

Abstract
Background
Fibroblast growth factor–23 (FGF‐23) is a phosphaturic factor previously associated with left ventricular hypertrophy and systolic dysfunction among individuals with chronic kidney disease. Whether FGF‐23 acts directly to induce left ventricular hypertrophy, potentially independent of its klotho coreceptor, remains uncertain. We investigated associations of FGF‐23 with cardiac structural abnormalities among individuals with a broad range of kidney function and explored potential biological mechanisms using cardiac magnetic resonance imaging and histology in klotho‐null mice, an established model of constitutively elevated FGF‐23.
Methods and Results
Among 887 participants with coronary artery disease in the Heart and Soul Study, FGF‐23 was modestly associated with worse left ventricular ejection fraction (−1.0% per standard deviation increase in lnFGF‐23; standard error, 0.4%), but was not associated with the overall prevalence of concentric hypertrophy (odds ratio, 1.5; CI, 0.9 to 2.4) or eccentric hypertrophy (odds ratio, 1.1; CI, 0.9 to 1.3). FGF‐23 was only associated with concentric hypertrophy among individuals with diminished kidney function (eGFR <60 mL/min per 1.73 m2; odds ratio, 2.3; CI, 1.0 to 5.3; P‐interaction=0.28). Comparing klotho‐null with wild‐type mice, null mice did not have greater left ventricular mass (P=0.37) or a lower ejection fraction (P=0.94).
Conclusions
Together, our results suggest that FGF‐23 is unlikely to have major effects on cardiovascular structure and function among patients free of substantial chronic kidney disease, and these effects may not be independent of the klotho coreceptor.
Keywords: chronic kidney disease, hypertrophy, structure
Introduction
Fibroblast growth factor–23 (FGF‐23) is a circulating hormone secreted primarily by osteocytes. In the renal tubules, FGF‐23 binds to the FGF receptor 1c and the klotho coreceptor to promote phosphaturia and lower circulating levels of 1,25‐dihydroxyvitamin D.1 Circulating levels of FGF‐23 can rise 100‐ to 1000‐fold in patients with a diminished capacity for phosphorous excretion, such as those with chronic kidney disease (CKD).2 In patients with CKD, supraphysiologic levels of FGF‐23 are strongly associated with left ventricular mass index (LVMI), left ventricular hypertrophy (LVH), cardiovascular events, and mortality.3–5 Recently, these findings have also been variably extended to individuals with intact renal function,6–8 but associations have not been consistent throughout the population.6,9
Given these inconsistencies, one of the key open questions that remains in the field is whether the association between FGF‐23 and LVH is likely to be causal.10 Several studies have failed to replicate associations of FGF‐23 and LVMI,11 and others have suggested that associations with cardiovascular disease may be context dependent, manifesting primarily among individuals with renal tubular dysfunction.9 If the latter is true, FGF‐23 could simply be a biomarker of underlying kidney damage among individuals with CKD, rather than being directly toxic to the cardiovascular system.10
To address this question, several research groups have investigated whether FGF‐23 has direct effects on cardiac myocytes in experimental models, but again results have been inconsistent.12–14 In 1 study, a 5/6‐nephrectomized rat model of CKD developed LVH that was significantly attenuated with administration of the FGF receptor antagonist PD‐173074.12 However, an FGF‐23‐neutralizing antibody did not reduce the hypertrophy in another investigation using the same animal model.13 In a third study, a Col4a3−/− mouse model of CKD with elevated FGF‐23 demonstrated increased gene expression of markers of pathological hypertrophy but no increase in cardiomyocyte size or ventricular wall thickness.14 The possibility of a klotho‐independent direct effect of FGF‐23 on cardiomyocytes has been raised by the observation that klotho‐deficient mice, an established animal model of constitutively elevated FGF‐23, develop echocardiographic LVH.12 This latter finding has not yet been widely replicated or tested in the context of other imaging modalities.
We investigated the relationships between FGF‐23, left ventricular mass, structure, and systolic function among patients in the Heart and Soul Study, a well‐characterized cohort of patients with stable coronary heart disease and a broad range of kidney function. We also explored the putative effects of FGF‐23 activity in homozygous and heterozygous klotho‐deficient mice using cardiac magnetic resonance imaging (MRI), a potentially more accurate method to quantify LVH than the echocardiography used previously. Together, our population and mouse results raise questions about the generalizability of the effect of FGF‐23 on cardiac structure.
Methods
Participants
The Heart and Soul Study is a longitudinal cohort study designed to investigate the relationship of psychosocial stressors and cardiovascular disease progression. Study methods have been described in detail elsewhere.15 Briefly, inclusion required ≥1 of the following: history of myocardial infarction, angiographic evidence of >50% stenosis in ≥1 coronary vessel, evidence of exercise‐induced ischemia by treadmill or nuclear testing, history of coronary revascularization, or documented diagnosis of coronary artery disease. Individuals were excluded if they were unable to walk 1 block, survived a myocardial infarction within the past 6 months, or planned to move out of the area within 3 years. The institutional review boards of all participating institutions approved the study protocol, and all study participants provided written informed consent.
