

Abstract
Patch-clamp electrophysiology is the technique of choice for the biophysical analysis of the function of nerve, muscle, and synapse in C. elegans nematodes. Considerable technical progress has been made in C. elegans electrophysiology in the decade since the initial publication of this technique. Today, most, if not all electrophysiological studies that can be done in larger animal preparations can also be done in C. elegans. This chapter has two main goals. The first is to present to a broad audience the many techniques available for patch-clamp analysis of neurons, muscles, and synapses in C. elegans. The second is to provide a methodological introduction to the techniques for patch-clamping C. elegans neurons and body-wall muscles in vivo, including emerging methods for optogenetic stimulation coupled with post-synaptic recording. We also present samples of the cell-intrinsic and post-synaptic ionic currents that can be measured in C. elegans nerve and muscle.
Introduction
Non-mammalian organisms continue to be effective experimental systems for the study of fundamental problems in the function of neurons and neural circuits. Non-mammalian neuroscience has coalesced around the three systems with a high level of genetic tractability: Drosophila melanogaster fruitflies, Danio rerio zebrafish, and Caenorhabditis elegans nematodes. Each animal has aspects in which it excels. The nematode, the focus of this chapter, has three main advantages: (i) a nervous system of only 302 neurons, (ii) neurons that are reidentifiable from one individual to the next, and (iii) a complete anatomical reconstruction of its nervous system.
The celebrated strengths of the C. elegans nervous system come at a price. The electrophysiologist must learn to record from neurons whose cell bodies are about 2 μm in diameter and protected by a tough, pressurized cuticle in an animal that is only 1 mm long. Our experience in training others to record from C. elegans neurons and muscles indicates that patch-clampers are able to master the aspects of the technique that are specific to C. elegans in days to weeks. The ease with which the technique can be learned is traceable to two factors. First, the approach involves simple modifications of standard patch clamping equipment and procedures (Goodman et al., 1998), mainly to compensate for the small size of the animals and their neurons and muscles. Second, the process of establishing and maintaining whole-cell recordings from C. elegans neurons is not especially difficult relative to other electrophysiological preparations.
With the advent of genetically encoded calcium indicators and techniques for in vivo calcium imaging in C. elegans, (Kerr et al., 2000; Kerr, 2006; Kerr and Schafer, 2006) it is reasonable to ask: Why bother with electrophysiology? The answer depends almost entirely on the type of information required for the problem under study. If one wants to observe neuronal activity in intact animals, then calcium imaging is indispensable, as electrophysiology requires dissection, whereas calcium imaging in C. elegans does not. For other problems, however, electrophysiology is the indispensable technique. Three main examples come to mind. First, voltage-clamp is an absolute requirement for recording single channel currents or isolating the macroscopic current flowing through a particular type of ion channel. Second, calcium imaging has neither the time resolution nor the signal-to-noise ratio to resolve voltage transients such as unitary postsynaptic potentials in neurons or single action potentials in muscle cells. Third, calcium imaging does not in general reveal synaptic inhibition, except in unusually favorable cases.
Patch-clamp electrophysiology has been available in C. elegans now for a little over a decade. During this time, patch-clamp recordings have been used to examine the muscles of the body wall and pharynx, and about a dozen types of neurons (Francis et al., 2003). Progress has been rapid along four main fronts including genetically identified, ligand-gated and voltage-gated channels, intrinsic electrical properties of C. elegans neurons, the electrical events of sensory transduction, and the physiology and molecular biology of the neuromuscular junction (NMJ).
This chapter has two main goals. The first is to present to a broad audience the many techniques available for patch-clamp analysis of neurons, muscles, and synapses in C. elegans. We hope this presentation will be useful in correcting the misapprehension that C. elegans remains mostly intractable to these types of studies. We would be pleased if it also helps to cultivate fruitful collaborations between C. elegans geneticists and electrophysiologists.
The second goal of this chapter is to provide a methodological introduction to the techniques for patch-clamping C. elegans neurons and body-wall muscles in vivo. Patch clamping in any organism requires a level of practical and analytical know-how that is not easily reduced to recipes and protocols. Thus, the methodological components of this chapter are written mainly for experimentalists who are already familiar with the basic practice and principles of patch-clamp electrophysiology. Accordingly, we emphasize the ways in which patch clamping in C. elegans is similar to and different from patch clamping neurons in other animals. Important techniques not covered in this chapter are sharp-electrode and patch-clamp recording in pharyngeal muscle (Shtonda and Avery, 2005) and patch-clamp recording from cultured neurons (Bianchi and Driscoll, 2006; Christensen et al., 2002).
Methods
in situ patch clamp recording from C. elegans neurons
Here, we provide an overview of a typical workflow followed for obtaining in vivo patch-clamp recordings from C. elegans neurons as well as detailed descriptions of each of the steps in the workflow (Figure 1). This work incorporates improvements devised in the ten years that have passed since reporting the first such experiments (Goodman et al., 1998).
Figure 1. Workflow and approximate timeline for in vivo recording from identified C. elegans neurons.
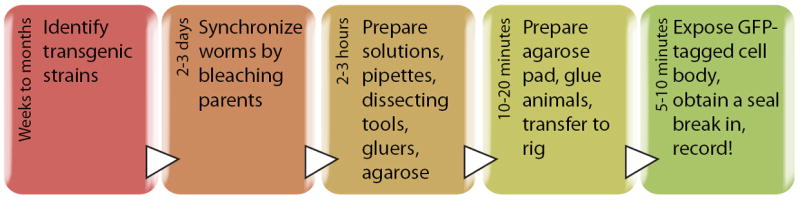
Each box represents a key step in the workflow, times given are prior to the moment of obtaining a whole-cell patch-clamp recording.
Immobilization
We immobilize worms on the surface of a thin agarose pad using veterinary-grade, cyanoacrylate adhesive (WormGlu, Glustich Inc., Delta, BC Canada). As the glue is somewhat toxic, we limit our experiments such that animals are exposed to the glue for no more than 1 hour. Thus, it is useful to glue animals quickly, accurately and efficiently. Four strategies streamline this step: 1) transfer tens-to-hundreds of animals to the agarose pad using a filter paper disk; 2) use age-synchronized animals; 3) cool the agarose pad to reduce movement; 4) arrange animals using a fine hair or glass fiber. Glue is applied from a glass pipette placed next to each animal. This can be done free-hand or with the aid of a mechanical manipulator (e.g. Mk1, Singer Instrument Co., Ltd, Somerset, UK). If animals must be rolled onto their dorsal or ventral sides, it is helpful to use a fluorescence stereomicroscope for gluing. In this way, the GFP label can be used to detect successful rolling: bilaterally symmetric pairs of target cells will appear as a single spot in animals lying on the left or right sides, but two spots will be visible when the animal is rolled to the dorsal or ventral side
Rolling is not the only maneuver that can improve dissection of neuronal cell bodies in the anterior ganglion. For instance, forming a slight bend in the body of the worm at the level of the pharynx can help move deep neurons more superficially towards the cuticle. A recent advance is the use of a stamp to create a grooved agarose pad; such pads improve the ease and reproducibility of the positioning step. We use grooved pads containing two arrays of channels (20μm wide) that intersect at 20°. The channels form a cradle that facilitates rotation about the worm’s long-axis and allows the pharyngeal region to be bent obliquely around the junction of two channels (Figure 2).
Figure 2. Grooved agarose pads with a worm glued to the surface.
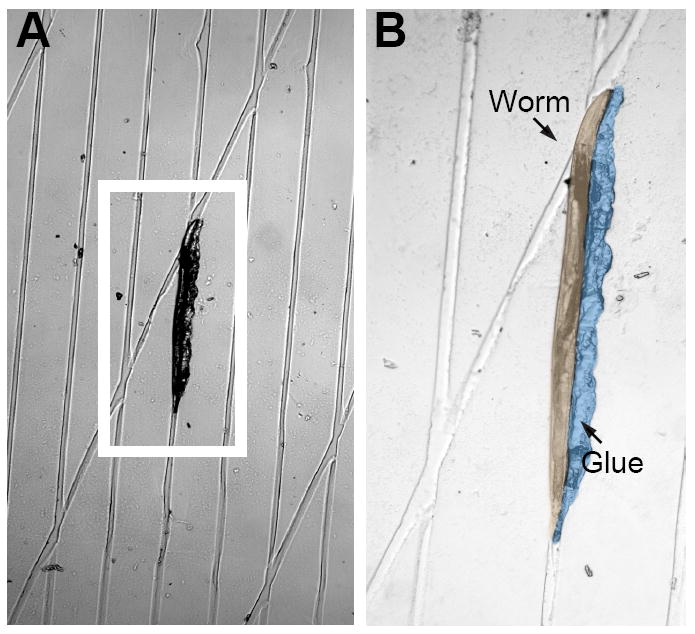
A young adult worm glued to a grooved agarose pad. (A) low-magnification (10X) micrograph and (B) high-magnification (20X) image. The glue and worm have been pseudocolored to enhance contrast. Anterior is up. Note that the animal’s head is bent around the intersection of two channels.
