

Abstract
Fetal growth restriction (FGR) increases the risk for perinatal complications and predisposes the infant to diabetes and cardiovascular disease later in life. No treatment for FGR is available, and the underlying pathophysiology remains poorly understood. Increased IGFBP-1 phosphorylation has been implicated as an important mechanism by which fetal growth is reduced. However, to what extent circulating IGFBP-1 is phosphorylated in FGR is unknown, and the molecular mechanisms linking FGR to IGFBP-1 phosphorylation have not been established. We used umbilical cord plasma of appropriate for gestational age (AGA) and growth–restricted human fetuses and determined IGFBP-1 and IGF-I concentrations (ELISA) and site-specific IGFBP-1 phosphorylation (Western blotting using IGFBP-1 phospho-site specific antibodies). In addition, we used a baboon model of FGR produced by 30% maternal nutrient restriction and determined mammalian target of rapamycin (mTOR)C1 activity, CK2 expression/activity, IGFBP-1 expression and phosphorylation, and IGF-I levels in baboon fetal liver by Western blot, enzymatic assay, and ELISA. HepG2 cells and primary fetal baboon hepatocytes were used to explore mechanistic links between mTORC1 signaling and IGFBP-1 phosphorylation. IGFBP-1 was hyperphosphorylated at Ser101, Ser119, and Ser169 in umbilical plasma of human FGR fetuses. IGFBP-1 was also hyperphosphorylated at Ser101, Ser119, and Ser169 in the liver of growth–restricted baboon fetus. mTOR signaling was markedly inhibited, whereas expression and activity of CK2 was increased in growth–restricted baboon fetal liver in vivo. Using HepG2 cells and primary fetal baboon hepatocytes, we established a mechanistic link between mTOR inhibition, CK2 activation, IGFBP-1 hyperphosphorylation, and decreased IGF-I–induced IGF-I receptor autophosphorylation. We provide clear evidence for IGFBP-1 hyperphosphorylation in FGR and identified an mTOR and CK2-mediated mechanism for regulation of IGF-I bioavailability. Our findings are consistent with the model that inhibition of mTOR in the fetal liver, resulting in increased CK2 activity and IGFBP-1 hyperphosphorylation, constitutes a novel mechanistic link between nutrient deprivation and restricted fetal growth.
In ∼6% to 8% of all pregnancies, oxygen and nutrient delivery to the fetus is impaired and fetal growth restriction (FGR) develops. FGR increases the risk of perinatal complications and long-term neurodevelopmental impairment (1) and predisposes infants to the development of diabetes and cardiovascular disease in both childhood and adult age (2). The mechanisms underlying the development of FGR are poorly understood. Transgenic mice with a homozygous deletion of the IGF-I (Igf-1) gene show marked FGR (3), demonstrating that IGF-I is a key regulator of fetal growth. Fetal circulating levels of IGF-I are decreased in human FGR (4), indicating an important role for IGF-I in the pathophysiology of this condition.
The bioavailability of IGFs is regulated by IGF binding proteins (IGFBP-1–6). Binding of IGF-I to IGFBPs prevents the interaction of IGF-I with its receptors, thereby inhibiting IGF-I action (5). During fetal development, the predominant IGF-I binding protein is IGFBP-1, which is primarily secreted by the fetal liver (6). The levels of IGFBP-1 in the fetal circulation are increased in FGR pregnancies (7). Transgenic mice overexpressing IGFBP-1 in the liver develop FGR, establishing a cause-and-effect relationship between changes in IGFBP-1 levels and fetal growth (8). The regulation of IGF-I action also occurs through IGFBP-1 phosphorylation, which markedly increases the binding affinity of IGFBP-1 for IGF-I (9), resulting in a more powerful inhibition of IGF-I action (10, 11). Our previous studies indicated that IGFBP-1 is hyperphosphorylated at three specific serine residues in the amniotic fluid of fetal growth–restricted fetuses (12, 13); however, whether FGR is associated with increased phosphorylation of IGFBP-1 at these three residues in the fetal circulation remains to be established.
Decreased cellular amino acid levels and hypoxia, which induce IGFBP-1 secretion (14) and phosphorylation (15), are potent inhibitors of mammalian target of rapamycin (mTOR) signaling (16). We rationalized that mTOR signaling in the fetal liver may regulate IGFBP-1 phosphorylation and thereby IGF-I bioavailability in FGR. Consensus sequence analysis suggests that IGFBP-1 is a potential substrate for CK2 (17), a highly conserved pleiotropic protein kinase. The hallmark of the CK2 phosphorylation consensus site is the presence of acidic amino acids surrounding the phosphoacceptor residue (18). The 3 hyperphosphorylated serine residues (Ser101, Ser119, and Ser169) on IGFBP-1 that we have identified in amniotic fluid from human FGR (12, 19) conform to this general recognition motif. In addition, we have shown that phosphorylation at these three sites can effectively inhibit IGF-I receptor autophosphorylation in vitro due to tighter binding to IGF-I (19). Furthermore, hypoxia, which inhibits mTOR signaling, activates CK2 (20). CK2 can phosphorylate IGFBP-1 in vitro (21), but the role of CK2 in IGFBP-1 phosphorylation in vivo or in live cultured cells remains to be established. In addition, whether CK2 is a key protein kinase linking mTOR inhibition to IGFBP-1 phosphorylation has not been explored previously. Thus, the molecular mechanisms linking decreased oxygen and nutrient availability to IGFBP-1 hyperphosphorylation in human FGR or in any animal model of FGR are largely unknown.
Here we hypothesized that inhibition of mTOR signaling and increased CK2 activity in the fetal liver constitutes a key molecular link between nutrient deprivation, increased IGFBP-1 secretion and phosphorylation, and decreased IGF-I bioavailability in cultured cells in vitro and in FGR in vivo. To address our hypothesis, we determined IGFBP-1 phosphorylation (at Ser101, Ser119, and Ser169) in umbilical cord plasma of appropriate for gestational age (AGA) and growth–restricted human fetuses. Furthermore, we studied mTOR signaling, CK2 activity, total IGFBP-1 expression, and IGFBP-1 phosphorylation in the livers of control and growth–restricted baboon fetuses. Finally, we performed mechanistic studies in cultured HepG2 cells and baboon fetal primary hepatocytes to investigate the causative link between mTOR and CK2 activities, IGFBP-1 phosphorylation, and subsequent IGF-I–induced IGF-I receptor autophosphorylation in vitro.
Materials and Methods
Additional experimental procedures are provided in the Supplemental Methods published on The Endocrine Society's Journals Online web site at http://end.endojournals.org.