Between September 2000 and December 2002, 1024 participants from 12 outpatient clinics in the San Francisco Bay area were enrolled. In primary analyses, we excluded 11 individuals (1.1%) with missing values of echocardiographic data, 34 individuals (3.3%) with missing values of FGF‐23, and 92 individuals (9.0%) with missing values of other demographic or clinical measurements, resulting in a final sample size of 887 participants. We performed sensitivity analyses using multiple imputation with the Markov Chain Monte Carlo method and 10 imputations to include all individuals, which produced essentially identical results as the complete case approach.
Exposure and Outcome Assessment
We obtained serum and plasma samples from all participants at their baseline visit after a 12‐hour fast. FGF‐23 was measured in duplicate (and averaged) from frozen samples in 2007 using a carboxy‐terminal human enzyme‐linked immunosorbent assay (Immutopics).16 The intra‐assay coefficient of variation was 5.0%; the interassay coefficient of variation was 9.9% at a concentration of 36.4 RU/mL and 12.6% at a concentration of 379 RU/mL.7
Participants underwent a complete 2‐dimensional echocardiogram at baseline. We obtained standard 2‐dimensional parasternal short axis and apical 2‐ and 4‐chamber views during suspended inspiration. A cardiologist blinded to all other clinical information interpreted the echocardiograms.
We determined left ventricular mass on the basis of wall thickness measurements using a truncated ellipsoid technique and indexed results to body surface area to generate LVMI.17 We evaluated whether associations with LVMI were sensitive to further adjustment by body mass index (BMI). We considered participants who had LVMI >90 g/m2 to have LVH.18 We calculated relative wall thickness as: (posterior wall thickness+septal wall thickness)/end‐diastolic cavity dimension.17 We further categorized LVH into mutually exclusive categories of eccentric and concentric LVH. We classified LVH as eccentric if relative wall thickness was ≤0.43 and concentric if relative wall thickness was >0.43. Because concentric and eccentric LVH result from different modes of cardiomyocyte growth, differences in their respective associations with FGF‐23 may provide insight about the underlying mechanism of FGF‐23 activity in the heart. We determined LVEF by the American Society of Echocardiography recommended biplane method of disks.19
Ascertainment of Demographic and Clinical Data
At baseline, participants reported age, sex, ethnicity, prevalent hypertension and diabetes, current smoking status, smoking history (used to calculate pack‐years of smoking), and level of physical activity (relative to others of the same age and sex) by questionnaire. Trained study personnel measured weight and height (used to calculate BMI) and measured blood pressure after 5 minutes of rest in the supine position. We measured total cholesterol, high‐density lipoprotein cholesterol, triglycerides, serum phosphorous, and serum calcium using standard clinical chemistry analyzers on fasting serum samples taken at baseline. We measured urine creatinine using the rate Jaffé method.20 We measured urine albumin using nephelometry, and indexed urine albumin to urine creatinine levels.21 We measured cystatin C using a BNII nephelometer (Siemens)–based particle‐enhanced immunonephelometric assay (N Latex Cystatin‐C).22 We calculated estimated glomerular filtration rate (eGFR) using the equation eGFR=76.7×(serum cystatin C)−1.19,23
Statistical Analysis
We examined the distribution of the above‐listed covariates by quartile of FGF‐23 and examined Spearman correlation coefficients. We evaluated dose‐response relationships of FGF‐23 with LVMI and LVEF and used multivariable‐adjusted linear regression with least‐square means24 to estimate adjusted LVMI and resting LVEF by quartile of FGF‐23. After ln‐transforming FGF‐23 because of its skewed distribution, we used multivariable‐adjusted linear regression to examine continuous relationships per standard deviation (SD) of lnFGF‐23. Finally, we used multinomial logistic regression to evaluate the simultaneous odds ratios of concentric or eccentric hypertrophy compared with no hypertrophy. We used multinomial logistic regression for this analysis, as these forms of LVH were, by definition, mutually exclusive.
We derived 3 sequential models for multivariable linear and logistic regression analyses. Model 1 adjusted for age, sex, ethnicity, and BMI. Model 2 further adjusted for systolic blood pressure, prevalent diabetes, prevalent hypertension, physical activity score, current smoking status, pack‐years, total cholesterol (quintiles), high‐density lipoprotein cholesterol (log), and triglycerides (log). Model 3 in addition adjusted for serum phosphorous and calcium, albumin‐to‐creatinine ratio (ACR; log), and eGFR. Covariate forms were chosen after graphical examination and comparison of model likelihood statistics.
