

Abstract
The gut is hypothesized to play a central role in the progression of sepsis and multiple organ dysfunction syndrome. Critical illness alters gut integrity by increasing epithelial apoptosis and permeability and by decreasing epithelial proliferation and mucus integrity. Additionally, toxic gut-derived lymph induces distant organ injury. Although the endogenous microflora ordinarily exist in a symbiotic relationship with the gut epithelium, severe physiologic insults alter this relationship, leading to induction of virulence factors in the microbiome, which, in turn, can perpetuate or worsen critical illness. This review highlights newly discovered ways in which the gut acts as the motor that perpetuates the systemic inflammatory response in critical illness.
Keywords: Gut, sepsis, critical illness intestine, epithelium, microbiome
The gut as the driving force of critical illness
The gut has long been characterized as the motor of multiple organ dysfunction syndrome (MODS) (1). This has been hypothesized to be due to dysregulated crosstalk between the gut’s epithelium, immune system and endogenous microflora (2) in which loss of balance between these highly interrelated systems leads to the development of systemic manifestations of disease, whose reach extends far beyond the intestine. Recent dramatic advances in our understanding of how the gut initiates and propagates critical illness have led to novel investigations into how it may be targeted for therapeutic gain in the intensive care unit (ICU). This is highly significant considering that critical illness accounts for up to 39% of total hospital costs (3) and sepsis kills 229,000–360,00 patients a year in the United States alone (4), yet a paucity of effective therapies exist for the sickest ICU patients. The purpose of this review is to highlight new insights into the complex balance that exists between the gut epithelium and the intestinal microbiome, and how perturbations in this relationship can lead to significant morbidity or even death (Figure 1).
Figure 1.
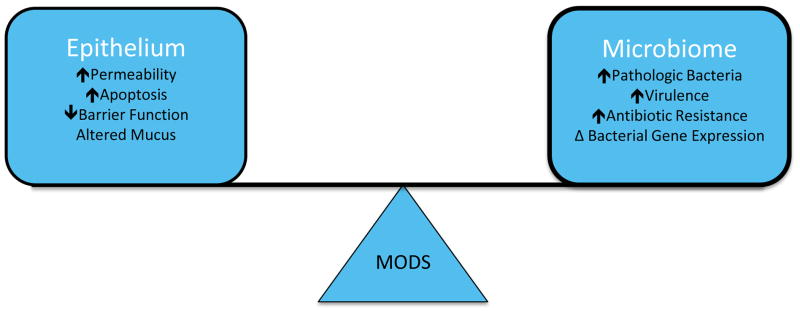
Relationship between the intestinal epithelium and microbiome. A complex but mutually beneficial relationship exists within the gut under homeostatic conditions. However, critical illness disrupts this balance, leading to pathologic changes in both the intestinal epithelium and microbiome, which can induce and perpetuate multiple organ dysfunction syndrome (MODS).
Evolution of the concept of the gut as the motor of MODS
In the 1980s, the predominant theory connecting the gut to MODS was bacterial translocation, a process whereby intestinal hyperpermeability allowed intact bacteria to leave the gut lumen, entering previously sterile environments and causing systemic illness (1;5;6). This theory was certainly plausible given the fact that multiple investigators over the past 30 years have demonstrated that critical illness – whether caused by sepsis, trauma, burn, or ischemia/reperfusion (I/R) – causes overgrowth of pathogenic bacteria within the gut and increased intestinal permeability (7–9). One theoretical mechanism through which intestinal contents were assumed to gain access to the rest of the body was through hematogenous spread via the mesenteric circulation to the portal circulation (2;6;10). However, although bacterial translocation was easy to detect in animal models of critical illness, this was not the case in human trials. A classic study sought to prove the concept of hematogenous spread of bacteria by placing portal vein catheters in patients following abdominal trauma. Contrary to prevailing thought, bacteria were detected in portal or systemic blood in only 2% of patients (and endotoxin in no patients) despite a 30% incidence of multiple organ failure (11). Multiple other studies over the last two decades have also not been able to confirm a direct relationship between intestinal hyperpermeability and bacterial translocation during critical illness. More recently, numerous studies have provided alternative routes through which the gut can both drive and perpetuate critical illness.
The gut-lymph hypothesis
An alternate route through which intestinal contents could initiate systemic injury is via the lymph (6;10). The gut-lymph hypothesis states that following injury, toxic mediators are released from the intestine, are transported through the mesenteric lymph nodes and go on to cause damage remote from the intestine.
Several lines of evidence support this theory. First, there is direct evidence that interrupting the flow of mesenteric lymph prevents distant organ damage. Anatomically, gut-derived lymph flows from the mesenteric lymphatic duct to the pulmonary circulation, and mesenteric lymph duct ligation attenuates both lung injury and neutrophil activation following endotoxemia in rats (12). Further, mesenteric lymph collected from mice following trauma-hemorrhagic shock (T/HS) and injected into trauma-sham shock (T/SS) mice induces lung injury and demonstrates the same toxic properties of intestinal lymph seen in T/HS (13). Additionally, ligation of the mesenteric lymph duct in either T/HS or in burn injury prevents injury-induced decreases in cardiac contractility and cardiac output (14–16). Perhaps most importantly, ligation of the mesenteric lymphatic duct improves survival in multiple animal models of critical illness (5;17).
Although the precise factors in gut-derived lymph that induce injury remain to be determined, several recent studies have shed light on this. A number of different proteins and lipid factors are responsible for the damage caused by mesenteric lymph while endotoxin and cytokines have been examined as candidate mediators and found not to be associated with its toxicity (18–21). Pancreatic proteases appear to be important in lymph toxicity, because ligation of the pancreatic duct in T/HS prevents production of bioactive mesenteric lymph (22). Further, T/HS-derived lymph has increased lipoprotein lipase activity, which induces endothelial toxicity (23;24). Lipase, in turn, generates free fatty acids (mainly palmitic, stearic, oleic and linoleic acid), which have been demonstrated to have the same cytotoxicity as T/HS-derived lymph and have been speculated to be the key components in lymph-induced damage (25). Of note, T/HS-derived lymph leads to activation of toll-like receptor (TLR) 4, which is necessary for mediation of the inflammatory cascade and priming of neutrophils required for lung injury (26;27). Lung injury is fully abrogated in TLR-domain-containing adapter-inducing interferon-β (TRIF) deficient mice and partially abrogated in Myd88 deficient mice (Myd88, like TRIF, is a TLR adapter protein), although no effect is seen in TLR2 deficient animals.