Human fetal plasma collection and sample preparation
The protocol for collection of blood samples from pregnant women attending London Health Sciences Centre (London, ON, Canada) was approved by the UWO Health Sciences Research Ethics Board. The samples were collected at the London Health Sciences Centre after informed consent was obtained. Relevant infant and maternal data procurement and details of selection criteria, sample collection, and sample preparation have all been reported previously (22).
Animals
Baboons (Papio species) from the Southwest National Primate Research Center (San Antonio, TX) were studied. Housing and environmental enrichment have been described in detail previously (23). All procedures related to the collection of baboon samples were approved by the Texas Biomedical Research Institute Institutional Animal Care and Use Committee and were conducted in Association for Assessment and Accreditation of Laboratory Animal Care–approved facilities. Maternal nutrient-restricted animals were fed 70% of food (Purina Monkey Diet 5038) eaten by contemporaneous controls. Pregnancy was confirmed at 30 days of gestation when the feeding regimen was initiated. Fetal liver tissue (left lobe from control animals only) was collected at gestation day (GD) 165 (term 185 days) and used directly for isolation of fetal hepatocytes or frozen at −80°C.
Cell culture
Human hepatoma HepG2 cells were cultured as described (22). P6 mouse embryo fibroblast cells that overexpress human IGF-I receptor (IGF-IR) (24) (a kind gift from Dr Renato Baserga, Thomas Jefferson University, Philadelphia, Pennsylvania) were cultured in DMEM, and treatments were performed in fetal bovine serum (FBS)–free conditions as described previously (19).
Primary baboon fetal hepatocyte cell culture
Hepatocytes were isolated and characterized from GD 165 control baboon fetuses (n = 3) as described previously (25).
RNA interference–mediated silencing
Silencing of individual subunits of human CK2 (α, α′, and β) in HepG2 cells was achieved by using SMARTpool siRNA (100 nM) and DharmaFECT transfection reagent 4 (Thermo Fisher Scientific) following the manufacturer's instructions. Raptor and/or rictor were silenced in HepG2 cells using transfection with 100 nM small interfering RNA (siRNA) (Sigma-Aldrich) and DharmaFECT transfection reagent 4 (Thermo Scientific) for 96 hours as described previously (26). The transfection reagents used for primary hepatocytes were same as those for HepG2 cells.
Two-dimensional (2-D) immunoblot of IGFBP-1
The 2-D immunoblot analysis with HepG2 cell media (∼150 μL) using polyclonal IGFBP-1 antibody and subsequent three-dimensional (3-D) image analysis of the 2-D spot intensity were all performed as described previously (12, 15, 19).
IGF-IR activation assay
The details of the bioassay using P6 cells have been described previously (19).
Protein kinase CK2 activity assay
The CK2 substrate peptide RRRDDDSDDD (DSD) (100 μM) was used to measure CK2 activity (27).
Results
IGFBP-1 concentration and phosphorylation are increased in umbilical plasma of human growth–restricted fetuses
To determine the site-specific phosphorylation of circulating IGFBP-1, we collected umbilical plasma from growth–restricted and gestational age–matched normal, AGA fetuses. The clinical characteristics of the study subjects are provided in Table 1. Fetal growth–restricted pregnancies had no complication other than FGR, and mothers had no concurrent disease (22), which is sometimes referred to as “idiopathic” FGR. Normal fetal growth (AGA) was defined as a birth weight between the 10th and 90th percentiles, using regional sex-specific growth charts. FGR was defined as a birth weight less than the third percentile (28). Similar to our previous amniotic fluid sample analysis (12, 13), an equal volume of umbilical cord plasma was loaded on gels. Total IGFBP-1 was markedly increased in FGR (Figure 1A), consistent with previous studies in other populations (29).
Table 1.
Clinical Characteristics of the Study Subjects
| Control (n = 12) | FGR (n = 12) | P Value | |
|---|---|---|---|
| Gestational age, wk, mean (range) | 32.5 (27–38) | 32.5 (26–39) | |
| Birth weight, g | 2072 ± 209 | 1335 ± 173 | .0126 |
| Placental weight, g | 504.9 ± 43 | 352.3 ± 46 | .0242 |
| pO2, mm Hg | 35.4 ± 1.87 | 21.0 ± 1.28 | .0001 |
| pCO2, mm Hg | 32.4 ± 1.78 | 46.3 ± 1.14 | .0054 |
Abbreviations: pCO2, partial pressure of carbon dioxide; pO2, partial pressure of oxygen.
Figure 1.
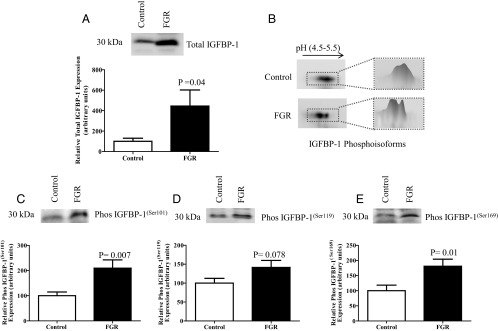
Total and phosphorylated IGFBP-1 in umbilical cord blood of fetal growth–restricted pregnancies. A, Representative Western blot of total IGFBP-1 in umbilical cord blood of FGR (n = 12) and gestational age–matched AGA controls (n = 12). Equal volumes of plasma samples were loaded. Fetal circulating IGFBP-1 was elevated in human FGR umbilical cord blood. B, Representative 2-D immunoblot of IGFBP-1 phosphoisoforms in umbilical cord blood from control (top) and growth–restricted fetuses (bottom) with a 3-D image analysis of spot intensity (adjacent), using equal aliquots of plasma samples. A marked increase in the intensity of spots and a shift toward the left (positive) was seen, representing increased IGFBP-1 phosphorylation in FGR. C–E, Representative Western blots of IGFBP-1 phosphorylated at Ser101, Ser119, and Ser169 in umbilical cord blood from FGR (n = 12) and gestational age–matched AGA control (n = 12). An equal volume of plasma was used as a loading control. Phosphorylation of circulating fetal IGFBP-1 was increased at the three serine residues in FGR. Values are given as means ± SEM. P < .05 vs control is considered significant as per unpaired Student t test.
Next we examined IGFBP-1 phosphorylation in human umbilical cord plasma using 2-D immunoblot analysis. IGFBP-1 phosphorylation was increased in cord plasma of fetal growth–restricted fetuses as shown by a clear shift in spots on the 2-D immunoblot toward pH 4.5 (acidic) compared with that for the gestational age–matched AGA controls (Figure 1B). In accordance with our previous studies (12, 13, 15, 19), we used 2-D immunoblot analysis as a qualitative approach for visual assessment of changes in IGFBP-1. To determine the site–specific changes in IGFBP-1 phosphoisoforms, we performed one-dimensional immunoblotting with antibodies raised against the three specific IGFBP-1 phosphorylation sites (Ser101, Ser119, and Ser169). The two phospho site–specific antibodies targeting pSer101 and pSer169 were validated in our previous study (19). A similar validation for the antibody raised against pSer119 is shown in Supplemental Figure 1. Using the three phosphorylation site–specific antibodies, we found that phosphorylation was increased at all three sites in umbilical cord plasma (Figure 1, C–E), which is the first report demonstrating increased phosphorylation at specific serine residues of circulating IGFBP-1 in human FGR.