These grooved pads are formed using a stamp to create channels in an agarose pad formed on a glass coverslip. The stamp is fabricated in the lab by casting a thin layer of polydimethylsiloxane (PDMS or Sylgard™) on a photolithographically-generated SU-8 master mold (Lindsay et al, 2011; Xia et al, 1996). A PDMS stamp can also be generated from a master mold created by cutting grooves into a piece of scotch tape attached to a glass slide with a razor blade. The stamp is used as follows. First, coverslips must be coated so that the grooved agarose pad adheres to the coverslip and is released from the PDMS stamp. This is accomplished by coating coverslips with a thin layer of 4% (w/w) agarose that is allowed to dry completely in an oven (80°C) or on a hot plate. Second, a drop of molten agarose (2% w/w) is transferred to the coated coverslip and the PDMS stamp is pressed into the molten agarose as it sets. After the agarose has set, the stamp is gently removed from the grooved pad.
Dissection: the ‘slit-worm’ preparation
The dissection procedure must be optimized for each neuron, a process that may take a few days of exploration. For example, the AFD neurons lie along the lateral midlines in the anterior ganglion in the head (see http://www.wormatlas.org/neurons/Individual%20Neurons/AFDframeset.html). This complicates dissection because worms normally lie on their left or right sides, positioning AFD in the center of each animal and increasing the likelihood that if exposed, AFD will be surrounded by other neurons. To circumvent this difficulty, animals are rolled onto their dorsal or ventral surfaces prior to gluing (Ramot et al., 2008). The ASE and PLM neurons, by contrast, are positioned closer to the ventral surface. Thus, whereas animals must be glued along their dorsal sides, there is no need to roll them prior to gluing. The PLM neurons are most easily dissected in animals in which the glue extends all the way down the dorsal aspect of the tail.
We make dissecting tools from solid glass rods pulled on a horizontal electrode puller (Sutter P-97 or P-2000). The ideal tool is not too long, but tapers to a tip that is no more than 1 μm in diameter. Two steps are needed to expose neuronal cell bodies for recording while maintaining their integrity. First, a small incision is made at some distance away from the target neuron. This incision allows a portion of the intestine and gonad to emerge. This maneuver decreases the internal hydrostatic pressure. A second incision is made near the target neuron, which allows this cell body to emerge intact (see Video 1).
Recording
We record only from dissections that meet the following three criteria. First, the neuron of interest should be near the outside edge of the bouquet of neuronal cell bodies. Second, no Brownian movement should be visible within the cell body. Third, the cell body should not have split in two. (For reasons that remain unclear, such splitting or blebbing is an all too frequent event). If a given dissection fails these quality control checks, it is wiser to move on to another animal than to make do with a poor dissection. Once a suitable dissection is achieved, a recording pipette is lowered into the recording chamber, applying positive pressure (3-4kPa) to the pipette. Positive pressure is maintained until the pipette contacts the cell body. Releasing pressure upon contact is generally sufficient to obtain a GΩ seal and to form a visible, GFP-tagged membrane bleb. At this point, we compensate pipette capacitance and apply a family of voltage pulses that will be subtracted from whole-cell records in order to minimize residual capacity transients (Goodman et al., 1998; Lockery and Goodman, 1998). Next, we apply a combination of suction by mouth and a brief voltage ‘zap’ (0.5-0.9 V for 0.5ms) to achieve the whole-cell recording configuration. Because the whole-cell capacitance of C. elegans neurons is typically less than 4 pF, the change in capacity transients is not a reliable indicator of a successful break-in. The rupture of the bleb and dilution of soluble GFP that occurs upon break-in is a better indicator.
Two key factors for successful recording from tiny C. elegans neurons are the shape of the recording pipette (blunt is ideal) and the stability of the pipette holder to limit movement during suction. Below, we describe improvements to standard patch-clamp recording techniques that address these issues. In principle, suction-induced pipette movement can be avoided by using the perforated-patch clamp technique, in which a pore-forming anti-fungal agent (e.g. nystatin or amphotericin) is used to provide low-resistance electrical access to the cell interior. Although this practice has been applied to C. elegans neurons and is indispensible for events that rely on soluble factors (Nickell et al., 2002; Ward et al., 2008), the general utility of the recordings is compromised by the very high access resistance of such recordings.
Electrodes for recording from C. elegans neurons and other small cells
Recording pipettes must have tip openings that are smaller than C. elegans neuronal cell bodies (1-3 μm in diameter). It is difficult to make pipettes this small that have resistances less than 20 MΩ with conventional fabrication techniques. To solve this problem, Goodman and Lockery invented “pressure polishing” which uses high-pressure air (40 PSI) to shape the pipette tip (Goodman and Lockery, 2000). Briefly, we mount freshly pulled pipettes on a microforge equipped with a long-working distance, high-power objective lens (100X) and fire-polish while applying pressure to the lumen. This creates a blunt shape. The tip opening is reduced by a second fire-polishing step without pressure. This method is applicable to a variety of glass types (borosilicate, aluminasilicate, soda lime) and dimensions; we use thick-walled borosilicate glass with a filament (Sutter BF110-86-10) pulled to an initial tip opening of ~1-2 μm on a horizontal puller (Sutter P-97 or P-2000). When filled with our standard intracellular solution, our pipettes have resistances of 8-12 MΩ and tip openings less than 1 μm in diameter. A visualized experiment demonstrating pressure polishing is available (Johnson et al., 2008).
At the rig
A typical recording session follows this script. First, prepare solutions, agarose, recording pipettes, dissecting tools, and gluers. Next, wash worms of the desired genotype from their growth plate in distilled water. Let the animals settle to the bottom of a conical, 1.5mL microfuge tube or spin them briefly to the bottom. Make a thin agarose pad (smooth or grooved) and transfer a large number of worms to the pad. [In the Goodman lab, we pipette 4μL of worm solution to a small circle of Whatman No. 1 filter paper and invert the filter paper on the agarose pad.] Use a fine hair or brush to distribute and arrange the worms. Apply glue alongside ~8-12 worms. Flood the pad with extracellular recording solution to re-hydrate worms and remove unglued animals. Transfer the pad/recording chamber to the recording rig. Start superfusion. Mount a dissecting tool and dissect animals until a good-quality dissection is achieved. Fill a recording pipette with intracellular saline, mount it on the headstage of the patch-clamp amplifier, and insert the pipette in the bath with positive pressure. Measure and record the pipette resistance in your notebook. Attempt to seal on to the cell body of the desired neuron. If successful, attempt to break in using a combination of mouth suction and electrical zap. Success is monitored either by looking for an increase in the amplitude of the capacity transient evoked by a small voltage step (hard) or by looking for dilution of cytoplasmic GFP (easy).
Types of recordings
The ‘slit-worm’ preparation can be used to investigate many aspects of neuronal function. Below is a brief summary of the types of information that can be obtained, and the recording configurations used to obtain the data.
1. Whole-cell ionic currents from identified neurons (voltage-activated)
Each type of neuron in C. elegans expresses an apparently unique ensemble of voltage-activated currents that include both voltage-activated outward and inward currents (Goodman et al., 1998). Such currents are studied by applying a series of voltage pulses during a whole-cell recording. In control saline, net membrane current is close to zero in the operating range of most C. elegans neurons (-70 to -20 mV), inward at more negative potentials, and outward at more depolarized potentials (Figure 3). This observation is consistent with the high input resistance (>1 GΩ) of most C. elegans neurons (Goodman et al., 1998).
Figure 3. Voltage-activated currents in AWC and AWA chemosensory neurons.
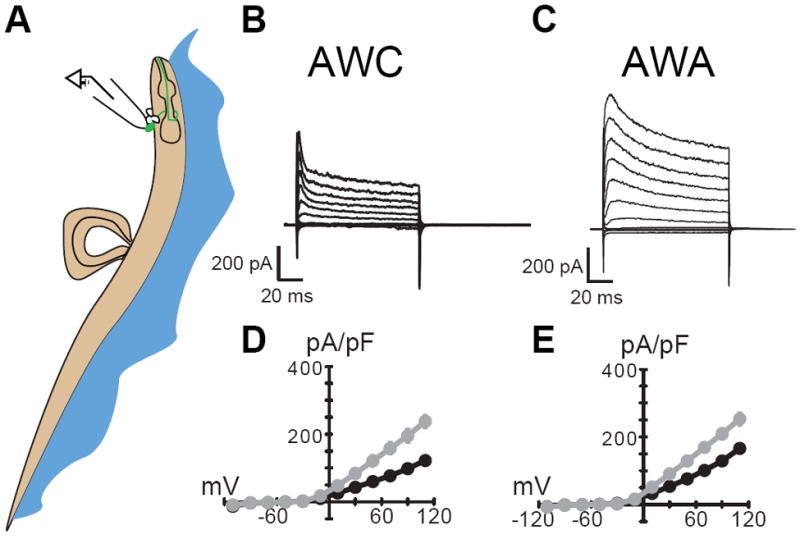
Schematic of recording from identified C. elegans neurons in the anterior ganglion (A) and voltage-activated currents recorded from the AWCON (B) and AWA (C) neurons. Traces in panels B and C are the response to a family of voltage pulses between -110 and +100 mV (in 20 mV) increments from a holding potential of -60 mV. Current-voltage curves are the average of 5 and 8 recordings; gray and black symbols are the mean of the peak and steady-state current, respectively. Error bars (±sem) are smaller than the symbols. Data for AWC reprinted with permission (Ramot et al., 2008). All recordings used solutions as described (Ramot et al., 2008).
