalyi A, Kyama CM, Simsa P, Debrock S, Mwenda JM, D'Hooghe TM. Role of immunologic factors in the development of endometriosis: indications for treatment strategies. Therapy. 2005;2:623–639 [Google Scholar]
- 54. Trukhacheva E, Lin Z, Reierstad S, Cheng YH, Milad M, Bulun SE. Estrogen receptor (ER) β regulates ERα expression in stromal cells derived from ovarian endometriosis. J Clin Endocrinol Metab. 2009;94:615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics. 2006;1:106–111 [DOI] [PubMed] [Google Scholar]
- 56. Misao R, Iwagaki S, Fujimoto J, Sun W, Tamaya T. Dominant expression of progesterone receptor form B mRNA in ovarian endometriosis. Horm Res. 1999;52:30–34 [DOI] [PubMed] [Google Scholar]
- 57. Chan RW, Schwab KE, Gargett CE. Clonogenicity of human endometrial epithelial and stromal cells. Biol Reprod. 2004;70:1738–1750 [DOI] [PubMed] [Google Scholar]
- 58. Spitzer TL, Rojas A, Zelenko Z, et al. Perivascular human endometrial mesenchymal stem cells express pathways relevant to self-renewal, lineage specification, and functional phenotype. Biol Reprod. 2012;86:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chan RW, Ng EH, Yeung WS. Identification of cells with colony-forming activity, self-renewal capacity, and multipotency in ovarian endometriosis. Am J Pathol. 2011;178:2832–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee K, Jeong J, Kwak I, et al. Indian Hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006;38:1204–1209 [DOI] [PubMed] [Google Scholar]
- 61. Simon L, Spiewak KA, Ekman GC, et al. Stromal progesterone receptors mediate induction of Indian Hedgehog (IHH) in uterine epithelium and its downstream targets in uterine stroma. Endocrinology. 2009;150:3871–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Smith K, Alnifaidy R, Wei Q, Nieman LK. Endometrial Indian Hedgehog expression is decreased in women with endometriosis. Fertil Steril. 2011;95:2738–2741.e1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kyama CM, Overbergh L, Debrock S, et al. Increased peritoneal and endometrial gene expression of biologically relevant cytokines and growth factors during the menstrual phase in women with endometriosis. Fertil Steril. 2006;85:1667–1675 [DOI] [PubMed] [Google Scholar]
- 64. Chae SJ, Kim H, Jee BC, Suh CS, Kim SH, Kim JG. Tumor necrosis factor (TNF)-TNF receptor gene polymorphisms and their serum levels in Korean women with endometriosis. Am J Reprod Immunol. 2008;60:432–439 [DOI] [PubMed] [Google Scholar]
- 65. Martínez S, Garrido N, Coperias JL, et al. Serum interleukin-6 levels are elevated in women with minimal-mild endometriosis. Hum Reprod. 2007;22:836–842 [DOI] [PubMed] [Google Scholar]
- 66. Schröder W, Gaetje R, Baumann R. Interleukin-6 and soluble interleukin-6 receptor in peritoneal fluid and serum of patients with endometriosis. Clin Exp Obstet Gynecol. 1996;23:10–14 [PubMed] [Google Scholar]
- 67. Drosdzol-Cop A, Skrzypulec-Plinta V. Selected cytokines and glycodelin A levels in serum and peritoneal fluid in girls with endometriosis. J Obstet Gynaecol Res. 2012;38:1245–1253 [DOI] [PubMed] [Google Scholar]
- 68. Lim YT, Schenken RS. Interleukin-6 in experimental endometriosis. Fertil Steril. 1993;59:912–916 [PubMed] [Google Scholar]
- 69. Mitchell DL, DiMario JX. Bimodal, reciprocal regulation of fibroblast growth factor receptor 1 promoter activity by BTEB1/KLF9 during myogenesis. Mol Biol Cell. 2010;21:2780–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tetreault MP, Yang Y, Katz JP. Krüppel-like factors in cancer. Nat Rev Cancer. 2013;13:701–713 [DOI] [PubMed] [Google Scholar]