IGFBP-1 expression and phosphorylation are increased in fetal growth–restricted baboon liver
To explore changes in total IGFBP-1 expression and IGFBP-1 phosphorylation in fetal liver, we used a well-established baboon model of moderate FGR induced by global maternal nutrient restriction (animals given 70% of the control diet). At GD 165 (term 184 days) fetal weight was reduced by 13% (control, 812 ± 34 g; FGR, 704 ± 33 g; P = .0004) and placental weight was decreased by 23% (control, 207 ± 15 g; FGR, 160 ± 13 g; P = .03) in the FGR group. As shown by Western blot analysis (Figure 2A), total IGFBP-1 expression in baboon fetal liver, normalized to actin, was markedly increased in the FGR group. Furthermore, using immunoblotting with phosphorylation site–specific antibodies, we showed that fetal liver IGFBP-1 phosphorylation was increased at Ser101, Ser119, and Ser169 in FGR (Figure 2, B–D).
Figure 2.

FGR in baboon fetal liver causes changes to IGFBP-1, mTOR signaling, and CK2 activity. A, Representative Western blot of IGFBP-1 in control (n = 4) and FGR (n = 9) baboon fetal livers. FGR resulted in increased total IGFBP-1 in fetal liver tissue. B–D, Representative Western blots of phosphorylated IGFBP-1 at Ser101, Ser119, and Ser169 in livers from control (n = 4) and growth–restricted fetuses (n = 9). FGR resulted in increased baboon fetal liver IGFBP-1 phosphorylation at the three serine sites. E and F, Representative Western blots of phos 4E-BP1(Thr70) and phos Akt(Ser473) in livers of control (n = 4) and growth–restricted fetuses (n = 9). mTORC1 and mTORC2 signaling was inhibited in FGR fetal liver tissue. G, Liver CK2 activity in control (n = 4) and growth–restricted baboon fetuses (n = 9). The kinase activity assays were performed in triplicate using equal amounts of protein from whole tissue extracts. CK2 activity was enhanced in growth–restricted baboon liver. Values are given as means ± SEM. P ≤ .05 vs control significant as per unpaired Student t test.
mTOR signaling is inhibited in fetal growth–restricted baboon liver
Given the decreased circulating fetal levels of essential amino acids in human FGR (30) and in our fetal growth–restricted baboon model (our unpublished data), we determined mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) signaling activity in baboon fetal liver using phosphorylation of 4E-BP1 at Thr70 (mTORC1) and Akt at Ser473 (mTORC2) as functional readouts. We found that the phosphorylation of 4E-BP1 (Thr70) was markedly reduced in the growth–restricted fetal liver (Figure 2E). Furthermore, liver mTORC2 signaling was also inhibited in FGR compared with that in controls as indicated by reduced phosphorylation of Akt (Ser473) (Figure 2F). These data demonstrate that the activity of both mTORC1 and mTORC2 signaling pathways is decreased in the fetal liver of fetal growth–restricted baboons and associated with increased total IGFBP-1 expression and IGFBP-1 phosphorylation.
Protein kinase CK2 activity and expression are increased in growth–restricted baboon fetal liver
Because IGFBP-1 contains consensus phosphorylation sites for CK2 (17) and given that CK2 can phosphorylate IGFBP-1 in vitro (21), we determined CK2 activity in baboon fetal liver extracts. Using a 32P kinase assay with the CK2 substrate peptide DSD (27), we demonstrate that CK2 activity was increased in fetal growth–restricted baboon fetal liver (Figure 2G). Furthermore, Western blot analysis using CK2 subunit-specific antibodies demonstrated that the expression of all 3 CK2 subunits (α, α′, and β) was higher in the fetal growth–restricted liver than in control liver (Supplemental Figure 2, A–C).
mTOR inhibition in HepG2 cells increases total expression and phosphorylation of IGFBP-1
To test the hypothesis that mTOR signaling is mechanistically linked to IGFBP-1 secretion and phosphorylation, we used cultured human HepG2 cells as a model for human fetal hepatocytes. HepG2 cells were treated with rapamycin (100 nM) for 24 hours to inhibit mTOR activity. First, we determined the phosphorylation of mTORC1 and C2 downstream targets as functional readouts in response to rapamycin. As shown in Supplemental Figure 3, A–C, rapamycin caused a profound decrease in the phosphorylation of 4E-BP1 at Thr70, p70S6K at Thr389, and Akt at Ser473, confirming that rapamycin efficiently inhibited both mTORC1 and C2 signaling in HepG2 cells after 24 hours. We determined the time-dependent effects of rapamycin on mTORC1 and mTORC2 signaling in HepG2 cells. Rapamycin inhibited mTORC1 signaling after 1 hour of treatment (Supplemental Figure 3D), but mTORC2 was not inhibited until 6 hours (Supplemental Figure 3E).
Next, we examined the effect of mTOR inhibition in HepG2 cells on IGFBP-1 secretion and phosphorylation. Rapamycin treatment increased secretion of total IGFBP-1 in the HepG2 cell media (Figure 3A). We measured total secreted IGFBP-1, which therefore includes both the phosphorylated and nonphosphorylated forms. In the absence of a recognized loading control for secreted proteins (equivalent to actin for nonsecreted protein), we used an equal volume of cell media (15) as a loading control. IGFBP-1 secretion was apparent already after 1 hour of rapamycin treatment (data not shown), suggesting that this effect may primarily be mediated by mTORC1, which was inhibited by 1 hour of rapamycin treatment (Supplemental Figure 3D). Rapamycin also induced IGFBP-1 phosphorylation as shown by the shift in spots on 2-D immunoblot toward pH 4.5 and confirmed by a shift toward pH 5.5 after dephosphorylation by alkaline phosphatase (Figure 3B). Further, immunoblot analysis with phosphorylation site–specific IGFBP-1 antibodies demonstrated that rapamycin caused a pronounced increase in phosphorylation at the three serine sites Ser101, Ser119, and Ser169 (Figure 3, C–E). Collectively, these data demonstrate that mTOR is a negative regulator of IGFBP-1 secretion and phosphorylation.
Figure 3.