Given the strong prior hypothesis of effect modification by underlying kidney disease,25 we performed analyses stratified by the presence or absence of CKD, defined as estimated GFR <60 versus ≥60 mL/min per 1.73 m2. We subsequently tested the multiplicative interaction of FGF‐23 with CKD. We also conducted sensitivity analyses restricting the range of FGF‐23 to levels observed among patients with normal kidney function (<350 RU/mL). We tested multiplicative interaction by sex and race, but no interactions were observed.
We performed analyses using SAS software, version 9.2, and Stata Statistical Software, version 10.1. P values <0.05 were considered statistically significant.
Murine Experiments
We crossbred heterozygous klotho‐null mice to obtain homozygous klotho(−/−) and heterozygous klotho(−/+) mutants. We identified genotypes by polymerase chain reaction using genomic DNA extracted from tail clips.26 Klotho‐deficient mice lack the klotho coreceptor required for FGF‐23 to promote phosphaturia in the kidney; as a result, they display significantly elevated circulating levels of FGF‐23 accompanied by soft‐tissue and vascular calcifications by 3–6 weeks of age.26 Mice were fed with a standard diet ad libidum and maintained in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental protocols were approved by the institutional care and use committee at Harvard Medical School.
Cardiac MRI in Mice
We performed MRI on wild‐type (n=4), klotho(−/+) (n=5), and klotho(−/−) (n=5) mice at 4 weeks of age. As positive controls for LVH,27 we also imaged 2 homozygous and 1 heterozygous 1‐α hydroxylase–null mice.
We conducted MRI with a Bruker 9.4 Tesla Biospec 94/20 system (Bruker Instruments, Billerica, MA) using a quadrature 40‐mm mouse body volume coil. We anesthetized animals with 0.5% to 3% vaporized isoflurane and monitored ECG and respiratory signals during imaging. We used a cardiac‐ and respiratory‐gated fast gradient echo sequence (FLASH‐cine) with the following parameters: 30‐degree flip angle; time to echo, 2.8 ms; time to repeat, 20 ms; matrix size, 192×192; field of view, 25.6×25.6 mm; and 4 averages. We acquired contiguous image slices (1 mm thick) of the heart's short axis from the apex covering the heart's full volume.
For each image slice, we manually traced the left ventricular wall region and internal chamber region using Image J software. We used the pixel‐to‐area conversion factor 1 pixel/0.018 mm2 to estimate the left ventricular wall area, left ventricular chamber area at systole, and left ventricular chamber area at diastole. We multiplied the area measurements by slice thickness to obtain volume measurements for each slice and summed the volume measurements to determine total volume.28 We determined LVEF on the basis of the estimated left ventricular chamber volume at systole and diastole. After ln‐transformation to achieve normality and similar variances across genotypes, we compared left ventricular wall volumes and ejection fractions between genotypes using ANOVA. For left ventricular wall volume, we adjusted our results for left ventricular internal chamber volume to account for differences in mouse size.
Heart Weight and Histology
We obtained murine data on heart weight and histology in a distinct set of mice to confirm the results we obtained via cardiac MRI. We anesthetized mice at 6 weeks of age and perfused with 0.1 mol/L phosphate‐buffered saline and fixed in 10% buffered formalin. We measured wet heart weight and calculated the ratio of wet heart weight to body weight to determine the index of cardiac hypertrophy. After embedding hearts in paraffin blocks, we cut 5‐μm sections and stained them with hematoxylin/eosin solution. Cross‐sectional images were documented using a DP controller and Manager (Olympus Soft Imaging Solutions).
We determined heart weight/body weight ratio in wild‐type (n=6), klotho(−/+) (n=5), and klotho(−/−) (n=12) mice. After checking normality and similar variances, we compared heart weight/body weight ratio between genotypes using ANOVA.
We obtained 1 to 3 histological cross‐sections at the midventricular level of the heart from wild‐type (n=4) and klotho(−/−) (n=4) mice. Using Image J software, we manually traced the total area of the LV in each section and compared LV area in wild‐type (n=4) and klotho(−/−) (n=4) mice using generalized estimating equations with a Gaussian link and a compound symmetry covariance structure to account for the repeated sections within individual mice.
Results
Participant Characteristics
Compared with participants in the lowest quartile of FGF‐23, participants in the higher quartiles were more frequently female, hypertensive, diabetic, and current smokers (Table 1). Those in higher quartiles of FGF‐23 were also less physically active, had higher triglycerides, higher serum phosphorous, higher ACR, and lower eGFR.
Table 1.