2. Whole-cell ionic currents from identified neurons (ligand-gated)
Ligand-gated currents such as those activated by neurotransmitters can also be studied in vivo, by applying ligands to dissected preparations (Mellem et al., 2002) (Figure 4).
Figure 4. Ligand-gated currents in AVA neurons.
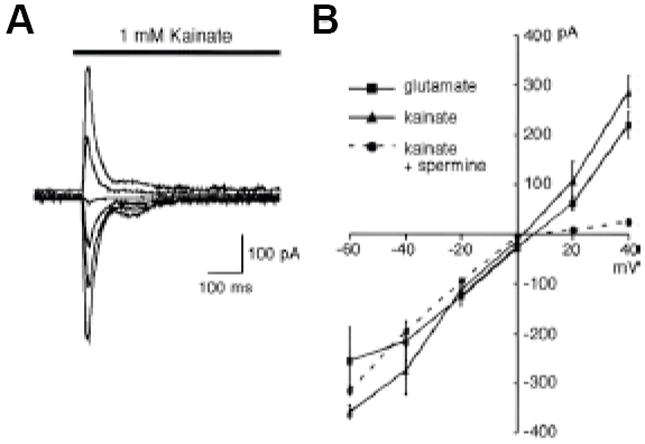
Currents activated by application of the indicated ionotropic glutamate receptor agonists. (A) kainate-gated membrane current recorded from AVA in vivo. Membrane potential was varied between -60 and +40 mV (in 20mV increments). (B) Peak current-voltage relationship for glutamate- and kainate-gated currents. As found in mammalian neurons, intracellular spermine blocks outward current. Reprinted with permission (Mellem et al., 2002).
Patch clamp recording from central synapses
Whereas the above methodology allows for routine recordings from single, identified C. elegans neurons, simultaneous patch clamp recording of identified pairs of neurons remains impractical. An alternative approach for investigating synaptic transmission is to record from post-synaptic cells while stimulating pre-synaptic neurons. Conventionally, two approaches have been considered: electrical stimulation and sensory stimulation. The former approach offers temporal precision and has been used to analyze synaptic transmission at the C. elegans neuromuscular junction (see below), but lacks the ability to restrict the stimulus to identified presynaptic neurons. The latter has been used to evoke synaptic currents in patch-clamped interneurons and provide the first physiological demonstration of synaptic connectivity in the C. elegans nervous system (Mellem et al., 2002), but can applied only to the synaptic partners of sensory neurons.
Optogenetic stimulation techniques (Boyden et al., 2005; Nagel et al., 2005) have emerged as a third approach that provides an elegant way to selectively activate identified pre-synaptic neurons while simultaneously recording from identified post-synaptic neurons (Lindsay et al., 2011) and muscles (Liewald et al. 2008; Liu et al., 2009). This technique involves the expressing a light-sensitive cation channel such as channelrhodopsin2 (ChR2) selectively in pre-synaptic neurons. Optogenetics enables temporal precision as well as control of stimulus intensity, as currents carried by ChR2 activate within milliseconds and are proportional to the power of the simulating light. Below, we describe the adaptations needed to combine optical stimulation of identified neurons with traditional C. elegans neuronal recordings for investigation of neuronal synapses between C. elegans neurons. Figure 5 shows a post-synaptic potential evoked by optical stimulation of a presynaptic neuron.
Figure 5. Light-activated post-synaptic potentials in AVA neurons.
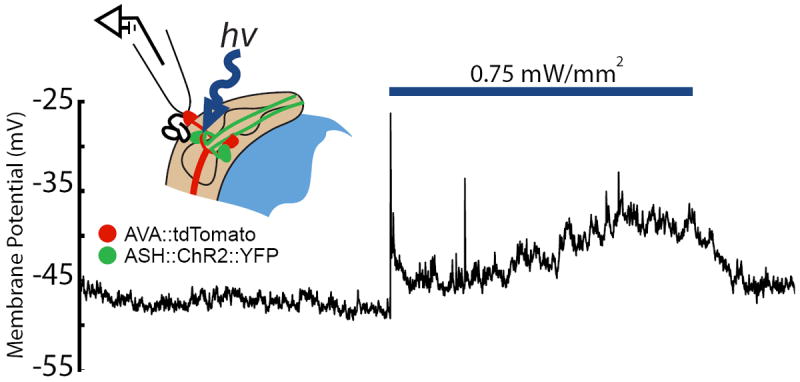
A light pulse (blue bar) activates the ASH neuron, which is pre-synaptic to AVA and expresses ChR2∷YFP, and induces a post-synaptic potential in the AVA neuron (labeled with tdTomato). The configuration for recording light-activated synaptic currents is illustrated in the inset. (T. Lindsay, S. Lockery, unpublished).
Strain Maintenance
C. elegans lacks endogenous all trans-retinal (ATR), an essential cofactor required for ChR2 activity. Accordingly, animals must be maintained with their diet supplemented with this co-factor (Nagel et al., 2005). Note that worms grown without ATR supplementation (ATR-) are useful since they can serve as controls for non-specific effects of blue-light stimulation. To prepare ATR-supplemented culture plates, we make a stock solution of 100 mM ATR in 100% EtOH (stored in the dark at -20°C). We combine this stock 1:100 with the OP50 E. coli when seeding bacterial lawns on standard NGM plates. Likewise we prepare ATR- control plates by adding EtOH 1:100 to the OP50 E. coli before seeding. Plates are used within 5 days and stored in the dark. We have observed that retinal can be transmitted from parent to progeny so it is best that ATR- control worms are not the progeny of worms grown on ATR-supplemented plates.
Preparation
Identical to single neuron recordings
Immobilization
Identical to single neuron recordings.
At the Rig
Dissection
The details of the dissection procedure depend on the anatomical position of the target postsynaptic neuron and achieving the best dissections requires practice. Since it is possible to disturb the neuropil during dissection, we advise inspecting the integrity and health (see recording, above) of both the pre-synaptic and post-synaptic neurons when practicing. It is critical that the neurites of the pre- and postsynaptic neurons remain intact. Whereas gross inspection does not ensure the integrity of all synaptic pathways, it provides a useful indication of the general condition of each dissection. We also advise assessing the condition of unlabeled neurons as a secondary measure of preparation viability.
Optical Stimulation
Three main requirements define the equipment needed to combine optical stimulation with in vivo recording from C. elegans neurons. First, it is necessary to control both the timing and the magnitude of the optical stimulus. Second, optical stimuli should be synchronized with the electrophysiological data acquisition system. Third, because it is convenient to activate ChR2 by delivering blue (470nm) light through the same microscope objective used to identify and patch-clamp the postsynaptic target (with green, 530nm light), a suitable arrangement is one that provides a method to switch between different wavelengths of excitation light without vibrations that can destroy a patch-clamp seal.
Fortunately, it is possible to equip an epifluorescent microscope with optics that meet the above specifications: a dual-wavelength LED system that delivers both 470 and 530 nm light (KSL-70, Rapp OptoElectronic, Germany). This system allows light to be switched using a digital TTL signal and for intensity to be modulated via an analogue control signal. We fitted our microscope filter cube with a 565DCXR dichroic mirror (Chroma Technology, VA, USA) and HQ645/75 excitation filter (Chroma).
Because the magnitude of light-evoked ChR2 currents depends on stimulus intensity (Nagel et al., 2003), it is important to measure the power of stimulus light delivered by the microscope objective to the preparation. We report irradiance (mW/mm2) since this is a measure of the optical power incident per unit surface area of the preparation. Power can be measured using a hand-held optical power meter (model 840-C Newport, CA, USA) and the area irradiated can be measured using a stage micrometer with intrinsic autofluorescence (MA285 Meiji Techno, Japan). Briefly, with the objective focused on the stage micrometer, we adjust the region to be irradiated by the stimulus light using the microscope field diaphragm and measure its dimensions using the stage micrometer. Next, we measure the total light energy directed at the specimen plane by focusing the objective on the photodetector of the power meter. Because the photodetector is large and placed close to the objective, we may assume that all light emitted by the objective is collected by the photodetector. We divide the total light power by the area of the irradiated field to calculate stimulus irradiance.