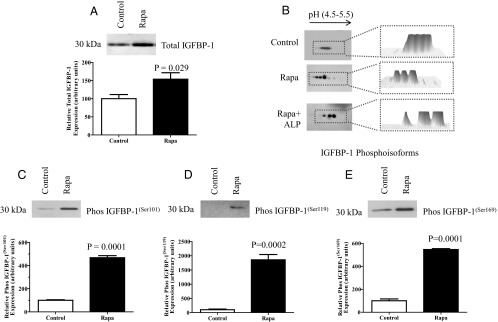
Effect of rapamycin (Rapa) on IGFBP-1 secretion and phosphorylation in HepG2 cells. A, Representative Western blot of IGFBP-1 secreted by HepG2 cells from control (n = 6) and rapamycin-treated cells (24 hours) (n = 6) using an equal volume of cell medium as a loading control. Rapamycin treatment of HepG2 cells significantly increased IGFBP-1 secretion. B, Representative 2-D Western blot of IGFBP-1 phosphoisoforms in cell medium from control cells (n = 3) and cells treated with rapamycin (n = 3) or treated with rapamycin and alkaline phosphatase (ALP) (n = 3) before 2-D immunoblot analysis as indicated. Equal loading (cell media) was performed. Phosphoisoform spot intensity for each blot is represented as a 3-D image in the adjacent panel. Rapamycin caused a marked increase in the number of spots and a shift toward the left (positive), indicating increased phosphorylation. C–E, Representative Western blots of phosphorylated IGFBP-1 at Ser101, Ser119, and Ser169 in cell media of control (n = 4) and rapamycin-treated cells (n = 4) using equal aliquots of cell media. Rapamycin treatment of HepG2 cells significantly increased IGFBP-1 phosphorylation at 3 serine sites. Values are given as means ± SEM. P < .05 vs control is significant as per unpaired Student t test.
mTOR inhibition in HepG2 cells stimulates CK2 activity
We have demonstrated that the increase in IGFBP-1 phosphorylation in fetal growth–restricted baboon liver is associated with inhibition of mTOR signaling and higher activity of CK2. These findings provided the basis to explore the role of CK2 in IGFBP-1 hyperphosphorylation in cultured HepG2 cells. The involvement of protein kinase CK2 in phosphorylation of IGFBP-1 in live intact cells has not been determined previously. First, we measured endogenous CK2 activity in cell lysate prepared from HepG2 cells in the 32P phosphorylation assay using the substrate peptide DSD (27). Treatment of HepG2 cells with rapamycin (100 nM) resulted in a marked stimulation of CK2 activity (Figure 4A). To confirm that our assays were specific for CK2 activity, we incubated cells with 4,5,6,7-tetrabromobenzotriazole (TBB), a selective CK2 inhibitor (31). TBB inhibited CK2 activity in a dose-dependent manner (data not shown). TBB (10 μM) significantly decreased CK2 activity and completely prevented the increase in CK2 activity that we observed in response to rapamycin treatment (Figure 4A). Next, we examined the effect of rapamycin on the expression of all 3 subunits of CK2 (CKα, CK2α′, and CK2β) in cell lysate prepared from HepG2 cells. As shown on immunoblots in Supplemental Figure 4, A–C, we did not detect any change in the relative levels of three CK2 subunits after rapamycin treatment, suggesting that mTOR signaling does not regulate CK2 at the transcriptional or translational levels in HepG2 cells.
Figure 4.
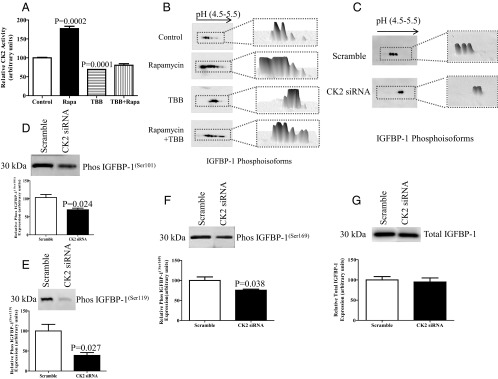
Effect of rapamycin (Rapa) and TBB on CK2 activity and IGFBP-1 phosphorylation, and the effect of CK2 siRNA silencing on IGFBP-1 phosphorylation. A, Summary of CK2 activity in HepG2 cell lysate after 24 hours of rapamycin and TBB treatments. The kinase activity assays were performed using equal amounts of protein from whole-cell lysates (n = 3). Rapamycin treatment of HepG2 cells resulted in mTOR inhibition and stimulation of CK2 activity that was prevented by treatment of cells with the CK2 inhibitor TBB. Values are means ± SEM. P < .05 vs control (n = 3) is significant as per Student t test. B, Representative 2-D Western blots (n = 3) provided qualitative assessments of IGFBP-1 phosphoisoforms in HepG2 cell media from control and rapamycin- and TBB-treated cells and from cells treated with both rapamycin and TBB together. An equal volume of cell medium was used as a loading control. Rapamycin treatment increased IGFBP-1 phosphorylation as is indicated by a marked increase in the number of spots and a shift toward the left (positive), representing increased phosphorylation. On the other hand, inhibition of CK2 by TBB attenuated IGFBP-1 phosphorylation induced by rapamycin (n = 3). C, Representative 2-D Western blots of IGFBP-1 phosphoisoforms in the media of HepG2 cells transfected with scramble or CK2 siRNA. The adjacent panels represent the 3-D isoform intensity profile in each sample. IGFBP-1 phosphorylation was markedly decreased in CK2 silenced (α + α′ + β combined) HepG2 cells. D, Representative Western blot of IGFBP-1 (n = 3) secretion in the media of HepG2 cells transfected with scramble or CK2 (α + α′ + β combined) siRNA using equal aliquots of cell media. E–G, Representative Western blots of IGFBP-1 phosphorylation at Ser101, Ser119, and Ser169 in the cell media of HepG2 cells transfected with scramble or CK2 (α + α′ + β combined) siRNA-silenced HepG2 cells showed a decrease in IGFBP-1 phosphorylation at all three phosphorylation sites. Values are given as means ± SEM. P < .05 vs control is significant as per unpaired Student t test.