Characteristics of Heart and Soul Study Subjects by Quartile of FGF‐23 (RU/mL)*
| Variables | Quartile 1 (≤27.7), n=222 | Quartile 2 (27.8 to 43.0), n=221 | Quartile 3 (43.1 to 71.4), n=222 | Quartile 4 (≥71.5), n=222 |
|---|---|---|---|---|
| Age, y | 66 (10) | 67 (11) | 67 (11) | 67 (12) |
| Male, n (%) | 193 (87) | 187 (85) | 182 (82) | 167 (75) |
| White, n (%) | 133 (60) | 124 (56) | 137 (62) | 149 (67) |
| Blood pressure | ||||
| Systolic, mm Hg | 129 (19) | 134 (22) | 133 (20) | 135 (23) |
| Comorbidities | ||||
| Diabetes, n (%) | 47 (21) | 58 (26) | 62 (28) | 64 (29) |
| Hypertension, n (%) | 140 (63) | 160 (72) | 160 (72) | 163 (73) |
| Lifestyle factors | ||||
| BMI, kg/m2 | 27.9 (4.6) | 28.2 (4.8) | 28.9 (5.4) | 28.4 (6.0) |
| Above average physical activity, n (%) | 83 (37) | 89 (40) | 78 (35) | 52 (23) |
| Current smoker, n (%) | 36 (16) | 35 (16) | 38 (17) | 68 (31) |
| Pack‐years | 20 (21) | 19 (21) | 20 (22) | 24 (22) |
| Laboratory values | ||||
| Total cholesterol, mg/dL | 175 (40) | 176 (38) | 181 (43) | 181 (49) |
| HDL‐C, mg/dL | 46 (13) | 47 (13) | 45 (14) | 46 (16) |
| Triglycerides, mg/dL* | 96 (66) | 106 (92) | 122 (90) | 119 (109) |
| Serum phosphorous, mg/dL | 3.5 (0.5) | 3.6 (0.5) | 3.7 (0.6) | 3.8 (0.7) |
| Serum calcium, mg/dL | 9.5 (0.5) | 9.5 (0.5) | 9.5 (0.5) | 9.5 (0.6) |
| Renal function | ||||
| Albumin‐to‐creatinine ratio ([ACR]×1000) | 30 (118) | 33 (118) | 53 (210) | 146 (497) |
| eGFR, mL/min per 1.73 m2 | 80 (18) | 75 (19) | 71 (24) | 58 (21) |
Mean (SD) for continuous variables except where specified; n (%) for dichotomous variables. BMI indicates body mass index; eGFR, estimated glomerular filtration rate; FGF‐23, fibroblast growth factor–23; HDL‐C, high‐density lipoprotein cholesterol.
Median (interquartile range).
The participants included in these analyses had a broad range of kidney function. In the entire study population, 167 participants had normal kidney function (eGFR ≥90 mL/min per 1.73 m2), 449 participants had eGFR between 60 and 90 mL/min per 1.73 m2, and 271 participants had eGFR <60 mL/min per 1.73 m2.
FGF‐23 concentrations varied markedly among patients with different levels of kidney function. Median FGF‐23 levels were 32.3 RU/mL (interquartile range 26.7) in participants with normal kidney function, 39.1 RU/mL (interquartile range 30.8) among those with eGFR between 60 and 90 mL/min per 1.73 m2, and 68.5 RU/mL (interquartile range 90.2) in participants with eGFR <60 mL/min per 1.73 m2.
FGF‐23, LVMI, and LVEF
In our crude model, FGF‐23 was positively associated with LVMI (Table 2). These results were attenuated and no longer statistically significant after adjustment for cardiovascular risk factors, including serum phosphorous, serum calcium, ACR, and eGFR. FGF‐23 was inversely associated with LVEF in both crude and multivariable‐adjusted models, but with only a modest decrease in LVEF across quartiles of FGF‐23 (Table 2).
Table 2.
Least‐Squares Mean Estimates of Left Ventricular Mass Index (g/m2) and Left Ventricular Ejection Fraction (%) by Quartile of FGF‐23 (RU/mL)
| Quartile 1 (≤27.7) | Quartile 2 (27.8 to 43.0) | Quartile 3 (43.1 to 71.4) | Quartile 4 (≥71.5) | Increase per SD lnFGF‐23 | P Value | |
|---|---|---|---|---|---|---|
| Left ventricular mass index, g/m2 | ||||||
| Model 1* | 93.6 (90.2 to 96.9) | 96.1 (92.7 to 99.4) | 96.9 (93.6 to 100.3) | 105.3 (101.9 to 108.7) | 3.9 (0.9) | <0.001 |
| Model 2* | 94.6 (91.3 to 98.0) | 96.3 (93.0 to 99.6) | 97.1 (93.8 to 100.4) | 104.7 (101.3 to 108.1) | 3.1 (0.9) | <0.001 |
| Model 3* | 95.9 (92.5 to 99.3) | 97.0 (93.7 to 100.3) | 97.3 (94.0 to 100.6) | 102.5 (99.0 to 106.1) | 1.4 (1.0) | 0.15 |
| Left ventricular ejection fraction, % | ||||||
| Model 1* | 62.9 (61.7 to 64.2) | 62.5 (61.2 to 63.7) | 62.2 (60.9 to 63.5) | 59.3 (58.1 to 60.6) | −1.3 (0.3) | <0.001 |
| Model 2* | 63.1 (61.8 to 64.4) | 62.3 (61.0, 63.6) | 62.3 (61.0 to 63.6) | 59.2 (57.9 to 60.5) | −1.3 (0.4) | <0.001 |
| Model 3* | 62.7 (61.4 to 64.0) | 62.2 (60.9 to 63.5) | 62.3 (61.0 to 63.6) | 59.7 (58.3 to 61.1) | −1.0 (0.4) | 0.01 |
We provide least‐squares mean estimates and confidence intervals (by quartiles) and regression coefficients and standard errors per SD lnFGF‐23 (in the overall population). P values were estimated by linear regression to test for trend. ACR indicates albumin‐to‐creatinine ratio; BMI, body mass index; eGFR, estimated glomerular filtration rate; FGF‐23, fibroblast growth factor–23; HDL, high‐density lipoprotein; SBP, systolic blood pressure.