Recording synaptic potentials and currents at the neuromuscular junction
The time required to master recording from body wall muscles can be several months, mostly due to the challenging dissection, particularly the initial steps of gluing worms and cutting the cuticle. To facilitate and possibly accelerate the learning process, we provide the following description and refer readers to this visualized experiment (Richmond, 2009). Figure 6 outlines a typical workflow for obtaining in vivo patch-clamp recordings from C. elegans body wall muscles.
Figure 6. Workflow and approximate timeline for in vivo recording from the C. elegans NMJ.
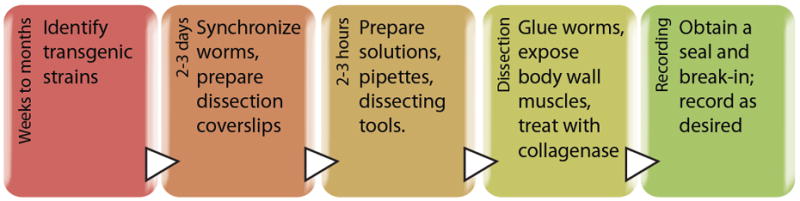
Each box represents a key step in the workflow, times given are prior to the moment of obtaining a whole-cell patch-clamp recording.
Gluing
Because older worms are larger and easier to dissect, we typically work with worms that have hatched at least 3-4 days earlier. In order to expose the body wall muscles, an incision running several hundred microns along the length of the worm is needed. To successfully make such a large incision, it is important that the worm is firmly attached to the underlying substrate during gluing, as the cuticle is relatively tough and the worm can easily detach during the incision step. For this reason, we are unable to glue worms to agarose pads. Instead the worms are glued to a PDMS-coated cover slip. The coating serves several functions: it provides a firm base to which the glue readily adheres, it acts as a cushion between the underlying glass cover slip and the worm, preventing shattering of the glue pipette during gluing, and provides a substrate upon which the glue pipette can be pressed to unplug polymerized glue from the tip during gluing. Because worms dry out immediately upon contact with PDMS, however, recording saline is used to keep the worms hydrated during gluing. When the worms are added to saline using a worm pick, they will exhibit thrashing behavior prior to immobilization with glue, presenting a challenge during the initial gluing. However, with practice it becomes routine to attach either the head or tail of the worm to the PDMS (Step 1, Figure 7). Once the worm is anchored the glue can be applied along the side of the cuticle to fully immobilize the animal (Steps 2-3, Figure 7). We use a cyanoacrylate glue (Histoacryl Blue, TissueSeal, Ann Arbor, MI) that polymerizes rapidly in solution. Gluing pipettes, pulled with the same settings to generate patch electrodes, are backfilled by dipping the tip into the glue containing PCR tube cap, and applying negative pressure by mouth suction. Polymerization of the glue is pH-sensitive and performs best around pH 7.4 (typical of physiological saline). Because the glue polymerizes in solution it takes practice to control the glue flow from the pipette without plugging up the tip or releasing too much glue. For best results, ensure that the pipette tip is not too big, that gentle positive pressure is maintained before and after lowering the glue pipette tip into saline and tap the pipette tip against the PDMS surface to maintain a stream of glue until you approach and glue the worm. The glue is relatively malleable for a few seconds after application to the worm, which provides a brief window to reshape the glue and to reposition the worm if the initial glue spot results in the wrong orientation. We typically glue the worm with the ventral side facing away from the glue and record from ventral neuromuscular junctions (NMJs), but this orientation can be reversed if the experiment requires dorsal muscle recordings. It is important to glue the entire length of the worm and to fill-in any regions along the cuticle where the glue is sparse. While gluing, be aware that tall peaks of glue will interfere with the placement of stimulating and recording pipettes, that come in at a shallow angle, so take the time to tamp peaks down during gluing. The advantage of gluing the dorsal side of the worm is that the eggs, which accumulate on the ventral side, protect the NMJs from damage while making the cuticle incision. It is advisable to glue five worms close to the center of the cover slip, prior to making any incisions, thus increasing the chance that at least one preparation meets the criteria for a healthy dissection.
Figure 7. The NMJ dissection.
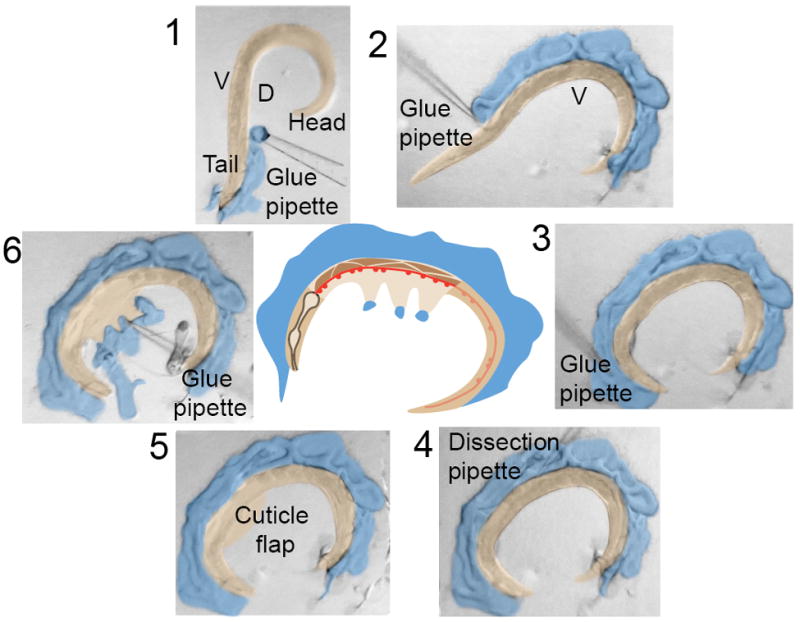
Micrographs 1-6 show serial images of a worm dissection, showing the gluing technique (1-3), the starting position of the incision near the vulva (4) producing a longitudinal incision toward the head (5) that is glued down to expose the anterior ventromedial neuromuscular junctions (6). The central cartoon depicts the position of the ventral nerve cord (red) and body wall muscles (brown) exposed by the dissection. The glue and worm have been pseudocolored to enhance contrast. For a visual demonstration, consult Richmond (2009).
Dissection
A dissecting scope with 20X eyepieces, a 1.5X objective and 4X range for a total magnification of 120X, with a black base, illuminated from above using a gooseneck light-source is optimal for the dissection (Figure 8). A cuticle incision is made with a hand-held glass pipette, pulled to a short tapered point (Step 4, Figure 7). Alternatively, a solid glass needle or sharpened tungsten wire can be substituted. The worm is positioned so that the dissection needle can be inserted parallel to the glued dorsal side of the cuticle, midway along the worm at the level of the vulva. The initial incision releases the worms’ hydrostatic pressure resulting in protrusion of viscera. A slit of several hundred micrometers is made by continuing to cut toward the head, along the cuticle/glue interface. During this procedure, stabilize your hands by resting them on the microscope base. A clean incision will leave the intact gut and eggs protruding from the cut. A dedicated suction pipette or extractor, which has an opening greater than an egg diameter, is used to clean out the viscera by mouth suction. This manipulation leaves the cuticle cylinder to which the ventral nerve cord and body wall muscles are attached. A new glue pipette is then used to spot glue the cut cuticle flap to the PDMS base and control saline is briefly (10 s) replaced with saline containing 0.4% collagenase to digest the basement membrane. We keep frozen 500μL aliquots of collagenase-containing saline for this purpose. The preparation is then ready for recording.
Figure 8. NMJ dissection apparatus.

The dissection microscope (total magnification 120X) is equipped with a black base and illuminated with gooseneck fiber optics for optimal visualization during dissection. The glue applicator and viscera extractor are similarly constructed and are mounted on dental wax between uses. Since the extractor becomes fluid filled during suction, where as the glue applicator must remain free of fluid for accurate glue application, the extractor is kept in the front position at all times to avoid confusion. The recording chamber, shown on the microscope base has a PDMS-coated coverslip inserted into the central chamber and held in place by a wax ring. The insert shows a close-up of the glue container, showing a drop of HistoacrylBlue glue, contained within a PCR tube lid, embedded in dental wax.
Recording from body wall muscles
The criteria used to judge the quality of the dissection are as follows: for ventral NMJ synaptic recordings, the ventral nerve cord needs to be intact along the entire length of the incision. The ventral cord motor neuron cell bodies and ventromedial muscle cells that directly contact the ventral nerve cord should appear healthy with minimal blebbing or vacuoles. A suitable worm is then orientated with the longitudinal body axis parallel to the front of the rig, head toward the left, to allow electrodes to be brought in from both the left and right sides, using micromanipulators. Typically two or three ventromedial body wall muscle cells are exposed between the head and the vulva and normally the most posterior muscle cell is patch-clamped using the right hand pipette. A fire-polished patch pipette is brought onto the muscle while applying positive pressure by mouth, until the pipette resistance starts to increase from a starting value of 4-7 MΩ, at which point negative pressure (suction) is applied immediately and held until a GΩ seal is achieved. The negative pressure is then relaxed briefly, the pipette capacitance is compensated and a holding potential of -60 mV is applied. Negative pressure is then reapplied while simultaneously applying a brief voltage pulse or ‘zap’ (0.4 V for 0.4 ms), which ruptures the membrane patch, resulting in a whole-cell recording configuration. Muscle cell capacitance (50-70 pF), is compensated and leak subtraction is performed. Typically, the data from a patch-clamped muscle takes only a few minutes to obtain, and usually only one recording is made per cover slip as the preparations deteriorate over time. However, even with this limitation, 4-6 recordings can be achieved per hour.