Inhibition of CK2 decreases IGFBP-1 phosphorylation and prevents IGFBP-1 hyperphosphorylation caused by mTOR inhibition
Inhibition of CK2 activity by TBB (10 μM) reduced phosphorylation of IGFBP-1 in HepG2 cells and strongly attenuated IGFBP-1 hyperphosphorylation induced by rapamycin (Figure 4B). To confirm that CK2 plays an important role in mediating IGFBP-1 phosphorylation, we performed silencing of the CK2 holoenzyme by targeting the two catalytic subunits (CK2α and CK2α′) as well as the regulatory subunit (CK2β) in combination. After transfection, there was a decrease in protein expression of each of the three CK2 subunits (CK2α, CKα′, and CKβ) (Supplemental Figure 5, A–C). Silencing of the holoenzyme also resulted in a marked decrease in IGFBP-1 phosphorylation as determined by 2-D immunoblot analysis (Figure 4C). Furthermore, CK2 silencing markedly decreased IGFBP-1 phosphorylation at all three phosphorylation sites, Ser101, Ser119, and Ser169 (Figure 4, D–F). In contrast to the effect on IGFBP-1 phosphorylation, CK2 holoenzyme silencing did not alter total protein expression of IGFBP-1 secreted by HepG2 cells (Figure 4G). Taken together, these data demonstrate for the first time that CK2 is a key kinase responsible for phosphorylation of IGFBP-1 in live cells.
Inhibition of mTORC1 and/or mTORC2 using gene silencing stimulates CK2 activity
To further explore the contributions of mTORC1 and/or mTORC2 in regulating CK2 activity, we silenced raptor or rictor individually (essential components of mTORC1 and mTORC2, respectively) and raptor plus rictor combined and then assayed CK2 activity in HepG2 cells. Using protein expression of raptor and rictor, we showed that silencing of these targets was highly efficient (Supplemental Figure 6, A and B). CK2 activity was stimulated in cells silenced with raptor or rictor alone, suggesting that mTORC1 and C2 independently modulate CK2 activity (Supplemental Figure 6C). Simultaneous silencing of both raptor plus rictor further stimulated CK2 activity (Supplemental Figure 6C).
mTOR inhibition markedly inhibits IGF-I function by stimulating CK2 activity
We examined the functional effects of mTOR inhibition on IGF-I bioavailability using an IGF-IRβ autophosphorylation assay with P6 cells as in our previous study (19). P6 cells are a BALB/c3T3 cell derivative that overexpress human IGF-IR (24). Incubation of P6 cells with IGF-I alone induced IGF-IRβ autophosphorylation (Figure 5, lane 2 [+IGF-I]), considered as 100% stimulation. We used equal concentrations of total IGFBP-1 (100 ng) secreted by HepG2 cells under different treatments to conduct the biological assays using P6 cells. We demonstrated that IGF-IRβ autophosphorylation in P6 cells was significantly reduced when IGFBP-1 from media of untreated HepG2 cells was used to treat P6 cells, representing the effect of total IGFBP-1 secreted by HepG2 cells under basal (control) conditions (Figure 5, lane 3 [Control]). IGF-IRβ autophosphorylation in P6 cells was markedly reduced (Figure 5, lane 4 [Rapa]) compared with that for basal (control) conditions, when medium with the same concentration of total IGFBP-1 from rapamycin-treated HepG2 cells was used to treat P6 cells. In contrast, when P6 cells were incubated with media from HepG2 cells treated with TBB, IGF-IRβ autophosphorylation was significantly stimulated (P = .003) beyond basal levels. Notably, HepG2 cell media from TBB and rapamycin treatments in combination showed IGF-IRβ autophosphorylation similar to the basal levels. Collectively, these data suggest that mTOR inhibition markedly inhibits IGF-I function mediated by activation of CK2 and increased IGFBP-1 phosphorylation. Because the concentration of total IGFBP-1 was consistent throughout different treatments, the data strongly suggest that changes in receptor autophosphorylation were due to altered IGFBP-1 phosphorylation that regulated IGF-I bioavailability for IGF-IR.
Figure 5.

Effect of IGFBP-1 phosphorylation on IGF-I–induced IGF-IR autophosphorylation. A representative Western blot of phospho IGF-IRβ in P6 cells after incubation with HepG2 cell media consisting of an equal concentration (100 ng) of total IGFBP-1 is shown. An immunoenzymometric assay (Medix Biochemica) was performed for the quantitative determination of IGFBP-1 as per the manufacturer's protocol. Samples from control (no treatment) and from HepG2 cells treated with rapamycin (Rapa), TBB, or TBB + rapamycin combined were preincubated with IGF-I. Rapamycin caused a pronounced inhibition of IGF-I–induced IGF-IR autophosphorylation in P6 cells, whereas TBB stimulated IGF-IRβ autophosphorylation, and TBB + rapamycin had no significant effect on IGF-IR activity compared with that in control cells (no treatment) as indicated. The comparison between treatments was done by Repeated Measures ANOVA and the Bonferroni multiple comparison test.
mTOR inhibition in primary fetal baboon hepatocytes increases the phosphorylation of IGFBP-1
To confirm the key findings in HepG2 cells, we used primary hepatocytes isolated from control baboon fetuses. Inhibition of mTOR signaling in baboon primary hepatocytes using rapamycin resulted in increased secretion of total IGFBP-1 (Figure 6A). Rapamycin treatment also induced phosphorylation of IGFBP-1 at Ser101, Ser119, and Ser169 (Figure 6, B–D). Next, we silenced raptor or rictor in fetal baboon primary hepatocytes using siRNA as we previously reported for cultured primary human trophoblast cells (26). Silencing raptor resulted in a pronounced decrease in raptor protein expression and inhibition of the ribosomal protein S6 phosphorylation (Supplemental Figure 7, A and B). Similarly, silencing rictor markedly decreased protein expression of rictor and inhibited the phosphorylation of Akt at Ser473 (Supplemental Figure 7, C and D). Specific inhibition of mTORC1 and C2 using raptor plus rictor silencing in three separate cell preparations resulted in an increased phosphorylation of IGFBP-1 at Ser101 (Figure 6E). However, because no signal was detectable in the control conditions, data could not be analyzed statistically. Raptor and/or rictor silencing also resulted in an increased phosphorylation of IGFBP-1 at Ser169, compared with scramble siRNA (Figure 6F). Importantly, silencing DEPTOR, an endogenous inhibitor of mTORC1 and mTORC2 appeared to decrease the phosphorylation of IGFBP-1 at Ser169 below the levels obtained after transfection with scramble siRNA (Figure 6F). Thus, the data obtained in primary fetal baboon hepatocytes are consistent with the results from HepG2 cells and confirmed a major role of mTORC1 and mTORC2 signaling in regulating fetal hepatocyte IGFBP-1 secretion and phosphorylation.
Figure 6.