Pathways involved in lipid absorption and processing may also play a role in transporting toxic lymph. Intestinal microsomal triglyceride transfer protein (Mttp) is crucial for chylomicron assembly within the lumen and absorption of lipids via the lymphatic system. Mttp deficient mice have a block in chylomicron assembly and lipid malabsorption (28). When these animals are subjected to sepsis from Pseudomonas aeruginosa pneumonia, mortality is prevented (29). Although lung injury is unaffected in these animals, gut integrity is improved as evidenced by protection against sepsis-induced changes in apoptosis, proliferation and villus length. Serum levels of interleukin (IL)-6–which are elevated 8-fold in septic wild type mice – are unaltered in knockout animals. This pathway may also be implicated in human sepsis. In a recent Phase II clinical trial, patients on long-term statin therapy (which is started to lower cholesterol) had lower baseline IL-6 levels, and continuation of atorvastatin in patients with severe sepsis was associated with a survival advantage (30).
Gut-derived lymph is sterile following experimental T/HS (6). However, there are multiple reports of bacterial translocation to the mesenteric lymph in both human studies and animal models of critical illness. In a burn model with acute alcohol ingestion, there is culture evidence of bacterial translocation within the mesenteric lymph nodes (31). Similarly, translocation of anaerobic bacteria to the mesenteric lymph nodes is noted following hepatobiliary surgical procedures when assayed by bacterium-specific ribosomal RNA-targeted reverse-transcriptase polymerase chain reaction (32), and translocation of gram negative bacteria to mesenteric lymph nodes is also implicated in the development of bacteremia and/or spontaneous bacterial peritonitis in cirrhotic patients (33). The literature thus supports a role for both gut-derived sterile toxic lymph as well as bacterial translocation to the lymph, in a complex and possibly disease-specific manner.
Intestinal integrity
The gut epithelium plays a number of important roles in maintaining homeostasis in the healthy host including but not limited to i) absorbing food, ii) acting as a barrier against invading pathogens, iii) producing hormones and cytokines, iv) producing antimicrobial peptides, and v) continuously communicating with both the gut associated lymphoid tissue (the largest lymphatic organ in the body) and the intraluminal bacteria. Despite these myriad roles, the gut is made up of only a single layer of columnar cells that are continuously renewed from multipotent stem cells originating in the crypts of Lieberkühn (Figure 2). Intestinal stem cells express TLR4, which regulates whether they proliferate or die by apoptosis (34;35). These give rise to the four major cell lineages, each of which plays a unique role in the host (2). As cells migrate up the villus, they differentiate into absorptive enterocytes, mucus-producing goblet cells and enteroendocrine cells where they die of apoptosis or are exfoliated whole into the lumen. The final lineage is the Paneth cells, which migrate toward the crypt base where they produce defensins. Remarkably, the intestine renews itself on a continual basis, replacing nearly all of its cells (except stem cells and longer-lived Paneth cells) every 3–5 days.
Figure 2.

Anatomy of the intestine. Epithelial proliferation is initiated in the stem cells and is restricted to the crypt. Epithelial cells migrate upwards to the villus (enterocytes, goblet cells, enteroendocrine cells) or downwards to the crypt base (Paneth cells) where they differentiate. The epithelium is lined by a mucus layer acting as the first line of defense against the greater than 100 trillion commensal bacteria that reside within the gut lumen. The gut has a complex immune system including intra-epithelial lymphocytes and elements of the gut-associated lymphatic tissue (GALT) present in Peyer’s patches.
Critically ill hosts have numerous alterations in gut integrity that are thought to be important in the progression of sepsis and MODS. Although no single definition of gut integrity exists, we believe it is appropriate to consider subcellular, cellular and organ-wide components when considering gut integrity, as alterations at any of these levels may play a crucial role in the progression of MODS. On a subcellular level, redox is altered with septic peritonitis oxidizing glutathione/glutathione disulfide redox (36). On a cellular level, crypt proliferation is markedly decreased (37) and both crypt and villus apoptosis are simultaneously increased (38) following sepsis. Although epithelial migration is slowed in critical illness in a TLR4-dependent manner (39), changes in proliferation and apoptosis overwhelm this slowed progression of cells, resulting in a marked diminution of villus length (40). Simultaneously, critical illness induces global alterations in the mucus layer (reduced thickness, diminished luminal coverage, and poor adherence) (41) and gut barrier function (42).
Dysregulated apoptosis
The gut and spleen are the only solid organs in which apoptosis is reliably found on autopsy in human studies and animal models of critical illness, and intestinal epithelial cells are the only epithelial cells consistently found to undergo apoptosis in this setting (43;44). Notably, multiple co-morbidities including cancer, chronic alcohol abuse and aging are all associated with increased gut epithelial apoptosis in sepsis (9;45;46).
The relationship between gut apoptosis and survival is complex in animal models of critical illness with varying degrees of cell death as well as timing of apoptosis dependent on model analyzed (47). Overexpression of the anti-apoptotic protein Bcl-2 improves survival in murine sepsis models of either peritonitis or P. aeruginosa pneumonia models, but does not confer a survival advantage in acute lung injury (48–50).
The circuitry controlling apoptosis is complex (51), and a number of mechanisms have been shown to be involved in increased gut epithelial apoptosis (Figure 3). Methicillin resistant Staphylococcus aureus (MRSA) pneumonia-induced sepsis induces increases in pro-apoptotic proteins Bax and Bid and the anti-apoptotic protein Bcl-xL in the mitochondrial pathway (38). This is consistent with results demonstrating these three mediators are elevated in cecal ligation and puncture (CLP), a model of septic peritonitis (52). MRSA pneumonia also simultaneously induces an increase in Fas ligand but a decrease in Fas, fas-associated protein with death domain (FADD), phospho-FADD (pFADD), tumor necrosis factor receptor-1 (TNF-R1) and tumor necrosis factor receptor type 1-associated death domain protein (TRADD) in the receptor-mediated pathway. These changes appear to be at least partially organism-specific because P. aeruginosa pneumonia-induced increases in gut apoptosis are associated with increased Bcl-2 in the mitochondrial pathway and increased TNF-R1 and decreased Fas in the receptor-mediated pathway. Additionally, aged (but not young) mice deficient in heat shock protein 70 have increased mortality compared to age-matched wild type mice, and this is associated with increased gut apoptosis in the knockout mice (53).