Types of recordings
The NMJ preparation can be used to study many physiological processes. Below is a brief summary of the types of information that can be obtained, and the recording configurations used to obtain the data (Figure 9).
Figure 9. Recording configurations and sample data obtained from the NMJ.
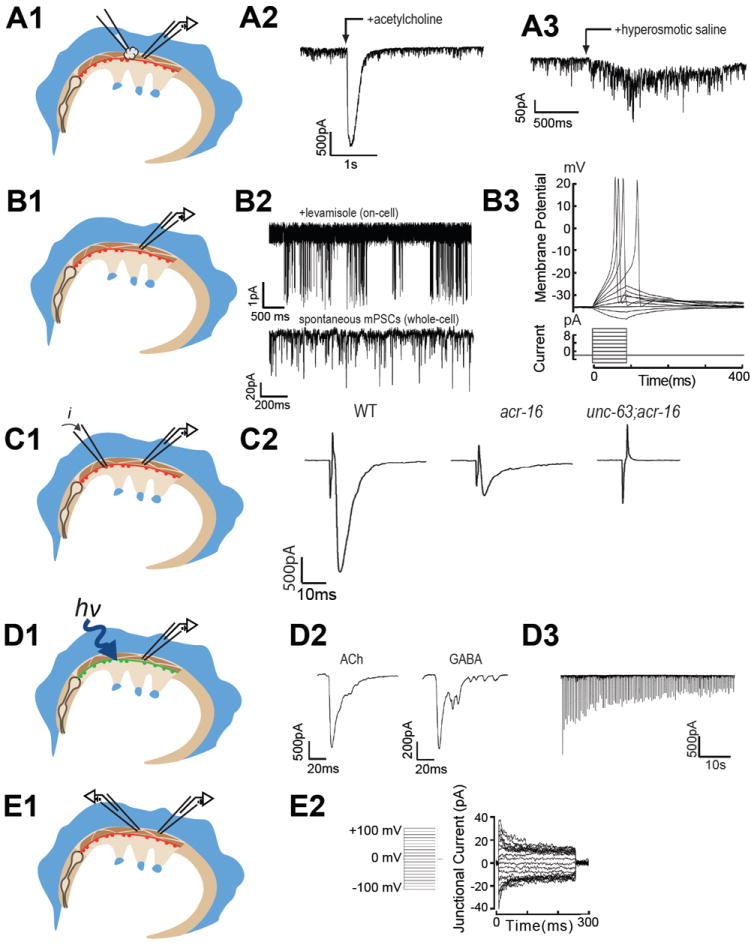
(A1) Applying agonists to muscle cells via pressure-ejection pipette. (A2) Acetylcholine-gated current (J.E.R, unpublished). (A3) Action potentials in body wall muscle recorded in current clamp mode via a whole-cell patch clamp recording. (B1) On-cell and whole-cell patch-clamp recording configuration from body wall muscle. (B2) Single levamisole-sensitive nicotinic receptor channels recorded on-cell (top) and spontaneous miniature post-synaptic currents recorded in whole-cell voltage clamp (bottom). Whole-cell recordings from individual muscles in the voltage-clamp mode can also be used to examine voltage-gated currents (see Liu, et al., 2011; Gao and Zhen, 2011). (B3) Hyperosmotic saline stimulated release of the primed vesicle pool. (C1) Electrical stimulation of synaptic release (via a loose patch-clamp electrode on the nerve cord). While effective for initial measures of release, this method has a higher propensity for nerve damage upon sequential stimulation. (C2) Electrically-evoked post-synaptic currents from wild type (left), acr-16 mutants (center), and unc-63;acr-16 double mutants (right). (D1) Channelrhodopsin (ChR2)-mediated stimulation of subsets of motor neurons via targeted expression of ChR2 in transgenic animals fed all-trans retinal and activated with blue-light. (D2) Optical stimulation of cholinergic (left) and GABAergic (right) motor neurons evoke robust post-synaptic currents. (D3) Trains of optical stimuli can be delivered with high-frequency, blue-light flashes and evoked post-synaptic currents (downward deflections) without damaging the ventral nerve cord. (E1) Whole cell voltage-clamp of adjacent muscles can be used to study electrical coupling between cells. (E2) Current flowing across gap junctions that connect adjacent muscle cells. Data collected by: J.E.R. (A2; B2 bottom; B3; C2; D2); Shangbang Gao and Mei Zhen (A3); Alan Robertson and Richard Martin (B2); Jana Leiwald and Alexander Gottschalk (D3); Ping Liu and Zhao-Wen Wang (E2).
1. Whole-cell membrane currents (ligand and voltage-gated)
The body wall muscles are innervated by cholinergic and GABAergic motorneurons and, therefore, respond to both ACh and GABA (Richmond and Jorgensen, 1999). The functional receptor field of a whole-cell voltage-clamped muscle cell can be examined by pressure-ejecting the relevant agonist from a pipette similar in size to the polished patch pipette (Figure 9A1, 9A2). To prevent receptor desensitization the preparation is continuously perfused via a gravity flow delivery system, in which fresh saline is streamed from the left to the right of the preparation and collected using a vacuum line.
Although the physiologically relevant ionotropic GABA current of the muscle would normally be outward, the intracellular recording solution typically used to measure GABA and ACh currents, has a high Cl- concentration that produces inward currents in response to either ligand at a holding potential of -60mV. This intracellular solution can be adjusted to produce outward GABA currents if desired (Sieburth et al., 2007). The identity and pharmacology of the muscle receptor subunits has been established. There are two classes of nicotinic receptors: one is sensitive to the nematocide levamisole and contains five subunits [UNC-38, UNC-63, UNC-39, LEV-1 and LEV-8 (Culetto et al., 2004; Lewis et al., 1987; Richmond and Jorgensen, 1999; Towers et al., 2005)] and a second is sensitive to nicotine and contains the ACR-16 subunit (Francis et al., 2005; Touroutine et al., 2005). All subunits of the ionotropic GABA receptor are encoded by the gene unc-49 (Bamber et al., 1999).
The body wall muscles also express voltage-gated ion channels and generate calcium-dependent action potentials (Gao and Zhen, 2011; Liu et al., 2011). Depolarizing voltage-steps from -60mV produce large outward currents composed of multiple K+ channel types, the majority of which can be blocked with a combination of tetraethylammonium chloride and 4-aminopyridine (Richmond and Jorgensen, 1999). Genetic dissection reveals that the Kv1 SHK-1 K+ channel and the calcium- and chloride-activated K+ channel, SLO-2 account for the majority of outward K+ current in C. elegans body wall muscle (Gao and Zhen, 2011; Liu et al., 2011). Voltage-gated Ca2+ currents are dependent on the L-type calcium channel, EGL-19 (Jospin et al., 2002; Gao and Zhen, 2011; Liu et al., 2011).
2. Muscle cell excitability
Although the C. elegans genome lacks canonical voltage-gated Na+ channels, EGL-19-dependent Ca2+-channel spiking activity has been recorded in the dissected muscle preparation in the whole-cell current-clamp mode (Jospin et al., 2002) and can be all-or-none in nature, as shown (Figure 9A3).
3. Single channel currents (ligand and voltage-gated)
While few single channel studies have been conducted on dissected body wall muscles, this approach has been applied to study large-conductance, calcium-activated potassium channels with the excised patch-clamp technique (Carre-Pierrat et al., 2006). Because body wall muscle receptors are clustered at synapses that localize to inaccessible muscle arms, mutants that disrupt the synaptic localization of the levamisole-sensitive ACh receptor have been used to study their single channel properties (Qian et al., 2008) (Figure 9B2, top). No such de-clustering mutants are yet available to study the nicotine-sensitive receptor, or the GABA receptor.
4. Synaptic responses: endogenous, spontaneous synaptic activity
Several synaptic parameters can be measured from the dissected muscle preparation. In the whole-cell voltage-clamp configuration, endogenous miniature post-synaptic currents (mPSCs) can be readily observed (Figure 9B2, bottom). Using the original pipette and external recording solutions, both cholinergic and GABAergic mPSCs appear as inward currents at a holding potential of -60 mV (Richmond and Jorgensen, 1999). Lowering Cl- concentration of the intracellular solution allows GABA and cholinergic mPSCs to be examined separately by changing holding potential in the same patch-clamp recording (Sieburth et al., 2007). Cholinergic and GABAergic mPSCs can also be isolated pharmacologically (Richmond and Jorgensen, 1999; Bamber et al., 2005). Alternatively, mutants in receptor subunit genes can be used to eliminate any or all of the receptor subtypes.