Effect of rapamycin (Rapa) on IGFBP-1 expression and phosphorylation, and mTORC1 and mTORC2 siRNA silencing on IGFBP-1 phosphorylation in fetal baboon hepatocytes. A, Representative Western blot of IGFBP-1 expression in baboon fetal hepatocytes treated with rapamycin (25 nM as indicated) (n = 3). Equal loading was performed. B–D, Representative Western blots of phosphorylated IGFBP-1 at Ser101, Ser119, and Ser169 in baboon fetal hepatocytes treated with rapamycin (n = 3). Equal loading was performed. E and F, Representative Western blots of IGFBP-1 phosphorylation at Ser101 and Ser169 in cell media of baboon hepatocytes transfected with scramble, rictor, raptor, rictor + raptor, and DEPTOR siRNA (n = 3 each). Values are given as means ± SEM. P < .05 vs control is significant as per unpaired Student t test. Rapamycin treatment (25 nm) of primary fetal baboon hepatocytes in 24 hours significantly enhanced IGFBP-1 secretion and phosphorylation.
Discussion
We provide novel evidence for an important role of fetal liver mTOR signaling in regulating IGF-I bioavailability by modulating CK2 activity and IGFBP-1 phosphorylation in FGR. Our in vitro data show that mTOR negatively regulates protein kinase CK2 that causes hyperphosphorylation of IGFBP-1 at three specific serine residues and inhibits IGF-I–induced IGF-IR signaling. The data suggest that CK2 constitutes an important mechanistic link between mTOR and IGFBP-1 phosphorylation. We further demonstrate that IGFBP-1 in umbilical cord blood is hyperphosphorylated at all three serine residues in human FGR. In addition, we show in vivo, that mTOR signaling is inhibited, whereas CK2 activity and expression as well as total IGFBP-1 expression and phosphorylation at the three sites are increased in the liver of fetal growth–restricted baboon fetuses. Based on this evidence, we propose a model in which inhibition of mTOR signaling with activation of CK2 in the fetal liver constitutes a key molecular link between nutrient deprivation, increased IGFBP-1 secretion and phosphorylation, and decreased IGF-I bioavailability in FGR (Figure 7). These findings provide new insights into the role of fetal IGFBP-1 phosphorylation in the pathophysiology of fetal growth restriction.
Figure 7.

The proposed model. Inhibition of mTOR signaling and activation of protein kinase CK2 in the fetal liver constitutes a key molecular link between nutrient deprivation, increased IGFBP-1 secretion and phosphorylation, and decreased IGF-I bioavailability in FGR.
Transgenic mice overexpressing human IGFBP-1 in the liver have elevated circulating IGFBP-1 and develop FGR, demonstrating a cause-and-effect relationship between changes in IGFBP-1 and fetal growth (8). In contrast, establishing a direct link between altered IGFBP-1 phosphorylation and fetal growth has been technically challenging (29–31). Fetal growth–restricted fetuses are often hypoxemic (32) and have decreased fetal circulating levels of essential amino acids (30, 33). Hypoxia and nutritional deprivation induce IGFBP-1 mRNA and protein expression in HepG2 cells (14), a cell line that exhibits many characteristics of fetal hepatocytes. Using the same cell culture model, we reported that hypoxia and leucine deprivation also caused increases in phosphorylation of IGFBP-1 at discrete sites. Greater phosphorylation of IGFBP-1 at three sites in leucine deprivation (at Ser101, 1.8-fold; Ser119, 4.5-fold; and Ser169, 2.5-fold) led to 30-fold higher affinity for IGF-I whereas hyperphosphorylation (at Ser101, 3-fold; Ser119, 2-fold; and Ser169, 4-fold) in response to hypoxia increased IGF-I affinity ∼300-fold and inhibited IGF-I–stimulated cell growth (15). Taken together, these data suggest the possibly that the synergistic effects of these phosphorylation events lead to differentially increased sequestration of IGF-I. These findings prompted us to explore the role of IGFBP-1 phosphorylation in regulating IGF-I bioavailability in FGR.
Our previous data using mass spectrometry demonstrated that human amniotic fluid IGFBP-1 is also hyperphosphorylated at three sites (Ser101, 4-fold; Ser119, 10-fold; and Ser169, 23-fold) in FGR (12, 13) with up to 4-fold higher affinity for IGF-I. The role of amniotic fluid IGFBP-1 phosphorylation in regulation of fetal growth is, however, yet to be determined. In the current study, using phosphorylation site–specific antibodies, we show for the first time a highly consistent increase in IGFBP-1 phosphorylation at all three sites, Ser101, Ser119, and Ser169 in umbilical plasma of human fetal growth–restricted fetuses. Although our data demonstrate that both total and phosphorylated IGFBP-1 were increased, we propose that the biological effects of IGFBP-1 in FGR are mainly due to the changes in sites and degree of IGFBP-1 phosphorylation. This line of reasoning is strongly supported by experimental data in our previous study (15) revealing that the predominant increase in IGF-I affinity occurs due to an increase in the abundance of specific phospho-IGFBP-1 isoforms (site and degree of phosphorylation) rather than increases in total IGFBP-1 or even total phosphorylated IGFBP-1.
Apart from what we have reported previously (12, 13, 15), little is known about the effects of different combinations of IGFBP-1 phosphoisoforms on IGF-I affinity. Although the synergistic effects of multiple phosphorylation sites and their relative IGF-I affinities are yet to be determined, Jones et al (9) have demonstrated by mutagenesis that phosphorylation at a single site (Ser101) led to a 3-fold reduction in IGF-I affinity. Using the same approach, we have reported that in addition to IGFBP-1 phosphorylation at Ser101, phosphorylation at Ser98, Ser119, and Ser169 profoundly but differentially decreases IGF-I bioavailability (19). We therefore speculate that site-specific increases in IGFBP-1 phosphorylation decrease IGF-I bioavailability and contribute to the decreased fetal growth in FGR (Figure 7). Furthermore, we also demonstrated in vivo that phosphorylation of IGFBP-1 at Ser101, Ser119, and Ser169 was consistently increased in baboon fetal growth–restricted liver, which has not been reported in any animal model. Because the liver is the primary source of circulating IGFBP-1 in the fetus (6), the data from the current study suggest that the changes in circulating total IGFBP-1 and IGFBP-1 phosphorylation in FGR are caused by altered fetal liver function.