Figure 3.

Pathways of apoptosis. Apoptosis occurs via the receptor-mediated pathway (green) or mitochondrial pathway (grey). Proapoptotic genes are shown in blue. Intracellular signals (black) activating caspase-2 may also initiate the mitochondrial pathway. Sepsis induces apoptosis in an organism and model-specific manner, in which multiple components of each pathway are affected. Both pathways converge at active caspase-3 which initiates cell death.
Sepsis-induced gut epithelial apoptosis is mediated by CD4+ lymphocytes (52). Although unmanipulated Rag−/− mice (that lack lymphocytes) and wild type mice have similar amounts of gut apoptosis, CLP induces a five-fold augmentation of gut apoptosis in Rag−/− mice, above and beyond the increase in gut apoptosis induced by sepsis. Subset analysis indicates that CD4+, but not CD8+, γδ or B cells are responsible for the anti-apoptotic effect of the adaptive immune system. Notably, the survival benefit conferred by gut overexpression of Bcl-2 is also dependent on lymphocytes because this intestine-specific transgene fails to alter mortality when overexpressed in Rag−/− mice, and adoptively transferring lymphocytes to these animals restores their survival benefit.
No current treatment exists which specifically targets gut epithelial apoptosis. However, targeting global gut integrity represents an attractive strategy in the treatment of sepsis (54). Epidermal growth factor (EGF) is a potent cytoprotective peptide that has healing effects on the intestinal mucosa. EGF reverses sepsis-induced changes in apoptosis, proliferation, villus length and permeability in both CLP and P. aeruginosa pneumonia (40;55). Notably, giving systemic EGF improves survival in both of these diseases, even when started 24 hours after pneumonia. In addition, overexpression of EGF in an intestine-specific manner improves both survival and gut integrity, suggesting that EGF may be a novel therapeutic for sepsis by targeting gut integrity (40;56). A similar survival advantage is seen after giving IL-15 to mice made septic via CLP or P. aeruginosa pneumonia (57). IL-15 is a pluripotent anti-apoptotic cytokine that decreases sepsis-induced gut apoptosis, although the relationship between gut apoptosis and survival in sepsis is associative in light of the fact that IL-15 also blocks apoptosis in NK cells, dendritic cells and CD8+ cells as well as altering systemic cytokines.
The mucus layer
The first line of defense against invading microorganisms is the intestinal mucus, and alterations in either the character or quantity of mucus can have a deleterious impact on the critically ill host. The unstirred mucus layer is hydrophobic and serves as an important barrier against the microflora in the gut lumen.
Mucus is the main barrier preventing digestive enzymes from entering the epithelium and causing epithelial disruption, a process that occurs, at least in part via degradation of E-cadherin and TLR4 (58). This process can be inhibited in shock or peritonitis using tranexamic acid, aprotinin, or 6-amidino-2-naphtyl p-guanidinobenzoate dimethanesulfate to inhibit proteases, and this approach leads to improved survival in pre-clinical models of critical illness (59;60). Because the presence of digestive enzymes in the wall of the intestine can cause autodigestion – a process that has been hypothesized to cause MODS -- the role of mucus in preventing this cascade may be critical for the maintenance of homeostasis (61).
I/R induces an acute loss of the mucus layer with a decrease in its hydrophobicity, which, in turn, is closely correlated to increased gut permeability (62). Similarly, T/HS induces breakdown of the mucus layer, and there is a direct correlation between loss of mucus layer and severity of villus injury (63). Interestingly, this is gender-dependent, as mucus is better preserved in female rats, associated to some degree with the estrus cycle (64). Although the mechanisms underlying mucus degradation are not fully elucidated, it is known that I/R breaks down mucus, at least in part, by degrading mucin 2 and fragmenting mucin 13 (58). Additionally, T/HS induces increased reactive oxygen species (ROS)-mediated mucus damage and loss of total antioxidant capacity immediately after shock, which is ameliorated by dimethyl sulfoxide (a free radical scavenger). T/HS also induces delayed reactive nitrogen intermediate-mediated mucus damage three hours later (65).
Digestive enzymes do not disturb mucin integrity in isolation. However, pancreatic proteases have been shown to play a synergistic role in mucus injury, and when combined with a mucolytic, they worsen mucus injury, over a mucolytic alone (66;67). However, although mucus injury is associated with villus injury, it is not sufficient in normal rats to cause the production of toxic gut-derived lymph or cause secondary lung injury, suggesting that mucus injury is additive to other insults but not sufficient to cause systemic injury from the gut.
Intestinal epithelium – a selective barrier
Although the intestinal tract is lined with only a single layer of epithelial cells, it acts as a selective barrier, that when functioning properly, allows physiologic movement of important elements but prevents pathophysiologic movement of potentially dangerous ones. The gut barrier allows paracellular movement of water, solutes, and immune modulating factors while preventing movement of large luminal molecules and microorganisms (68). This is done in large part via the apical tight junction and junctional adherens molecules (JAM), which prevent luminal contents from escaping into the local extraluminal environment and potentially from systemic spread (Figure 4).
Figure 4.

The gut barrier. The single layer of columnar cells in the intestinal epithelium are held together by tight junctions and adherens junctions (inset), which regulate the paracellular movement of intraluminal contents. Extracellular proteins, JAM, occludins, and claudins, interact with intracellular ZO proteins and the perijunctional actin-myosin ring to regulate permeability. MLCK modulates contraction of the actin-myosin ring. Extracellular cadherins along with intracellular catenins interact with the actin-myosin ring to form the adherens junction. Abbreviations: JAM: junctional adherens molecule; ZO: zonula occludens; MLCK: myosin light chain kinase.