5. Readily releasable vesicle pool
Pressure-ejection of hyperosmotic saline (800 mOsm) for several seconds (Figure 9B3, right) can be used to measure the size of the readily releasable pool in whole-cell voltage-clamped muscles (Richmond et al., 1999). This protocol causes the asynchronous release of the fusion-competent vesicle pool, which is measured as total charge integral for the duration of the hyperosmotic application.
6. Evoked synaptic currents
Until recently, the only method available to study evoked synaptic vesicle release at the C. elegans NMJ entailed positioning a stimulating pipette on the ventral nerve cord anterior to the whole-cell patch clamped muscle (Figure 9C1). For convenience, the pipette holder used for this experiment carries two devices: a pressure-ejector for ligand application and an electrical simulator connected to a silver wire inserted into the pipette. To evoke release, the stimulating electrode must be pressed firmly against the nerve cord and short (1-2ms) stimuli in the range of 20-40V are used to maximally stimulate the nerve via this loose patch arrangement (Figure 9C2). The disadvantages of this approach are multifold and include the following: The stimulating pipette cannot be placed too close to the patch-clamped muscle, otherwise the recording is lost during stimulation, therefore the incision must be large enough to expose at least two body wall muscles in the worms longitudinal axis, increasing the risk of nerve damage during dissection. The stimulating pipette must be maneuvered against the nerve after the muscle cell has been whole-cell patch clamped which is challenging, and the approach can be easily blocked by excess glue near the head of the worm. Because the voltage used to stimulate the nerve often causes damage, repeated evoked responses are difficult, although not impossible to acquire. Another limitation of this approach is that anterior nerve cord stimulation activates only cholinergic motorneurons.
With the development of optogenetic tools in C. elegans, all of these difficulties have been overcome (Liewald et al., 2008). Using specific promoters, transgenic lines expressing channelrhodopsin in cholinergic or GABAergic motorneurons allows for selective, repeat stimulation of evoked release from the NMJ using blue light pulses (Figure 9D1-D3). Since the worm is transparent the incision need only expose a single ventromedial muscle cell, thereby reducing the risk of damage to the preparation during dissection. The primary limitations of this approach are those imposed by channelrhodpsin itself, since this protein undergoes inactivation with repeat stimulation (Liu et al., 2009; Nagel et al., 2003). Another minor disadvantage is that for any mutant of interest, the channelrhodopsin transgene must be crossed into the mutant strain. Fortunately, this is relatively simple to do, since the channelrhodopsin is fused to a fluorescent protein, which can be followed during the cross. Conversely, halorhodopsins can be expressed in motorneurons to inhibit release (Liu et al., 2009).
7. Muscle cell electrical coupling
Application of dual whole cell recordings to adjacent body wall muscles in the dissected C. elegans muscle preparation (Figure 9E1-E2), has been used to demonstrate that body wall muscles are weakly electrically coupled, via innexin based gap junctions that require both UNC-9 and UNC-1 proteins (Chen et al., 2007; Liu et al., 2006).
Materials
Transgenic C. elegans strains that label target cells
In order to identify the desired neuron or muscle for patch clamp recording, we use transgenic C. elegans strains in which target cells are labeled with a soluble fluorescent protein such as GFP (Chalfie et al., 1994). A soluble, rather than a membrane-bound construct is preferred, since dilution of soluble GFP is a useful indicator of successful break-in for whole-cell recording. In building a transgenic strain suitable for in situ electrophysiology (or choosing a published strain from the literature), the choice of co-injection marker is important. For example the dominant marker, rol-6, which causes animals to corkscrew around their long axis while crawling is impractical (Cox et al., 1980; Kramer et al., 1990) as this behavior leads animals to encase themselves in glue during the immobilization procedure. We also recommend verifying that the transgene does not significantly alter behavior or neuronal morphology. Generally speaking, fluorescent proteins are benign. By contrast, over-expression of some gene promoters can alter neuronal function and development (Toms et al., 2001).
When building a new strain, a good practice is to create a set of transgenic animals using different promoters active in the target neuron (if available) and a variety of expression levels and to screen each strain for behavioral and morphological defects. Retain only those strains with little, if any effect on either behavior or neuronal morphology. Continuing challenges include identifying neuron-specific gene promoters and minimizing ‘side effects’ of over-expression. Recent advances in C. elegans molecular genetics reduce the need for neuron-specific promoters, introducing several strategies to achieve combinatorial control of fluorescent protein expression (Davis et al., 2008; Zhang et al., 2004; Macosko et al., 2009; Voutev and Hubbard, 2008) and minimize potential deleterious effects of over-expression by using transposons to create integrated, single-copy transgenic animals (Frokjaer-Jensen et al., 2008).
Transgenic C. elegans strains for optogenetic control of neuronal activation
There are several additional points that should be considered when designing strains for investigation of C. elegans central synapses using ChR2 photostimulation of presynaptic neurons. First, it is useful to express the ChR2 and a fluorescent reporter protein as a fusion protein, which can be used to confirm that ChR2 expression is restricted to the expected neurons, targeted to the cell membrane, and expressed at reasonable levels. YFP, and dsRED have been successfully fused to ChR2 (Liu et al., 2009; Nagel et al., 2005). Although fluorescence can be used as a preliminary method of vetting transgenic lines, the ability of ChR2 to control neuronal activation can be tested directly using patch clamp recordings to measure light-induced currents and changes in membrane potential in the ChR2-expressing neuron.
A second point to consider is the choice of fluorescent protein used to identify the postsynaptic neuron. It is important that the fluorescence be intense since it is used in four essential ways: (1) identification of the target postsynaptic neuron; (2) assessment of the integrity of the neurites of the target neuron; (3) determination of the precise moment when the electrode contacts the target neuron; and (4) exclusion of mis-targeted seals. A third consideration is to avoid inadvertent stimulation of the ChR2-expressing presynaptic cell while visualizing the post-synaptic target neuron. This is best achieved by choosing a post-synaptic reporter protein that is not excited by the same wavelength of light as ChR2 (450-500 nm). We find that tagging pre-synaptic ChR2 with GFP or YFP and the postsynaptic neuron with soluble tdTomato (Shaner et al., 2004) is a good choice. The red fluorescent protein tdTomato is an excellent choice for labeling post-synaptic target neurons because it is very bright and excited by green (525-575 nm) light.
Equipment needed for gluing animals for neuronal recordings
-
Cooling Pad (two options)
Cooled (4°C) 75mL tissue culture flask filled with water and sealed with Parafilm
Cooled (4°C), 10-cm Petri dish filled with 5% gelatin and 0.1% NaN3 (w/v).
-
Agarose pads
2% agarose (w/v) in physiological saline, pH adjusted to 7.5-8.0 to accelerate polymerization of the glue
Glue: WormGlu™ (Glustitch, Inc) or Histoacryl Blue (TissueSeal, Ann Arbor, MI).
-
Glue pipettes
Similar to worm injection pipettes or intracellular recording electrodes
Break the tip and fill by capillary action from a small puddle on a cover slip
Equipment needed for gluing animals for muscle recordings
The tools needed to glue and dissect adult worms to expose the body wall muscles are shown in Figure 8. The glue applicator is made from a borosilicate patch electrode, connected to a 2-foot length of polyethylene tubing with an inner diameter that matches the outer diameter of the electrode. A replaceable pipette tip attached to the other end of the tubing, serves as a mouthpiece. The extractor is similarly constructed and used to clear viscera following incision of the body wall. A working supply of glue is kept in an inverted PCR tube lid stabilized with dental wax (Figure 8, insert). The lip of the lid is removed using a razor blade. The recording chamber can be constructed from a thin magnetic sheet (1.59 mm or 1/16”, see Figure 8) or plexiglass, in which a centered 22mm bore hole, is made. A 48×66mm rectangular cover glass is attached to the back of the chamber with low melt Paraplax tissue embedding wax, to make a leak-free chamber. Glass chambers like those used for neuronal recording can also be used. A PDMS-coated cover slip, to which worms will be glued, is held in place within the chamber, by making a wax ring on the glass base of the chamber with a wax pen before tamping down the cover slip. The PDMS coat is generated by applying a small drop, off-centre on a cover glass that fits your recording chamber, and then smearing PDMS across the centre of the cover slip using the edge of a razor blade. Since the coated cover slips are only used once, we coat large batches of cover slips, place them in covered boxes and cure them overnight in an oven at 65°C.