Adjusted for age, sex, ethnicity, BMI.
Adjusted for age, sex, ethnicity, BMI, SBP, diabetes, hypertension, physical activity score, smoking history, total cholesterol, HDL cholesterol, triglycerides.
Adjusted for age, sex, ethnicity, BMI, SBP, diabetes, hypertension, physical activity score, smoking history, total cholesterol, HDL cholesterol, triglycerides, serum phosphorous, serum calcium, ACR, and eGFR.
FGF‐23 and Concentric and Eccentric Hypertrophy
In multinomial logistic regression, FGF‐23 was not statistically significantly associated with the prevalence of concentric hypertrophy (odds ratio, 1.5; CI, 0.9 to 2.4 per SD increase in lnFGF‐23) or eccentric hypertrophy (odds ratio, 1.1; CI, 0.9 to 1.3) compared with no hypertrophy. The differential associations of FGF‐23 with concentric versus eccentric hypertrophy (with no hypertrophy as the referent) were not statistically significant (P=0.27).
Interactions With Kidney Function
In stratified analyses adjusting for eGFR as a continuous variable (Table 3), FGF‐23 was strongly related to concentric hypertrophy in patients with eGFR <60 mL/min per 1.73 m2 but not among patients with eGFR ≥60 mL/min per 1.73 m2. To determine if the greater association among participants with CKD reflected their greater range in FGF‐23 levels, we performed analyses restricted to individuals with FGF‐23 <350 RU/mL. Associations in this restricted population were similar to our previous results. In the total population, kidney function did not appear to modify the relationship between FGF‐23 and LVMI (P‐interaction=0.87) or LVEF (P‐interaction=0.31).
Table 3.
Estimated Odds Ratios and Confidence Intervals Per Standard Deviation (SD) Increase in lnFGF‐23, Stratified by Chronic Kidney Disease (eGFR <60 mL/min per 1.73 m2)
| eGFR <60 mL/min per 1.73 m2 | eGFR ≥60 mL/min per 1.73 m2 | |||||
|---|---|---|---|---|---|---|
| n=271 | Odds Ratio | P Value | n=616 | Odds Ratio | P Value | |
| No hypertrophy | 87 | 1.0 (ref) | — | 305 | 1.0 (ref) | — |
| Eccentric hypertrophy | 171 | 1.0 (0.7 to 1.4) | 0.93 | 300 | 1.1 (0.9 to 1.4) | 0.37 |
| Concentric hypertrophy | 13 | 2.3 (1.0 to 5.3) | 0.04 | 11 | 1.0 (0.4 to 2.5) | 0.94 |
ACR indicates albumin‐to‐creatinine ratio; BMI, body mass index; eGFR, estimated glomerular filtration rate; FGF‐23, fibroblast growth factor–23; HDL, high‐density lipoprotein; SBP, systolic blood pressure. Adjusted for age, sex, ethnicity, BMI, SBP, diabetes, hypertension, physical activity score, smoking history, total cholesterol, HDL cholesterol, triglycerides, serum phosphorous, serum calcium, ACR, and eGFR. P‐interaction=0.28.
Cardiac MRI in Mice
As expected, 1‐α hydroxylase–deficient mice used as positive controls had significantly greater wall volumes than did wild‐type mice (P=0.04). In contrast, klotho‐deficient mice did not differ significantly from wild‐type mice (−0.28 SD smaller ln‐tranformed wall volume in klotho‐deficient mice; CI ,−0.87 to 0.32 SD; P=0.37). Similar to wild‐type mice, klotho‐deficient mice had significantly lower wall volumes than 1‐α hydroxylase–deficient mice (−1.14 SD; CI, −2.13 to −0.14 SD; P=0.02). Left ventricular function among klotho‐deficient mice also did not differ significantly from wild‐type mice (0.04 SD higher ln‐transformed LVEF in klotho‐deficient mice; CI, −0.99 to 1.06 SD; P=0.94).