Changes in intestinal permeability are seen in both sepsis and non-infectious (T/HS, I/R, burn) models of critical illness (7;41;69). Intestinal hyperpermeability occurs when any of these disease processes alters the expression of zonula occludens 1 (ZO-1), any of multiple claudin isoforms, or occludin in the tight junction or alternatively by altering expression of components of JAM (70). The paracellular route of transport is modulated by multiple intracellular and extracellular factors. Cytokines can act on junctional complexes to modulate permeability. Activation of myosin light chain kinase (MLCK) can also worsen paracellular permeability. MLCK phosphorylates the myosin light chain, which results in contraction or opening of the apical tight junction (70). MLCK activation is also associated with an increase in IL-6, TNFα, and IL-1β that further activates MLCK, which in turn increases intestinal permeability, at least in part via altered ZO-1 protein dynamics and occludin removal, ultimately leading to an amplification of the systemic inflammatory response in a feed forward response (31;71).
Numerous efforts at improving intestinal hyperpermeability – either in isolation or in conjunction with attempts to improve other components of gut integrity – have recently been described. One strategy involves targeting MLCK. Intraperitoneal injection of the MLCK inhibitor ML-9 attenuates burn-induced intestinal hyperpermeability, and this is associated with normalization of alterations in claudin-1, ZO-1 and occludin (69). Similarly, animals subjected to binge ethanol exposure and burn injury have morphological damage in their intestines, elevated intestinal IL-6 and IL-1β, increased bacterial translocation and reduced ZO-1 and occludin localization in the tight junction compared to sham mice (31). However, mice that receive PIK, an inhibitor of MLCK, do not have any of these changes.
Growth factors represent potential therapeutic candidates that mediate their effect, at least in part, via modulation of the gut barrier. As outlined above, EGF improves survival in sepsis, whether given systemically following CLP or pneumonia or delivered in an intestine-specific manner via overexpression in transgenic mice (40;55;56). EGF treatment normalizes gut permeability to sham levels, and this is associated with decreases in claudin-2 as well as changes in subcellular localization of this tight junction mediator. Exogenous EGF also improves gut permeability in a rat model of I/R when given in a pre-treatment or post-treatment manner, associated with alterations in ZO-1 and occludin expression as well as reduced systemic and intestinal IL-6 and TNFα (72). Exogenous heparin binding epidermal growth factor-like growth factor (HB-EGF) also induces beneficial effects. Animals subjected to CLP given exogenous HB-EGF have improved survival, associated with prevention of sepsis-induced intestinal hyperpermeability, increased villus length, gut apoptosis and bacterial counts (73). Of note, exogenous HB-EGF is effective in reversing these deficits in septic HB-EGF deficient animals, which have worsened gut integrity compared to septic wild-type animals. HB-EGF likely promotes its beneficial effects in multiple fashions, including promoting integrin-extracellular matrix interactions as well as intercellular adhesions (74) and preventing intestinal stem cells from injury under pathological conditions (75). Additionally, milk fat globule-EGF factor 8 also improves survival in both CLP and hemorrhagic shock and decreases both inflammation and tissue injury although its impact on intestinal permeability is unknown (76;77).
Microbiome
The intestinal microbiome is made up of more than 100 trillion microorganisms, and is continuously changing over the life of the host, based on diet, age, drug intake and presence or absence of disease (78–80). Increasingly, it is recognized that the microbiome plays a crucial role in the maintenance of health and that alterations in both the number and function of microorganisms in the microbiome can have a critical role on survival –both for good and for ill -- in critical illness.
The most reductionist method of assessing the role of the microbiome in critical illness is using germ-free mice, which entirely lack an endogenous microflora. Consistent with the complexity of the microbiome, germ-free mice have widely varying responses in preclinical models of critical illness. Germ-free mice have markedly higher mortality following P. aeruginosa pneumonia and have altered intestinal epithelial apoptosis (81). Further, sepsis-induced lymphocyte-dependent increases in gut epithelial apoptosis appear to be mediated by the microbiome in light of the fact that septic germ-free Rag−/− mice have similar levels of apoptosis as septic germ-free wild type mice (unlike in conventional animals where gut apoptosis is markedly higher following sepsis in Rag−/− mice). Alterations in mortality in septic germ-free mice are associated with lower TNF and IL-1β levels without alterations in systemic cytokines or intestinal proliferation or permeability. Similar results are found in germ-free mice subjected to Klebsiella pneumoniae pneumonia, which have significantly higher mortality than conventional mice, mediated in an IL-10 dependent fashion, modulatable by TLR activation (82). In contrast, germ-free mice have improved survival compared to animals with an intact microflora when subjected to hemorrhagic shock (83). Additionally, germ-free animals have 100% survival in a model of I/R that is 100% lethal in conventional animals, associated with an abrogation of the systemic inflammatory response (84). These disparate responses highlight the importance of disease model (and accompanying timing and pathophysiology) in mediating mortality in germ-free mice. They also suggest that while germ-free mice represent an important tool in understanding the microbiome, there may also be limitations to conclusions that can be drawn since the very strength of these animals (total absence of endogenous flora) is quite distinct from ICU patients that have a lifetime of host-microbial interactions.
Microbes alter their own virulence
An important aspect of the microbiome is its interaction with the host and the role this plays in the progression of disease (85). Although host and bacteria live under commensal or symbiotic conditions under homeostasis, alterations in this relationship drive bacteria to a more pathogenic phenotype (Figure 5). The concept that bacteria are able to sense their environmental state, including density of surrounding bacteria, and adjust behavior/virulence accordingly is a new direction in critical illness research and represents an evolution in our understanding of how the gut can function as the motor of critical illness. This has profound clinical implications because pathogen identification alone without attention to its virulence may not be sufficient for treating critically ill patients.
Figure 5.

Microbiome virulence induction. Commensal bacteria are able to sense the host microenvironment and other bacterial populations around them. Under basal conditions, the microbiome has a colonizing and/or symbiotic relationship with the host. During critical illness, microorganisms receive signals from both the host and surrounding bacteria that drive them to a virulent phenotype. Newly pathogenic bacteria can then further injure the already compromised host in a maladaptive positive feedback loop.