Recording chamber for neuronal recordings
We use a very simple chamber consisting of a 1 mm-thick glass plate with a 20 mm hole drilled in the center. The surface of the chamber is treated with silane (RainX™) to make it hydrophobic. A glass coverslip carrying the thin agarose pad (and glued worms) is sealed over the hole with beeswax (available from any art supply store), forming a shallow pool. Glass is better than plastic because it resists thermal expansion. We use beeswax, since it has a low melting point and is non-toxic. Once mounted on the microscope for electrical recording, the preparation should be supplied continuously with fresh saline. Influx to our superfusion system is powered by gravity, using a flowmeter (Dial-a-flow, Becton-Dickson) to regulate the flow rate. Saline is removed from the chamber using a peristaltic pump, connected to the bath using a small tube and fiber wick. To minimize electrical noise conducted by the perfusion system, the tubing connecting the recording chamber to the pump is grounded in-line by a platinum wire. Figure 10 shows the arrangement for perfusion influx, efflux, and a typical recording chamber.
Figure 10. Recording chamber and superfusion tubing.
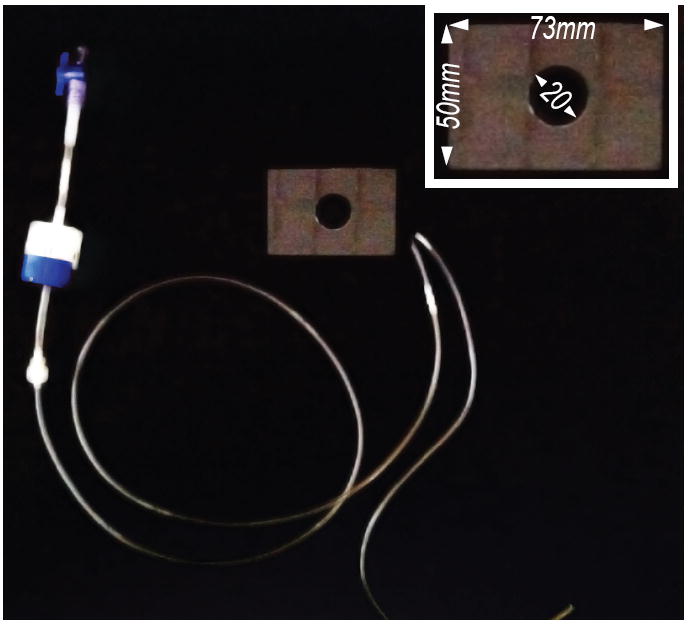
Solution is delivered to the recording chamber by gravity with the flow rate controlled by a Dial-a-flow meter (blue/white valve on the left) and removed by a peristaltic pump (not shown). The glass perfusion chamber (inset) is custom-fabricated by drilling a hole in a 1mm-thick glass plate.
Microforge for pressure polishing
The microforge we use is adapted from the CPM-2 kit (ALA Scientific Instruments, Farmingdale, NY) mounted on a simple inverted microscope equipped with a long-working distance, 100× metallurgical objective lens (Figure 11). As of this writing, four vendors sell equipment for pressure polishing. Apart from the CPM-2, we have no experience with these devices. They are (in no particular order): (1) Fine Point Microforge System w/ Pressure Polishing, The GlassWorx.com; (2) Coating and Polishing Microforge (CPM-2) with high-pressure regulator (PR-60) and pipette holder (IPH-THP), ALA Scientific Instruments; (3) DMF1000, World Precision Instruments; (4) Flyion Microforge.
Figure 11. Pressure-polishing microforge.

This inverted microscope is equipped with a long-working distance 100× objective (Leica), a CPM-2 microforge kit (ALA Science), and a custom-designed pressure polishing accessory. Compressed air is passed through a regulator/filter (not shown) to provide a high-pressure (40 PSI) air stream. This stream is directed to the pipette being shaped by means of a four-way valve and a pipette holder equipped with a luer-lock connector (WPI Instruments). For a visual demonstration of the process, see (Johnson et al., 2008).
The rig
1. Neuronal recordings
We have experience using both inverted and fixed-stage, upright compound microscopes for in vivo recordings from C. elegans neurons. Here, we focus on aspects of the rig that we think are critical for success. The microscope must be mounted on a vibration isolation table and be free of mechanical vibration. A stage capable of rotation is also needed, since animals will need to be repositioned in between the dissection and recording steps. Finally, it must be possible to observe samples in differential interference contrast (DIC) and epifluoresence simultaneously. A high-power, high numerical aperture (NA) lens is also critical. We have used a 63X/1.4NA oil-immersion lens on an inverted microscope (Goodman et al., 1998) as well as a 60X/1.0NA water-immersion lens on an upright microscope (O’Hagan et al., 2005). The minimum specification is magnification that exceeds 60X and numerical aperture (NA) that exceeds 0.9. We use an inexpensive analog video camera to enlarge the microscope image. The signal from this camera is viewed on a flat-panel liquid crystal display (LCD) computer monitor that accepts analog signals (Neovo USA) and can be positioned close to the recording microscope without introducing electrical noise.
To date, recordings have been obtained with patch-clamp amplifiers from Axon Instruments (Axopatch 200A, 200B; Molecular Devices, Sunnyvale, CA) and HEKA Instruments (EPC-9, EPC-10; Heka Instruments, Bellmore, NY), using either pClamp or Patchmaster software, respectively. Both configurations work well and the choice between the two likely rests more on personal preference than on any inherent features of the amplifiers and associated software. A mechanically stable micromanipulator capable of submicron steps is needed to hold the amplifier headstage. We have used manipulators based on piezoelectric movers (PCS-5000, Burleigh/Lumen Dynamics, Mississauga, Ontario Canada) and stepper motors (MP-225, MP-285, Sutter Instruments, Novato, CA) and both styles work well. The dissecting tool is most conveniently mounted on an oil hydraulic manipulator (MMO-3, Narishige Instruments International, East Meadow, NY), though other manipulators that allow for fine control over long travel distances will likely work.
2. Body wall muscle recordings
The recording equipment used to patch body wall muscles is similar to that used to record from neurons with the following additions. Due to the thickness of the PDMS pad, neither electrodes nor worms can be adequately resolved using an inverted scope. Therefore, we use an upright, fixed-stage, compound microscope equipped with DIC optics, mounted on a vibration isolation table. The recording microscope should have a 10× objective to find, center and focus on the dissected worms and a higher magnification, water immersion objective (40X/1.9mm working distance) provides the magnification needed to inspect and identify the best dissection and to visualize the approach of the recording pipettes.
Discussion
Considerable technical progress has been made in C. elegans electrophysiology in the decade since the initial publication of the technique for whole-cell patch-clamp recordings from C. elegans neurons (Goodman et al., 1998) and muscle (Richmond and Jorgensen, 1999). Notable advances include technical adaptations for quantitative analysis of sensory mechano-, thermo-, and phototransduction (O’Hagan et al., 2005; Ramot et al., 2008; Ward et al., 2008) and for studying the physiology of chemical (Mellem et al., 2002; Richmond et al., 1999; Richmond and Jorgensen, 1999) and electrical synapses (Liu et al., 2006). Today, most, if not all electrophysiological experiments that can be done in larger animals preparations can also be done in C. elegans. One exception may be whole-cell recording during behavior in intact or semi-intact animals. Technical advances in the next decade will almost certainly involve new and exciting combinations of optogenetics and electrophysiology in which a variety of genetically targeted probes will be utilized to excite or inhibit identified neurons during whole-cell patch-clamp experiments. Other advances are likely to result from novel combinations of calcium imaging with electrophysiology. The new techniques will almost certainly provide unforeseen insights into the function of neurons and networks in C. elegans and can be expected to accelerate progress toward an essentially complete understanding of its nervous system.
Summary
Here, we provide the methods and materials needed to obtain patch clamp recordings from C. elegans neurons and muscles in situ, including emerging methods for optogenetic stimulation coupled with post-synaptic recording. We emphasize procedures and equipment that are essential for successful patch clamp recordings with this small animal, including critical factors in choosing transgenic lines for study.
Acknowledgments
We thank the members of our laboratories for comments and many colleagues in the worm community for contributing unpublished data: Shangbang Gao, Alex Gottschalk, Jana Liewald, Ping Liu, Richard Martin, Daniel Ramot, Alan Robertson, Zhao-Wen Wang, Mei Zhen. Work supported by John Simon Guggenheim Foundation (S.R.L), McKnight, Alfred P. Sloan and Klingenstein Foundations (M.B.G) and by research grants from NIH (M.B.G., S.R.L., J.E.R.) and NSF (M.B.G.).