Heart Weight and Histology in Mice
No statistically significant differences could be observed in the heart weight/body weight ratio comparing wild‐type, klotho(−/+), and klotho(−/−) mice (P=0.97; Figure) or in left ventricular wall area measurements comparing wild‐type and klotho(−/−) mice (P=0.94).
Figure 1.
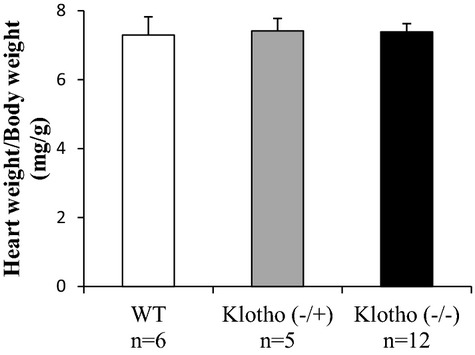
Heart weight/body weight ratio in wild‐type (WT) (n=6), klotho(−/+) (n=5), and klotho(−/−) (n=12) mice at 6 weeks of age. Values are expressed as mean ± standard error (SE) for each group.
Discussion
Our study in a well‐characterized population with stable coronary artery disease and a broad range of kidney function extends previously published evidence on the association between circulating levels of FGF‐23 and cardiac hypertrophy. In 887 individuals enrolled in the Heart and Soul Study, FGF‐23 was not associated with left ventricular mass or odds of concentric or eccentric hypertrophy. We observed a strong association between FGF‐23 and concentric hypertrophy only among individuals with CKD. In our murine experiments, we were unable to confirm previous findings of LVH in klotho‐deficient mice, an established animal model of elevated FGF‐23. Together, our results raise questions about the generalizability of the effect of FGF‐23 on cardiac structure and suggest that further research is needed to prove a direct effect of FGF‐23 on the cardiovascular system in humans.
Although FGF‐23 has been variably associated with abnormal cardiac structure and function3,6,8 and adverse cardiovascular outcomes7,25 in individuals with and without CKD, findings have been inconsistent,11 and when present, associations have generally been stronger among individuals with impaired renal function.6,9 At present, it remains controversial whether FGF‐23 has a direct effect on the cardiovascular system.
There are several possible mechanisms by which FGF‐23 may affect cardiac structure and function indirectly. It is possible that FGF23 provides an alternative measure of renal impairment (imperfectly captured by eGFR or ACR) that can indirectly lead to cardiovascular remodeling and adverse events via hemodynamic overload and pressure and volume expansion.29 Alternatively, FGF‐23 may be a marker of time‐averaged serum phosphorous and, by extension, vascular calcification—a known risk factor for cardiovascular remodeling.30 In this study, associations of FGF‐23 with LVEF were not significantly attenuated by adjustment for serum phosphorous levels measured at a single time. Nonetheless, a single serum phosphorus measurement may not accurately represent cumulative phosphorus burden, given the high intraindividual biological variability in serum phosphorus levels when measured repeatedly over time.31 Finally, FGF‐23 may affect the cardiovascular system indirectly by inducing hypovitaminosis D via inhibition of 1α‐hydroxylase, given the large body of evidence for vitamin D deficiency as a cardiovascular risk factor.32
Although a direct effect of FGF‐23 on the heart has been hypothesized, evidence to support this hypothesis is limited.12,14 In 1 murine experiment, intravenous or direct injection of recombinant FGF‐23 protein into the myocardium resulted in LVH within 5 or 14 days, respectively.12 However, confirmatory studies of LVH reduction by FGF‐23 neutralization13 have produced mixed results. Faul et al reported that klotho‐deficient mice, an established murine model of elevated FGF‐23, develop echocardiographic LVH12—and interpreted this as evidence that FGF‐23 action on cardiac tissue is independent of the klotho coreceptor. Our study using cardiac MRI, a potentially more accurate 3‐dimensional imaging modality than echocardiography, did not provide evidence of LVH or diminished ejection fraction in klotho‐deficient mice. Our MRI findings were substantiated by heart weight and histological data, which also showed no significant differences between wild‐type and klotho‐deficient mice.
Several caveats are necessary when comparing our results with those reported previously. First, mice in our study were 4 to 6 weeks old, whereas klotho‐deficient mice found to have echocardiographic LVH were 8 to 12 weeks old at the time that cardiac size was assessed. It is possible that differences in left ventricular size and systolic function do not manifest at an early age, although klotho‐deficient mice do display significantly elevated circulating levels of FGF‐23 accompanied by soft‐tissue and vascular calcifications as early as 3–6 weeks of age.26 Another possible explanation for our failure to detect LVH in klotho‐deficient mice is our small sample sizes (n=5 to 6 mice/genotype), although our sample sizes were similar to those in previous studies that observed LVH and were sufficient to observe LVH in our positive controls. A final possible explanation for our findings is that the use of cardiac MRI, a potentially more accurate imaging modality than echocardiography, reduced measurement error in assessing cardiac size and produced more accurate results. To validate our findings, further in vitro and in vivo experimentation with direct injection of FGF‐23 and inhibition of the klotho coreceptor, ideally in murine models with and without CKD at earlier and later ages, is needed.