The prototypical example of inducible bacterial virulence is P. aeruginosa, as during injury, compounds are released by the host that either bind to or are taken up by this microorganism, leading to activation of its quorum sensing system, which in turn leads to the development of a lethal phenotype (86;87). Importantly, not only do activated microorganisms employ a variety of virulence tactics that subvert normal clearance mechanisms but they are also able to cause sepsis and remote organ failure without systemic dissemination. This is relevant both in isolation and in combination with other injury as evidenced by the fact that mortality is significantly higher in a murine model of intestinal I/R with superimposed inoculation of P. aeruginosa with in vivo virulence expression of the barrier disrupting adhesin PI-IL but not in mutant bacterial strains lacking this virulence protein (88). Another direct example of the acquired virulence of P. aeruginosa is seen following surgical insult. When this microorganism is harvested after inoculation into the cecum of mice subjected to sham surgery, it causes no lethality when injected into the peritoneum of noninjured mice. However, when P. aeruginosa is harvested after inoculation into the cecum of mice subjected to 30% hepatectomy, it causes 100% lethality when injected into the peritoneum of noninjured mice, demonstrating that surgical injury alters the virulence of the organism (89). In addition, bacteria isolated from the gut of mice that undergo hepatectomy demonstrate a distinct morphological appearance (wrinkled) compared to bacteria isolated from sham animals (smooth phenotype). Notably, Candida albicans has the ability to alter its own virulence in response to surgical injury in the same model (90).
Targeting the microbiome for therapeutic gain
Recent work has demonstrated that phosphate depletion may be a key modulatable determinant of altered microbial virulence. In times of extreme physiologic stress, phosphate is depleted in the host. Local phosphate concentration functions as an important cue by which endogenous flora evaluate the resources and health status of a host, and determine whether they should colonize or invade a host (91;92). For example, phosphate limitation induces Candida albicans isolated from the gut of critically ill patients to express filaments and a lethal phenotype (90). Further, in the presence of opioids, virulence is induced in P. aeruginosa via quinolone signal production; however, this genetic shift toward virulence is prevented in the presence of abundant phosphate (93). These findings suggest the possibility that maintaining local phosphate abundance may prevent virulence within multiple components of the microbiome and represent a novel therapeutic in the fight against gut-derived sepsis (94).
In light of the observation that altered gut flora measured in stool are associated with an increased risk of death in patients with systemic inflammation (95), another strategy to target the microbiome is the use of probiotics, prebiotics and synbiotics. Probiotics are exogenous live organisms whereas prebiotics are non-digestible nutrients that stimulate commensal bacterial growth. Synbiotics are a combination of both probiotics and prebiotics. Although their clinical use is rising, data on efficacy and mechanisms induced by stimulating the microbiome are still emerging (96). Probiotics improve mortality, bacteremia, and prevent sepsis-induced changes in gut epithelial apoptosis and proliferation following CLP (97). Additionally, probiotics can attenuate growth of intestinal bacteria, potentially limiting endotoxin production and preventing bacteremia (96). As a corollary to probiotic/synbiotic administration, there is also increasing evidence that fecal microbiota transplant is significantly more effective in the treatment of recurrent Clostridium difficile infection than standard antibiotic therapy by increasing fecal bacterial diversity in recipients (98).
Concluding remarks
The concept of the gut as the motor of critical illness is now 30 years old. Although the original iteration of this theory is not strongly supported by experimental and observational data, the concept that the gut might drive critical illness is as relevant now as it ever has been. During critical illness, the combined effect of erosion of the mucus barrier, a shift in the composition and virulence of intestinal microbes, and the inability of the host epithelium to regulate its proliferative and apoptotic response may lead to a tipping point in gut function where cascading inflammation drives distant organ failure. Additionally, the formation of toxic gut-derived lymph and alterations in the microbiome resulting in destructive host-pathogen crosstalk further propagate the cascade of distal organ injury. While each of these abnormalities is likely detrimental in isolation, a feed forward loop likely exists in which damage to one component of the gut leads to local and systemic injury which, in turn, damages other components of the gut. Unless broken, a continuous cycle of injury/amplification/repeat can lead to devastating consequences. In theory, rationale therapies aimed at restoring gut integrity, the microbiome and the homeostatic balance between the two systems represents an exciting avenue in the battle against critical illness, representing a proximal target in the road to multiple organ dysfunction that can be impacted early in the course of critical illness, prior to global dysregulation. Further studies need to be performed in the context of the gut as a multi-component organ, with examination not only of the element targeted but other local and systemic processes as well. Which approach in targeting the gut will be most effective in revving down its role as a motor of critical illness represents a prime research challenge for the next decade.
Highlights.