In our previous report, we examined the effects of nonphosphorylated and phosphorylated IGFBP-1 secreted by HepG2 cells on IGF-I bioavailability using an immunodepletion strategy for IGFBP-1 (15). These studies demonstrated that the effect on IGF-1 activity was entirely dependent on IGFBP-1 and not on the other IGF-binding proteins that were secreted in only very small amounts (15). Using these findings as a basis, in our functional assays with P6 cells we showed that TBB treatment caused increased IGF-IR phosphorylation, compared with that for control media (Figure 5, lane 3 [Control]). Because we used equal concentrations of total IGFBP-1 in our assay, our data confirm that the functional effects (shown in Figure 5) in response to inhibition of mTOR and/or CK2 are caused by altered phosphorylation rather than total IGFBP-1. However, for more conclusive outcomes, it will be necessary to quantitatively identify functional endpoints (IGFBP-1 phosphorylation sites) linked with each of these treatments (events) using multiple reaction monitoring mass spectrometry and/or site-directed mutagenesis.
mTORC1 is activated by a multitude of upstream signals including insulin and amino acid, glucose, energy, and oxygen levels (16). IGFBP-1 secretion is inhibited by insulin and stimulated by cAMP (34–36), glucocorticoids (37), and hypoxia (14), whereas its phosphorylation is suggested to be regulated by hormones (38). The regulation of CK2, on the other hand, is complex and not well understood but involves expression and assembly, phosphorylation, regulatory interactions with small molecules such as polyamines, and protein-protein interactions with, for example, cyclin-dependent kinases (18). Here, we have demonstrated that inhibition of mTOR signaling stimulated CK2 activity in HepG2 cells, which is the first report of mTOR-mediated regulation of CK2 in any cell type. Furthermore, we also demonstrate that mTOR is a powerful negative regulator of liver IGFBP-1 phosphorylation. Notably, we show that mTOR inhibition significantly decreased IGF-I function, an effect that was prevented by inhibition of CK2 activity. Thus, these experiments identify CK2 and IGFBP-1 phosphorylation as a novel mechanistic and causative link between mTOR and IGF-I signaling, two critical pathways in regulation of cellular growth.
The fetal liver directly receives blood from the umbilical vein, which is normally nutrient rich and well oxygenated. However, in the case of placental insufficiency or maternal undernutrition, two common causes of FGR in human, the liver is perfused with blood with lower nutrient and/or oxygen content. A large number of nutritional and growth signals impinge on the mTOR signaling pathway, and we propose that mTOR in the liver constitutes a sensitive rheostat that links fetal nutrient availability to whole-body growth. The data provided in this study are consistent with the model that inhibition of mTOR signaling and activation of CK2 in the fetal liver constitutes a key molecular link between nutrient deprivation, increased IGFBP-1 secretion and phosphorylation, and decreased IGF-I bioavailability, leading to reduced fetal growth (Figure 7). As an interesting, albeit distant, analogy to this model Colombani et al (39) reported that in Drosophila, coordination of whole organism growth originates from the fat body, an insect organ with endocrine and storage functions similar to those of the vertebrate liver. TSC/TOR, homologous to the vertebrate mTOR, was identified as a fat body nutrient sensor that regulated body growth via a humoral mechanism that regulates peripheral phosphatidylinositol 3-kinase signaling.
In conclusion, through the combination of mechanistic approaches in vitro together with in vivo studies of liver tissues from a baboon FGR model and umbilical blood samples from well-characterized human fetal growth–restricted fetuses, we have shown that liver mTOR signaling negatively regulates IGF-I bioavailability by enhancing CK2 activity and stimulating IGFBP-1 phosphorylation. Further, by showing that mTOR regulates CK2, we have established a novel mechanistic link between mTOR and IGF-I signaling. These findings will allow us to construct a model for the regulation of secretion and phosphorylation of liver IGFBP-1 in the human fetus. In addition, because evolutionarily conserved mTOR and ubiquitous protein kinase CK2 can be present in diverse contexts, the mTOR-mediated CK2 regulation reported in this study may represent a general mechanism for regulation of cell growth.
Acknowledgments
We thank Dr. Laszlo Gyenis for his insightful discussions and for critical reading of the manuscript. We also gratefully acknowledge Dr. David W. Litchfield, Chair, Department of Biochemistry, University of Western Ontario (London, ON, Canada) for his interest and generous support in providing the necessary reagents and the facility for conducting studies with protein kinase CK2.
This work was supported by the Lawson Health Research Institute and Children's Health Research Institute (grants to M.B.G.), National Institutes of Health (Grants HD 078313 to M.B.G. and T.J. and HD 21350 to P.W.N.), and the VA (Merit Review Award 1I01BX001744–01A1 to A.K.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AGA
- appropriate for gestational age
- 2-D
- two-dimensional
- 3-D
- 3-dimensional
- FGR
- fetal growth restriction
- GD
- gestational day
- IGF-IR
- IGF-I receptor
- mTOR
- mammalian target of rapamycin
- siRNA
- small interfering RNA
- TBB
- 4,5,6,7-tetrabromobenzotriazole.
References
- 1. Pallotto EK, Kilbride HW. Perinatal outcome and later implications of intrauterine growth restriction. Clin Obstet Gynecol. 2006;49:257–269 [DOI] [PubMed] [Google Scholar]
- 2. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75:59–72 [PubMed] [Google Scholar]
- 4. Lassarre C, Hardouin S, Daffos F, Forestier F, Frankenne F, Binoux M. Serum insulin-like growth factors and insulin-like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res. 1991;29:219–225 [DOI] [PubMed] [Google Scholar]
- 5. Gibson JM, Aplin JD, White A, Westwood M. Regulation of IGF bioavailability in pregnancy. Mol Hum Reprod. 2001;7:79–87 [DOI] [PubMed] [Google Scholar]
- 6. Han VK, Matsell DG, Delhanty PJ, Hill DJ, Shimasaki S, Nygard K. IGF-binding protein mRNAs in the human fetus: tissue and cellular distribution of developmental expression. Horm Res. 1996;45:160–166 [DOI] [PubMed] [Google Scholar]
- 7. Giudice LC, de Zegher F, Gargosky SE, et al. Insulin-like growth factors and their binding proteins in the term and preterm human fetus and neonate with normal and extremes of intrauterine growth. J Clin Endocrinol Metab. 1995;80:1548–1555 [DOI] [PubMed] [Google Scholar]