This study has several strengths. Few previous studies have had the necessary data to examine whether FGF‐23 is associated with left ventricular mass and systolic dysfunction in patients without CKD. With 887 participants, we were able to study the association of FGF‐23 and cardiac structure and function with precision. We adjusted for important potential confounders not available in most previous studies, including serum phosphorous and calcium, ACR, and cystatin‐based eGFR, though we cannot exclude the possibility of residual confounding or confounding by other unknown characteristics. FGF‐23 was measured in duplicate, with low intra‐ and interassay coefficients of variations. Echocardiograms were examined by a blinded cardiologist before the FGF23 measurements were conducted, reducing the likelihood of measurement error and misclassification bias.
Our study also has important limitations. As a result of our cross‐sectional study design, we are unable to determine temporality. Because the participants in our study were mostly white men with stable coronary heart disease, caution must be taken in generalizing our results to other populations. Furthermore, we did not have information on variables such as erythropoietin levels or vasomotor responses in this cohort study, which could have shed light on the observed association among individuals with CKD.
Despite recent pharmacological advances, long‐term outcomes in patients with CKD remain poor. If the observed relationships between elevated FGF‐23 and LVH among individuals with CKD are causal, it is tempting to speculate that FGF‐23 may become a therapeutic target in these patients, and clinical trials are currently ongoing. However, our results suggest that FGF‐23 is unlikely to be a major target among patients without underlying CKD.
In conclusion, we observed a modest association of FGF‐23 with worse left ventricular systolic function in the entire population and an association of FGF‐23 with LVMI and concentric LVH only among individuals with CKD. Our murine experiments found no evidence of LVH in klotho‐deficient mice, using both cardiac MRI and histological analysis. Our results suggest that FGF‐23 is unlikely to have major direct effects on cardiovascular structure and function among patients free of substantial CKD; furthermore, if present, any direct effect on the myocardium may not be independent of the klotho coreceptor.
Sources of Funding
The Heart and Soul Study was supported by the Department of Veterans Affairs Epidemiology Merit Review Program and Health Services Research and Development service; National Heart, Lung, and Blood Institute; American Federation for Aging Research; Robert Wood Johnson Foundation; and Ischemia Research and Education Foundation. FGF‐23 measurements were supported by a grant from the American Heart Association and the NHLBI (1R01HL096851), and Drs Ix and Mukamal were supported by 1R01HL094555. Murine work was supported in part by R56 DK073944 (to Dr Lanske) at Harvard School of Dental Medicine and a Shared Instrumentation Grant S10 RR028792 at Beth Israel Deaconess Medical Center. Dr Kestenbaum has received grant funding from Amgen.
Disclosures
None.
Acknowledgments
We thank Michelle Farley and Michael Densmore for their invaluable contributions to the murine experiments.
References
- 1.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004; 19:429-435 [DOI] [PubMed] [Google Scholar]
- 2.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF‐23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003; 64:2272-2279 [DOI] [PubMed] [Google Scholar]
- 3.Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, Wang TJ, deFilippi C, Wolf M. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009; 119:2545-2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutierrez OM, Mannstadt M, Isakova T, Rauh‐Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, Wolf M. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008; 359:584-592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf MChronic Renal Insufficiency Cohort (CRIC) Study Group Fibroblast growth factor 23 and risks of mortality and end‐stage renal disease in patients with chronic kidney disease. JAMA. 2011; 305:2432-2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis. 2009; 207:546-551 [DOI] [PubMed] [Google Scholar]
- 7.Parker BD, Schurgers LJ, Brandenburg VM, Christenson RH, Vermeer C, Ketteler M, Shlipak MG, Whooley MA, Ix JH. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med. 2010; 152:640-648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiler S, Cremers B, Rebling NM, Hornof F, Jeken J, Kersting S, Steimle C, Ege P, Fehrenz M, Rogacev KS, Scheller B, Bohm M, Fliser D, Heine GH. The phosphatonin fibroblast growth factor 23 links calcium‐phosphate metabolism with left‐ventricular dysfunction and atrial fibrillation. Eur Heart J. 2011; 32:2688-2696 [DOI] [PubMed] [Google Scholar]
- 9.Dominguez JR, Shlipak MG, Whooley MA, Ix JH. Fractional excretion of phosphorus modifies the association between fibroblast growth factor‐23 and outcomes. J Am Soc Nephrol. 2013; 24:647-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolf M. Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol. 2010; 21:1427-1435 [DOI] [PubMed] [Google Scholar]