Critical illness alters apoptosis, permeability, proliferation and mucus integrity
Toxic gut-derived lymph induces distant organ damage
Critical illness alters the microbiome, bacterial virulence and pathogenicity
Critical illness damages the balance between the gut epithelium and microbiome
Acknowledgments
This work was supported by funding from the National Institutes of Health (GM072808, GM104323, GM095442)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Carrico CJ, et al. Multiple-organ-failure syndrome. The gastrointestinal tract: the “motor” of MOF. Arch Surg. 1986;121:196–208. doi: 10.1001/archsurg.1986.01400020082010. [DOI] [PubMed] [Google Scholar]
- 2.Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. 2007;28:384–393. doi: 10.1097/shk.0b013e31805569df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coopersmith CM, et al. A comparison of critical care research funding and the financial burden of critical illness in the United States. Crit Care Med. 2012;40:1072–1079. doi: 10.1097/CCM.0b013e31823c8d03. [DOI] [PubMed] [Google Scholar]
- 4.Gaieski DF, et al. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 5.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. 2012;10:350–356. doi: 10.1016/j.surge.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deitch EA. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci. 2010;21207(Suppl 1):E103–E111. doi: 10.1111/j.1749-6632.2010.05713.x. [DOI] [PubMed] [Google Scholar]
- 7.Fink MP, et al. Epithelial Barrier Dysfunction: A Unifying Theme to Explain the Pathogenesis of Multiple Organ Dysfunction at the Cellular Level. Crit Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Swank GM, et al. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. 1996;20:411–417. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- 9.Yoseph BP, et al. Chronic alcohol ingestion increases mortality and organ injury in a murine model of septic peritonitis. PLoS ONE. 2013;8:e62792. doi: 10.1371/journal.pone.0062792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Purohit V, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008;42:349–361. doi: 10.1016/j.alcohol.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore FA, et al. Gut bacterial translocation via the portal vein: a clinical perspective with major torso trauma. J Trauma. 1991;31:629–636. doi: 10.1097/00005373-199105000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Watkins AC, et al. Mesenteric lymph duct ligation attenuates lung injury and neutrophil activation after intraperitoneal injection of endotoxin in rats. J Trauma. 2008;64:126–130. doi: 10.1097/TA.0b013e3181574a8a. [DOI] [PubMed] [Google Scholar]
- 13.Senthil M, et al. Intravenous injection of trauma-hemorrhagic shock mesenteric lymph causes lung injury that is dependent upon activation of the inducible nitric oxide synthase pathway. Ann Surg. 2007;246:822–830. doi: 10.1097/SLA.0b013e3180caa3af. [DOI] [PubMed] [Google Scholar]
- 14.Lee MA. Role of gut-lymph factors in the induction of burn-induced and trauma-shock-induced acute heart failure. Int J Clin Exp Med. 2008;1:171–180. [PMC free article] [PubMed] [Google Scholar]
- 15.Sambol JT, et al. Mesenteric lymph duct ligation prevents trauma/hemorrhage shock-induced cardiac contractile dysfunction. J Appl Physiol. 2009;106:57–65. doi: 10.1152/japplphysiol.90937.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawai K, et al. Cellular mechanisms of burn-related changes in contractility and its prevention by mesenteric lymph ligation. Am J Physiol Heart Circ Physiol. 2007;292:H2475–H2484. doi: 10.1152/ajpheart.01164.2006. [DOI] [PubMed] [Google Scholar]
- 17.Badami CD, et al. Mesenteric lymph duct ligation improves survival in a lethal shock model. Shock. 2008;30:680–685. doi: 10.1097/SHK.0b013e318173edd1. [DOI] [PubMed] [Google Scholar]
- 18.Sarin EL, et al. Systemic neutrophil priming by lipid mediators in post-shock mesenteric lymph exists across species. J Trauma. 57:950–954. doi: 10.1097/01.ta.0000149493.95859.6c. [DOI] [PubMed] [Google Scholar]
- 19.Fang JF, et al. Proteomic analysis of post-hemorrhagic shock mesenteric lymph. Shock. 2010;34:291–298. doi: 10.1097/SHK.0b013e3181ceef5e. [DOI] [PubMed] [Google Scholar]
- 20.Mittal A, et al. Changes in the mesenteric lymph proteome induced by hemorrhagic shock. Shock. 2010;34:140–149. doi: 10.1097/SHK.0b013e3181cd8631. [DOI] [PubMed] [Google Scholar]
- 21.Peltz ED, et al. Proteome and system ontology of hemorrhagic shock: exploring early constitutive changes in postshock mesenteric lymph. Surgery. 2009;146:347–357. doi: 10.1016/j.surg.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caputo FJ, et al. Pancreatic duct ligation abrogates the trauma hemorrhage-induced gut barrier failure and the subsequent production of biologically active intestinal lymph. Shock. 2007;28:441–446. doi: 10.1097/shk.0b013e31804858f2. [DOI] [PubMed] [Google Scholar]
- 23.Deitch EA, et al. Anticoagulants influence the in vitro activity and composition of shock lymph but not its in vivo activity. Shock. 2011;36:177–183. doi: 10.1097/SHK.0b013e3182205c30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin Y, et al. Heparin use in a rat hemorrhagic shock model induces biologic activity in mesenteric lymph separate from shock. Shock. 2011;35:411–421. doi: 10.1097/SHK.0b013e31820239ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin X, et al. Role of lipase-generated free fatty acids in converting mesenteric lymph from a noncytotoxic to a cytotoxic fluid. Am J Physiol Gastrointest Liver Physiol. 2012;303:G969–G978. doi: 10.1152/ajpgi.00290.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reino DC, et al. Trauma hemorrhagic shock-induced lung injury involves a gut-lymph-induced TLR4 pathway in mice. PLoS ONE. 2011;6:e14829. doi: 10.1371/journal.pone.0014829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reino DC, et al. Activation of toll-like receptor 4 is necessary for trauma hemorrhagic shock-induced gut injury and polymorphonuclear neutrophil priming. Shock. 2012;38:107–114. doi: 10.1097/SHK.0b013e318257123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, et al. Compensatory increase in hepatic lipogenesis in mice with conditional intestine-specific Mttp deficiency. J Biol Chem. 2006;281:4075–4086. doi: 10.1074/jbc.M510622200. [DOI] [PubMed] [Google Scholar]
- 29.Dominguez JA, et al. Intestine-specific Mttp deletion decreases mortality and prevents sepsis-induced intestinal injury in a murine model of Pseudomonas aeruginosa pneumonia. PLoS ONE. 2012;7:e49159. doi: 10.1371/journal.pone.0049159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kruger P, et al. A multicenter randomized trial of atorvastatin therapy in intensive care patients with severe sepsis. Am J Respir Crit Care Med. 2013;187:743–750. doi: 10.1164/rccm.201209-1718OC. [DOI] [PubMed] [Google Scholar]