- 8. Watson CS, Bialek P, Anzo M, Khosravi J, Yee SP, Han VK. Elevated circulating insulin-like growth factor binding protein-1 is sufficient to cause fetal growth restriction. Endocrinology. 2006;147:1175–1186 [DOI] [PubMed] [Google Scholar]
- 9. Jones JI, Busby WH, Jr, Wright G, Clemmons DR. Human IGFBP-1 is phosphorylated on 3 serine residues: effects of site-directed mutagenesis of the major phosphoserine. Growth Regul. 1993;3:37–40 [PubMed] [Google Scholar]
- 10. Yu J, Iwashita M, Kudo Y, Takeda Y. Phosphorylated insulin-like growth factor (IGF)-binding protein-1 (IGFBP-1) inhibits while non-phosphorylated IGFBP-1 stimulates IGF-I-induced amino acid uptake by cultured trophoblast cells. Growth Horm IGF Res. 1998;8:65–70 [DOI] [PubMed] [Google Scholar]
- 11. Frost RA, Tseng L. Insulin-like growth factor-binding protein-1 is phosphorylated by cultured human endometrial stromal cells and multiple protein kinases in vitro. J Biol Chem. 1991;266:18082–18088 [PubMed] [Google Scholar]
- 12. Nissum M, Abu Shehab M, Sukop U, et al. Functional and complementary phosphorylation state attributes of human insulin-like growth factor-binding protein-1 (IGFBP-1) isoforms resolved by free flow electrophoresis. Mol Cell Proteomics. 2009;8:1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abu Shehab M, Khosravi J, Han VK, Shilton BH, Gupta MB. Site-specific IGFBP-1 hyper-phosphorylation in fetal growth restriction: clinical and functional relevance. J Proteome Res. 2010;9:1873–1881 [DOI] [PubMed] [Google Scholar]
- 14. Popovici RM, Lu M, Bhatia S, Faessen GH, Giaccia AJ, Giudice LC. Hypoxia regulates insulin-like growth factor-binding protein 1 in human fetal hepatocytes in primary culture: suggestive molecular mechanisms for in utero fetal growth restriction caused by uteroplacental insufficiency. J Clin Endocrinol Metab. 2001;86:2653–2659 [DOI] [PubMed] [Google Scholar]
- 15. Seferovic MD, Ali R, Kamei H, et al. Hypoxia and leucine deprivation induce human insulin-like growth factor binding protein-1 hyperphosphorylation and increase its biological activity. Endocrinology. 2009;150:220–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368 [DOI] [PubMed] [Google Scholar]
- 18. Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abu Shehab M, Iosef C, Wildgruber R, Sardana G, Gupta MB. Phosphorylation of IGFBP-1 at discrete sites elicits variable effects on IGF-I receptor autophosphorylation. Endocrinology. 2013;154:1130–1143 [DOI] [PubMed] [Google Scholar]
- 20. Mottet D, Ruys SP, Demazy C, Raes M, Michiels C. Role for casein kinase 2 in the regulation of HIF-1 activity. Int J Cancer. 2005;117:764–774 [DOI] [PubMed] [Google Scholar]
- 21. Ankrapp DP, Jones JI, Clemmons DR. Characterization of insulin-like growth factor binding protein-1 kinases from human hepatoma cells. J Cell Biochem. 1996;60:387–399 [DOI] [PubMed] [Google Scholar]
- 22. Seferovic MD, Chen S, Pinto DM, Gupta MB. Altered liver secretion of vascular regulatory proteins in hypoxic pregnancies stimulate angiogenesis in vitro. J Proteome Res. 2011;10:1495–1504 [DOI] [PubMed] [Google Scholar]
- 23. Li C, Schlabritz-Loutsevitch NE, Hubbard GB, et al. Effects of maternal global nutrient restriction on fetal baboon hepatic insulin-like growth factor system genes and gene products. Endocrinology. 2009;150:4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol. 2001;54:133–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li C, Shu ZJ, Lee S, et al. Effects of maternal nutrient restriction, intrauterine growth restriction, and glucocorticoid exposure on phosphoenolpyruvate carboxykinase-1 expression in fetal baboon hepatocytes in vitro. J Med Primatol. 2013;42:211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vilk G, Saulnier RB, St Pierre R, Litchfield DW. Inducible expression of protein kinase CK2 in mammalian cells. Evidence for functional specialization of CK2 isoforms. J Biol Chem. 1999;274:14406–14414 [DOI] [PubMed] [Google Scholar]
- 28. Zhang J, Merialdi M, Platt LD, Kramer MS. Defining normal and abnormal fetal growth: promises and challenges. Am J Obstet Gynecol. 2010;202:522–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ong K, Kratzsch J, Kiess W, Costello M, Scott C, Dunger D. Size at birth and cord blood levels of insulin, insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-1 (IGFBP-1), IGFBP-3, and the soluble IGF-II/mannose-6-phosphate receptor in term human infants. The ALSPAC Study Team. Avon Longitudinal Study of Pregnancy and Childhood. J Clin Endocrinol Metab. 2000;85:4266–4269 [DOI] [PubMed] [Google Scholar]
- 30. Cetin I, Corbetta C, Sereni LP, et al. Umbilical amino acid concentrations in normal and growth-retarded fetuses sampled in utero by cordocentesis. Am J Obstet Gynecol. 1990;162:253–261 [DOI] [PubMed] [Google Scholar]
- 31. Sarno S, Reddy H, Meggio F, et al. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 ('casein kinase-2'). FEBS Lett. 2001;496:44–48 [DOI] [PubMed] [Google Scholar]
- 32. Pardi G, Cetin I, Marconi AM, et al. Diagnostic value of blood sampling in fetuses with growth retardation. N Engl J Med. 1993;328:692–696 [DOI] [PubMed] [Google Scholar]
- 33. Cetin I, Marconi AM, Bozzetti P, et al. Umbilical amino acid concentrations in appropriate and small for gestational age infants: a biochemical difference present in utero. Am J Obstet Gynecol. 1988;158:120–126 [DOI] [PubMed] [Google Scholar]
- 34. Patel S, Van Der Kaay J, Sutherland C. Insulin regulation of hepatic insulin-like growth factor-binding protein-1 (IGFBP-1) gene expression and mammalian target of rapamycin (mTOR) signalling is impaired by the presence of hydrogen peroxide. Biochem J. 2002;365:537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patel S, Lochhead PA, Rena G, et al. Insulin regulation of insulin-like growth factor-binding protein-1 gene expression is dependent on the mammalian target of rapamycin, but independent of ribosomal S6 kinase activity. J Biol Chem. 2002;277:9889–9895 [DOI] [PubMed] [Google Scholar]
- 36. Mounier C, Dumas V, Posner BI. Regulation of hepatic insulin-like growth factor-binding protein-1 gene expression by insulin: central role for mammalian target of rapamycin independent of forkhead box O proteins. Endocrinology. 2006;147:2383–2391 [DOI] [PubMed] [Google Scholar]
- 37. Sugawara J, Suh DS, Faessen GH, Suen LF, Shibata T, Kaper F, Giaccia AJ, Giudice LC. Regulation of insulin-like growth factor-binding protein-1 by nitric oxide under hypoxic conditions. J Clin Endocrinol Metab. 2000;85:2714–2721 [DOI] [PubMed] [Google Scholar]
- 38. Westwood M, Gibson JM, Williams AC, et al. Hormonal regulation of circulating insulin-like growth factor-binding protein-1 phosphorylation status. J Clin Endocrinol Metab. 1995;80:3520–3527 [DOI] [PubMed] [Google Scholar]
- 39. Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Léopold P. A nutrient sensor mechanism controls Drosophila growth. Cell. 2003;114:739–749 [DOI] [PubMed] [Google Scholar]