- 11.Unsal A, Kose Budak S, Koc Y, Basturk T, Sakaci T, Ahbap E, Sinangil A. Relationship of fibroblast growth factor 23 with left ventricle mass index and coronary calcification in chronic renal disease. Kidney Blood Press Res. 2012; 36:55-64 [DOI] [PubMed] [Google Scholar]
- 12.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon‐Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro‐O M, Kusek JW, Keane MG, Wolf M. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011; 121:4393-4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, Tsai MM, Cattley RC, Wronski TJ, Xia X, Li X, Henley C, Eschenberg M, Richards WG. FGF23 neutralization improves chronic kidney disease‐associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012; 122:2543-2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touchberry CD, Green TM, Tchikrizov V, Mannix JE, Mao TF, Carney BW, Girgis M, Vincent RJ, Wetmore LA, Dawn B, Bonewald LF, Stubbs JR, Wacker MJ. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am J Physiol Endocrinol Metab. 2013; 304:E863-E873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruo B, Rumsfeld JS, Hlatky MA, Liu H, Browner WS, Whooley MA. Depressive symptoms and health‐related quality of life: the Heart and Soul Study. JAMA. 2003; 290:215-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juppner H, Wolf M, Salusky IB. FGF‐23: more than a regulator of renal phosphate handling? J Bone Miner Res. 2010; 25:2091-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJChamber Quantification Writing Group, American Society of Echocardiography's Guidelines and Standards Committee, European Association of Echocardiography Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005; 18:1440-1463 [DOI] [PubMed] [Google Scholar]
- 18.Nair D, Shlipak MG, Angeja B, Liu HH, Schiller NB, Whooley MA. Association of anemia with diastolic dysfunction among patients with coronary artery disease in the Heart and Soul Study. Am J Cardiol. 2005; 95:332-336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recommendations for quantitation of the left ventricle by two‐dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two‐Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989; 2:358-367 [DOI] [PubMed] [Google Scholar]
- 20.Ix JH, Wassel CL, Stevens LA, Beck GJ, Froissart M, Navis G, Rodby R, Torres VE, Zhang YL, Greene T, Levey AS. Equations to estimate creatinine excretion rate: the CKD epidemiology collaboration. Clin J Am Soc Nephrol. 2011; 6:184-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ix JH, Chertow GM, Shlipak MG, Brandenburg VM, Ketteler M, Whooley MA. Fetuin‐A and kidney function in persons with coronary artery disease—data from the Heart and Soul Study. Nephrol Dial Transplant. 2006; 21:2144-2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ix JH, Shlipak MG, Katz R, Budoff MJ, Shavelle DM, Probstfield JL, Takasu J, Detrano R, O'Brien KD. Kidney function and aortic valve and mitral annular calcification in the Multi‐Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis. 2007; 50:412-420 [DOI] [PubMed] [Google Scholar]
- 23.Stevens LA, Zhang Y, Schmid CH. Evaluating the performance of equations for estimating glomerular filtration rate. J Nephrol. 2008; 21:797-807 [PMC free article] [PubMed] [Google Scholar]
- 24.SAS/STAT. SAS/STAT(R) 9.2 User's Guide, Second Edition. Construction of Least Squares Means. http://support.sas.com/documentation/cdl/en/statug/63033/HTML/default/viewer.htm#statug_glm_a0000000866.htm [Google Scholar]
- 25.Ix JH, Katz R, Kestenbaum BR, de Boer IH, Chonchol M, Mukamal KJ, Rifkin D, Siscovick DS, Sarnak MJ, Shlipak MG. Fibroblast growth factor‐23 and death, heart failure, and cardiovascular events in community‐living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol. 2012; 60:200-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS. In vivo genetic evidence for klotho‐dependent, fibroblast growth factor 23 (FGF23)‐mediated regulation of systemic phosphate homeostasis. FASEB J. 2009; 23:433-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D. Calcium‐independent and 1,25(OH)2D3‐dependent regulation of the renin‐angiotensin system in 1alpha‐hydroxylase knockout mice. Kidney Int. 2008; 74:170-179 [DOI] [PubMed] [Google Scholar]
- 28.Loganathan R, Bilgen M, Al‐Hafez B, Alenezy MD, Smirnova IV. Cardiac dysfunction in the diabetic rat: quantitative evaluation using high resolution magnetic resonance imaging. Cardiovasc Diabetol. 2006; 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.London GM. Cardiovascular disease in chronic renal failure: pathophysiologic aspects. Semin Dial. 2003; 16:85-94 [DOI] [PubMed] [Google Scholar]
- 30.Masai H, Joki N, Sugi K, Moroi M. A preliminary study of the potential role of FGF‐23 in coronary calcification in patients with suspected coronary artery disease. Atherosclerosis. 2013; 226:228-233 [DOI] [PubMed] [Google Scholar]
- 31.Drueke TB, Massy ZA. Role of vitamin D in vascular calcification: bad guy or good guy? Nephrol Dial Transplant. 2012; 27:1704-1707 [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, O'Keefe JH, Bell D, Hensrud DD, Holick MF. Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor? J Am Coll Cardiol. 2008; 52:1949-1956 [DOI] [PubMed] [Google Scholar]