- 31.Zahs A, et al. Inhibition of long myosin light-chain kinase activation alleviates intestinal damage after binge ethanol exposure and burn injury. Am J Physiol Gastrointest Liver Physiol. 2012;303:G705–G712. doi: 10.1152/ajpgi.00157.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuno T, et al. Intraoperative bacterial translocation detected by bacterium-specific ribosomal rna-targeted reverse-transcriptase polymerase chain reaction for the mesenteric lymph node strongly predicts postoperative infectious complications after major hepatectomy for biliary malignancies. Ann Surg. 2010;252:1013–1019. doi: 10.1097/SLA.0b013e3181f3f355. [DOI] [PubMed] [Google Scholar]
- 33.Bellot P, et al. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33:31–39. doi: 10.1111/liv.12021. [DOI] [PubMed] [Google Scholar]
- 34.Neal MD, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem. 2012;287:37296–3308. doi: 10.1074/jbc.M112.375881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neal MD, et al. Intestinal stem cells and their roles during mucosal injury and repair. J Surg Res. 2011;167:1–8. doi: 10.1016/j.jss.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benton SM, et al. Differential regulation of tissue thiol-disulfide redox status in a murine model of peritonitis. J Inflamm (Lond) 2012;9:36. doi: 10.1186/1476-9255-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coopersmith CM, et al. Sepsis from Pseudomonas aeruginosa pneumonia decreases intestinal proliferation and induces gut epithelial cell cycle arrest. Crit Care Med. 2003;31:1630–1637. doi: 10.1097/01.CCM.0000055385.29232.11. [DOI] [PubMed] [Google Scholar]
- 38.Perrone EE, et al. Mechanisms of methicillin-resistant Staphylococcus aureus pneumonia-induced intestinal epithelial apoptosis. Shock. 2012;38:68–75. doi: 10.1097/SHK.0b013e318259abdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neal MD, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol. 2013;190:3541–3551. doi: 10.4049/jimmunol.1202264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez JA, et al. Epidermal Growth Factor Improves Survival and Prevents Intestinal Injury in a Murine Model of Pseudomonas Aeruginosa Pneumonia. Shock. 2011;36:381–389. doi: 10.1097/SHK.0b013e31822793c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rupani B, et al. Relationship between disruption of the unstirred mucus layer and intestinal restitution in loss of gut barrier function after trauma hemorrhagic shock. Surgery. 2007;141:481–489. doi: 10.1016/j.surg.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Fink MP. Intestinal epithelial hyperpermeability: update on the pathogenesis of gut mucosal barrier dysfunction in critical illness. Curr Opin Crit Care 2003. 2003;9:143–151. doi: 10.1097/00075198-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 43.Hotchkiss RS, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Takasu O, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. doi: 10.1164/rccm.201211-1983OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox AC, et al. Cancer causes increased mortality and is associated with altered apoptosis in murine sepsis. Crit Care Med. 2010;38:886–893. doi: 10.1097/CCM.0b013e3181c8fdb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turnbull IR, et al. Age disproportionately increases sepsis-induced apoptosis in the spleen and gut epithelium. Shock. 2004;22:364–368. doi: 10.1097/01.shk.0000142552.77473.7d. [DOI] [PubMed] [Google Scholar]
- 47.Vyas D, et al. Epithelial apoptosis in mechanistically distinct methods of injury in the murine small intestine. Histol Histopathol. 2007;22:623–630. doi: 10.14670/hh-22.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coopersmith CM, et al. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA. 2002;287:1716–1721. doi: 10.1001/jama.287.13.1716. [DOI] [PubMed] [Google Scholar]
- 49.Coopersmith CM, et al. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med. 2002;30:195–201. doi: 10.1097/00003246-200201000-00028. [DOI] [PubMed] [Google Scholar]
- 50.Husain KD, et al. BCL-2 Inhibits Gut Epithelial Apoptosis Induced by Acute Lung Injury in Mice but Has No Effect On Survival. Shock. 2003;20:437–443. doi: 10.1097/01.shk.0000094559.76615.1c. [DOI] [PubMed] [Google Scholar]
- 51.Andersen JL, et al. The tangled circuitry of metabolism and apoptosis. Mol Cell. 2013;49:399–410. doi: 10.1016/j.molcel.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stromberg PE, et al. CD4+ lymphocytes control gut epithelial apoptosis and mediate survival in sepsis. FASEB J. 2009;23:1817–1825. doi: 10.1096/fj.08-119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McConnell KW, et al. The role of heat shock protein 70 in mediating age-dependent mortality in sepsis. J Immunol. 2011;186:3718–3725. doi: 10.4049/jimmunol.1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dominguez JA, et al. Can we protect the gut in critical illness? The role of growth factors and other novel approaches. Crit Care Clin. 2010;26:549–565. doi: 10.1016/j.ccc.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark JA, et al. Epidermal growth factor treatment decreases mortality and is associated with improved gut integrity in sepsis. Shock. 2008;30:36–42. doi: 10.1097/shk.0b013e31815D0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark JA, et al. Enterocyte-specific epidermal growth factor prevents barrier dysfunction and improves mortality in murine peritonitis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G471–G479. doi: 10.1152/ajpgi.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inoue S, et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184:1401–1409. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang M, et al. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS ONE. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang M, et al. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeLano FA, et al. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. 2013;5:169ra11. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmid-Schonbein GW, et al. An elementary analysis of physiologic shock and multi-organ failure: the autodigestion hypothesis. Conf Proc IEEE Eng Med Biol Soc. 2012;2012:3114–3115. doi: 10.1109/EMBC.2012.6346623. [DOI] [PubMed] [Google Scholar]
- 62.Qin X, et al. Hydrophobicity of mucosal surface and its relationship to gut barrier function. Shock. 2008;29:372–376. doi: 10.1097/shk.0b013e3181453f4e. [DOI] [PubMed] [Google Scholar]
- 63.Lu Q, et al. The anatomic sites of disruption of the mucus layer directly correlate with areas of trauma/hemorrhagic shock-induced gut injury. J Trauma. 2011;70:630–635. doi: 10.1097/TA.0b013e3181e1221b. [DOI] [PubMed] [Google Scholar]