References
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM. The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci. 1999;19:5348–5359. doi: 10.1523/JNEUROSCI.19-13-05348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamber BA, Richmond JE, Otto JF, Jorgensen EM. The composition of the GABA receptor at the Caenorhabditis elegans neuromuscular junction. Br J Pharmacol. 2005;144:502–509. doi: 10.1038/sj.bjp.0706052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Driscoll M. Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses. WormBook. 2006:1–15. doi: 10.1895/wormbook.1.122.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Carre-Pierrat M, Grisoni K, Gieseler K, Mariol MC, Martin E, Jospin M, Allard B, Segalat L. The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J Mol Biol. 2006;358:387–395. doi: 10.1016/j.jmb.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chen B, Liu Q, Ge Q, Xie J, Wang ZW. UNC-1 regulates gap junctions important to locomotion in C. elegans. Curr Biol. 2007;17:1334–1339. doi: 10.1016/j.cub.2007.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M, Estevez A, Yin X, Fox R, Morrison R, McDonnell M, Gleason C, Miller DM, 3rd, Strange K. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron. 2002;33:503–514. doi: 10.1016/s0896-6273(02)00591-3. [DOI] [PubMed] [Google Scholar]
- Cox GN, Laufer JS, Kusch M, Edgar RS. Genetic and Phenotypic Characterization of Roller Mutants of CAENORHABDITIS ELEGANS. Genetics. 1980;95:317–339. doi: 10.1093/genetics/95.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culetto E, Baylis HA, Richmond JE, Jones AK, Fleming JT, Squire MD, Lewis JA, Sattelle DB. The Caenorhabditis elegans unc-63 gene encodes a levamisole-sensitive nicotinic acetylcholine receptor alpha subunit. J Biol Chem. 2004;279:42476–42483. doi: 10.1074/jbc.M404370200. [DOI] [PubMed] [Google Scholar]
- Davis MW, Morton JJ, Carroll D, Jorgensen EM. Gene activation using FLP recombinase in C. elegans. PLoS Genet. 2008;4:e1000028. doi: 10.1371/journal.pgen.1000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis MM, Evans SP, Jensen M, Madsen DM, Mancuso J, Norman KR, Maricq AV. The Ror receptor tyrosine kinase CAM-1 is required for ACR-16-mediated synaptic transmission at the C. elegans neuromuscular junction. Neuron. 2005;46:581–594. doi: 10.1016/j.neuron.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Francis MM, Mellem JE, Maricq AV. Bridging the gap between genes and behavior: recent advances in the electrophysiological analysis of neural function in Caenorhabditis elegans. Trends Neurosci. 2003;26:90–99. doi: 10.1016/S0166-2236(02)00041-3. [DOI] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Zhen M. Action potentials drive body wall muscle contractions in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2011;108:2557–2562. doi: 10.1073/pnas.1012346108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB, Hall DH, Avery L, Lockery SR. Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron. 1998;20:763–772. doi: 10.1016/s0896-6273(00)81014-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB, Lockery SR. Pressure polishing: a method for re-shaping patch pipettes during fire polishing. J Neurosci Methods. 2000;100:13–15. doi: 10.1016/s0165-0270(00)00224-7. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Brown AL, Goodman MB. Pressure-polishing pipettes for improved patch-clamp recording. J Vis Exp. 2008 doi: 10.3791/964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jospin M, Jacquemond V, Mariol MC, Segalat L, Allard B. The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J Cell Biol. 2002;159:337–348. doi: 10.1083/jcb.200203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR. Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron. 2000;26:583–594. doi: 10.1016/s0896-6273(00)81196-4. [DOI] [PubMed] [Google Scholar]
- Kerr RA. Imaging the activity of neurons and muscles. WormBook. 2006:1–13. doi: 10.1895/wormbook.1.113.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr RA, Schafer WR. Intracellular Ca2+ imaging in C. elegans. Methods Mol Biol. 2006;351:253–264. doi: 10.1385/1-59745-151-7:253. [DOI] [PubMed] [Google Scholar]
- Kramer JM, French RP, Park EC, Johnson JJ. The Caenorhabditis elegans rol-6 gene, which interacts with the sqt-1 collagen gene to determine organismal morphology, encodes a collagen. Mol Cell Biol. 1990;10:2081–2089. doi: 10.1128/mcb.10.5.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT, McLafferty S, Murphy H, Wu C. The levamisole receptor, a cholinergic receptor of the nematode Caenorhabditis elegans. Mol Pharmacol. 1987;31:185–193. [PubMed] [Google Scholar]
- Liewald JF, Brauner M, Stephens GJ, Bouhours M, Schultheis C, Zhen M, Gottschalk A. Optogenetic analysis of synaptic function. Nat Methods. 2008;5:895–902. doi: 10.1038/nmeth.1252. [DOI] [PubMed] [Google Scholar]
- Lindsay TH, Thiele TR, Lockery SR. Optogenetic analysis of synaptic transmission in the central nervous system of the nematode Caenorhabditis elegans. Nat Commun. 2011;2:306. doi: 10.1038/ncomms1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Gaier E, Joshi J, Wang ZW. Low conductance gap junctions mediate specific electrical coupling in body-wall muscle cells of Caenorhabditis elegans. J Biol Chem. 2006;281:7881–7889. doi: 10.1074/jbc.M512382200. [DOI] [PubMed] [Google Scholar]
- Liu P, Ge Q, Chen B, Salkoff L, Kotlikoff MI, Wang ZW. Genetic dissection of ion currents underlying all-or-none action potentials in C. elegans body-wall muscle cells. J Physiol. 2011;589:101–117. doi: 10.1113/jphysiol.2010.200683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hollopeter G, Jorgensen EM. Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc Natil Acad Sci. 2009;106:10823–10828. doi: 10.1073/pnas.0903570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockery SR, Goodman MB. Tight-seal whole-cell patch clamping of Caenorhabditis elegans neurons. Methods Enzymol. 1998;293:201–217. doi: 10.1016/s0076-6879(98)93016-6. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Pokala N, Feinberg EH, Chalasani SH, Butcher RA, Clardy J, Bargmann CI. A hub-and-spoke circuit drives pheromone attraction and social behaviour in C. elegans. Nature. 2009:1–6. doi: 10.1038/nature07886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellem JE, Brockie PJ, Zheng Y, Madsen DM, Maricq AV. Decoding of polymodal sensory stimuli by postsynaptic glutamate receptors in C. elegans. Neuron. 2002;36:933–944. doi: 10.1016/s0896-6273(02)01088-7. [DOI] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light Activation of Channelrhodopsin-2 in Excitable Cells of Caenorhabditis elegans Triggers Rapid Behavioral Responses. Current Biology. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell WT, Pun RY, Bargmann CI, Kleene SJ. Single ionic channels of two Caenorhabditis elegans chemosensory neurons in native membrane. J Membr Biol. 2002;189:55–66. doi: 10.1007/s00232-002-1004-x. [DOI] [PubMed] [Google Scholar]
- O’Hagan R, Chalfie M, Goodman MB. The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat Neurosci. 2005;8:43–50. doi: 10.1038/nn1362. [DOI] [PubMed] [Google Scholar]
- Qian H, Robertson AP, Powell-Coffman JA, Martin RJ. Levamisole resistance resolved at the single-channel level in Caenorhabditis elegans. FASEB J. 2008;22:3247–3254. doi: 10.1096/fj.08-110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramot D, MacInnis BL, Goodman MB. Bidirectional temperature-sensing by a single thermosensory neuron in C. elegans. Nat Neurosci. 2008;11:908–915. doi: 10.1038/nn.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond J. Dissecting and recording from the C. elegans neuromuscular junction. J Vis Exp. 2009 Feb 25;(24) doi: 10.3791/1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shtonda B, Avery L. CCA-1, EGL-19 and EXP-2 currents shape action potentials in the Caenorhabditis elegans pharynx. J Exp Biol. 2005;208:2177–2190. doi: 10.1242/jeb.01615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth D, Madison JM, Kaplan JM. PKC-1 regulates secretion of neuropeptides. Nat Neurosci. 2007;10:49–57. doi: 10.1038/nn1810. [DOI] [PubMed] [Google Scholar]
- Toms N, Cooper J, Patchen B, Aamodt E. High copy arrays containing a sequence upstream of mec-3 alter cell migration and axonal morphology in C. elegans. BMC Dev Biol. 2001;1:2. doi: 10.1186/1471-213X-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touroutine D, Fox RM, Von Stetina SE, Burdina A, Miller DM, 3rd, Richmond JE. acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J Biol Chem. 2005;280:27013–27021. doi: 10.1074/jbc.M502818200. [DOI] [PubMed] [Google Scholar]
- Towers PR, Edwards B, Richmond JE, Sattelle DB. The Caenorhabditis elegans lev-8 gene encodes a novel type of nicotinic acetylcholine receptor alpha subunit. J Neurochem. 2005;93:1–9. doi: 10.1111/j.1471-4159.2004.02951.x. [DOI] [PubMed] [Google Scholar]
- Voutev R, Hubbard EJ. A “FLP-Out” system for controlled gene expression in Caenorhabditis elegans. Genetics. 2008;180:103–119. doi: 10.1534/genetics.108.090274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Liu J, Feng Z, Xu XZ. Light-sensitive neurons and channels mediate phototaxis in C. elegans. Nat Neurosci. 2008;11:916–922. doi: 10.1038/nn.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Kim E, Zhao XM, Rogers JA, Prentiss M, Whitesides GM. Complex optical surfaces formed by replica molding against elastomeric masters. Science. 1996;273:347–349. doi: 10.1126/science.273.5273.347. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ma C, Chalfie M. Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell. 2004;119:137–144. doi: 10.1016/j.cell.2004.09.012. [DOI] [PubMed] [Google Scholar]