- 64.Sheth SU, et al. Intestinal mucus layer preservation in female rats attenuates gut injury after trauma-hemorrhagic shock. J Trauma. 2010;68:279–288. doi: 10.1097/TA.0b013e3181caa6bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fishman JE, et al. Oxidative modification of the intestinal mucus layer is a critical but unrecognized component of trauma hemorrhagic shock-induced gut barrier failure. Am J Physiol Gastrointest Liver Physiol. 2013;304:G57–G63. doi: 10.1152/ajpgi.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharpe SM, et al. Loss of the intestinal mucus layer in the normal rat causes gut injury but not toxic mesenteric lymph nor lung injury. Shock. 2010;34:475–481. doi: 10.1097/SHK.0b013e3181dc3ff5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qin X, et al. The mucus layer is critical in protecting against ischemia-reperfusion-mediated gut injury and in the restitution of gut barrier function. Shock. 2011;35:275–281. doi: 10.1097/SHK.0b013e3181f6aaf1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McConnell KW, et al. Epithelial cells. Crit Care Med. 2005;33:S520–S522. doi: 10.1097/01.ccm.0000187004.09189.1b. [DOI] [PubMed] [Google Scholar]
- 69.Chen C, et al. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE. 2012;7:e34946. doi: 10.1371/journal.pone.0034946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 71.Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. 2012;1258:9–18. doi: 10.1111/j.1749-6632.2012.06613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Geng Y, et al. Epidermal growth factor promotes proliferation and improves restoration after intestinal ischemia-reperfusion injury in rats. Inflammation. 2013;36:670–679. doi: 10.1007/s10753-012-9591-x. [DOI] [PubMed] [Google Scholar]
- 73.Yang J, et al. Heparin-binding epidermal growth factor-like growth factor improves intestinal barrier function and reduces mortality in a murine model of peritonitis. Surgery. 2013;153:52–62. doi: 10.1016/j.surg.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su Y, et al. HB-EGF promotes intestinal restitution by affecting integrin-extracellular matrix interactions and intercellular adhesions. Growth Factors. 2013;31:39–55. doi: 10.3109/08977194.2012.755966. [DOI] [PubMed] [Google Scholar]
- 75.Chen CL, et al. Heparin-binding EGF-like growth factor protects intestinal stem cells from injury in a rat model of necrotizing enterocolitis. Lab Invest. 2012;92:331–344. doi: 10.1038/labinvest.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shah KG, et al. Recombinant human milk fat globule-EGF factor 8 produces dose-dependent benefits in sepsis. Intensive Care Med. 2012;38:128–136. doi: 10.1007/s00134-011-2353-7. [DOI] [PubMed] [Google Scholar]
- 77.Zhang F, et al. Milk fat globule epidermal growth factor-factor 8 mitigates inflammation and tissue injury after hemorrhagic shock in experimental animals. J Trauma Acute Care Surg. 2012;72:861–869. doi: 10.1097/TA.0b013e318249a97c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schuijt TJ, et al. The intestinal microbiota and host immune interactions in the critically ill. Trends Microbiol. 2013;21:221–229. doi: 10.1016/j.tim.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 79.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cho I, et al. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fox AC, et al. The endogenous bacteria alter gut epithelial apoptosis and decrease mortality following Pseudomonas aeruginosa pneumonia. Shock. 2012;38:508–514. doi: 10.1097/SHK.0b013e31826e47e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fagundes CT, et al. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J Immunol. 2012;188:1411–1420. doi: 10.4049/jimmunol.1101682. [DOI] [PubMed] [Google Scholar]
- 83.Ferraro FJ, et al. A comparison of survival at different degrees of hemorrhagic shock in germ-free and germ-bearing rats. Shock. 1995;4:117–120. doi: 10.1097/00024382-199508000-00007. [DOI] [PubMed] [Google Scholar]
- 84.Souza DG, et al. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol. 2004;173:4137–4146. doi: 10.4049/jimmunol.173.6.4137. [DOI] [PubMed] [Google Scholar]
- 85.Seal JB, et al. Agent-based dynamic knowledge representation of Pseudomonas aeruginosa virulence activation in the stressed gut: Towards characterizing host-pathogen interactions in gut-derived sepsis. Theor Biol Med Model. 2011;8:33. doi: 10.1186/1742-4682-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patel NJ, et al. Recognition of intestinal epithelial HIF-1alpha activation by Pseudomonas aeruginosa. Am J Physiol Gastrointest Liver Physiol. 2007;292:G134–G142. doi: 10.1152/ajpgi.00276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu L, et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science. 2005;309:774–777. doi: 10.1126/science.1112422. [DOI] [PubMed] [Google Scholar]
- 88.Fink D, et al. Pseudomonas aeruginosa potentiates the lethal effect of intestinal ischemia-reperfusion injury: the role of in vivo virulence activation. J Trauma. 2011;71:1575–1582. doi: 10.1097/TA.0b013e31821cb7e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Babrowski T, et al. The intestinal environment of surgical injury transforms Pseudomonas aeruginosa into a discrete hypervirulent morphotype capable of causing lethal peritonitis. Surgery. 2013;153:36–43. doi: 10.1016/j.surg.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Romanowski K, et al. Candida albicans isolates from the gut of critically ill patients respond to phosphate limitation by expressing filaments and a lethal phenotype. PLoS ONE. 2012;7:e30119. doi: 10.1371/journal.pone.0030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zaborin A, et al. Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc Natl Acad Sci U S A. 2009;106:6327–6332. doi: 10.1073/pnas.0813199106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lamarche MG, et al. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev. 2008;32:461–473. doi: 10.1111/j.1574-6976.2008.00101.x. [DOI] [PubMed] [Google Scholar]
- 93.Zaborin A, et al. Pseudomonas aeruginosa overrides the virulence inducing effect of opioids when it senses an abundance of phosphate. PLoS ONE. 2012;7:e34883. doi: 10.1371/journal.pone.0034883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Romanowski K, et al. Prevention of siderophore-mediated gut-derived sepsis due to P. aeruginosa can be achieved without iron provision by maintaining local phosphate abundance: role of pH. BMC Microbiol. 2011;11:212. doi: 10.1186/1471-2180-11-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shimizu K, et al. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci. 2011;56:1171–1177. doi: 10.1007/s10620-010-1418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shimizu K, et al. Probiotic/synbiotic therapy for treating critically ill patients from a gut microbiota perspective. Dig Dis Sci. 2013;58:23–32. doi: 10.1007/s10620-012-2334-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khailova L, et al. Probiotic Administration Reduces Mortality and Improves Intestinal Epithelial Homeostasis in Experimental Sepsis. Anesthesiology. 2013;119:166–177. doi: 10.1097/ALN.0b013e318291c2fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van NE, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–15. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]