

Abstract
Background
Development of thoracic aortic aneurysms is the most significant clinical phenotype in patients with Marfan syndrome. An inflammatory response has been described in advanced stages of the disease. Because the hallmark of vascular inflammation is local interleukin‐6 (IL‐6) secretion, we explored the role of this proinflammatory cytokine in the formation of aortic aneurysms and rupture in hypomorphic fibrillin‐deficient mice (mgR/mgR).
Methods and Results
MgR/mgR mice developed ascending aortic aneurysms with significant dilation of the ascending aorta by 12 weeks (2.7±0.1 and 1.3±0.1 for mgR/mgR versus wild‐type mice, respectively; P<0.001). IL‐6 signaling was increased in mgR/mgR aortas measured by increases in IL‐6 and SOCS3 mRNA transcripts (P<0.05) and in cytokine secretion of IL‐6, MCP‐1, and GM‐CSF (P<0.05). To investigate the role of IL‐6 signaling, we generated mgR homozygous mice with IL‐6 deficiency (DKO). The extracellular matrix of mgR/mgR mice showed significant disruption of elastin and the presence of dysregulated collagen deposition in the medial‐adventitial border by second harmonic generation multiphoton autofluorescence microscopy. DKO mice exhibited less elastin and collagen degeneration than mgR/mgR mice, which was associated with decreased activity of matrix metalloproteinase‐9 and had significantly reduced aortic dilation (1.0±0.1 versus 1.6±0.2 mm change from baseline, DKO versus mgR/mgR, P<0.05) that did not affect rupture and survival.
Conclusion
Activation of IL‐6‐STAT3 signaling contributes to aneurysmal dilation in mgR/mgR mice through increased MMP‐9 activity, aggravating extracellular matrix degradation.
Keywords: extracellular matrix, interleukin‐6, Marfan syndrome, matrix metalloproteinases, mgR, thoracic aortic aneurysms and dissections, vascular inflammation
Introduction
Marfan syndrome (MFS), an inherited autosomal dominant disorder, affects connective tissues in the skeletal, ocular, respiratory, and cardiovascular systems.1 The most serious complication in patients with MFS is degenerative aortic lesions, including progressive aneurysmal dilatation of the thoracic aorta (TAA), leading to acute aortic dissection and rupture. The most important clinical approach for improving patient survival is surgical repair of the aneurysm once the aorta has enlarged to approximately twice normal size, and pharmaceutical interventions can slow the rate of enlargement of the aneurysms.2–3 Features of TAAs in MFS include significant pathological remodeling of extracellular matrix (ECM) in the tunica media and adventitia, smooth muscle cell apoptosis, and decreased arterial distensibility.1,4 MFS is caused by mutations in the fibrillin‐1 gene (Fbn‐1) on chromosome 15, a gene encoding a latent TGF‐β binding glycoprotein in microfibrils.1,5 In this syndrome, tonic TGF‐β signaling is linked to alveolar septation and aneurysmal formation.1 Through TGF‐β signaling cross‐talk, enhanced angiotensin II (Ang II) signaling has been implicated in MFS; the AT1 angiotensin receptor blocker losartan normalized TGF‐β levels and TGF‐β signaling, reduced aortic MMP expression, and prevented aortic dilation in mice with MFS.6–7
Recent studies indicate that enhanced IL‐6 signaling is associated with aneurysm development. IL‐6 is elevated systemically and locally in both patients and animal models of abdominal aortic aneurysmal (AAA) disease.8–12 Furthermore, meta‐analysis of human studies shows that a nonsynonymous variant (Asp358Ala) of the IL‐6 receptor (IL‐6R) decreases risk of AAA, suggesting that IL‐6 signaling is important in AAA formation.12 The Asp358Ala variant is associated with increased transcription of the soluble IL‐6R and a decreased presence of membrane‐bound IL‐6R indicating that signaling through the membrane IL‐6R may be important for development of AAA.13 IL‐6 is a growth‐promoting and monocyte‐activating cytokine that signals via activation of Janus kinase (JAK) and tyrosine phosphorylation of signal transducer and activator and transcription 3 (STAT3).14 Our studies have shown that local IL‐6 signaling plays a central role in Ang II‐induced aortic aneurysms,11 where its action promotes aortic monocyte recruitment and monocyte‐to‐macrophage activation via a monocyte chemotactic protein‐1 (MCP‐1)‐based mechanism.15–17 These activated macrophages produce pro‐inflammatory cytokines, chemokines, ROS, and matrix metalloproteinases (MMPs) in the vessel wall, further facilitating local inflammation and remodeling.
Histological analysis of TAA associated with MFS shows a less profound leukocytic infiltrate than that observed in AAA, although local inflammatory events have been observed.18–21 In the hypomorphic fibrillin‐1 mutant mouse model (mgR/mgR), an inflammatory‐fibroproliferative response consisting of intimal hyperplasia, monocyte infiltration and smooth muscle deposition has been described, where the enhanced monocyte/macrophage infiltration becomes pronounced at later stages of the disease.20 Mechanistically, aortas from these mice secrete a GxxPG‐containing fibrillin‐1 fragment that induces macrophage chemotaxis22 and MMP expression, creating an amplification loop for further matrix degeneration and product‐mediated chemotaxis.23–24 Taken together, these findings suggest that inflammation may participate in extracellular matrix degradation associated with fibrillin deficiency‐induced TAAs. However, the role of the inflammatory cytokine IL‐6 in the pathogenesis of TAAs remains unclear.
In this study, we tested the role of IL‐6 in the pathogenesis of TAA in the mgR/mgR homozygous mouse model of MFS. Using novel imaging techniques, we observed not only disruption of elastin fibers in the medial layer but the presence of thinly dispersed collagen sheets in the adventitial layer that was associated with increased MMP. We investigated the presence of local aortic inflammatory events and found that expression and secretion of IL‐6 and the chemokine MCP‐1 were both significantly increased in the medial layer of mgR/mgR mice versus their wild‐type littermates. Our findings of enhanced medial expression of SOCS3 and formation of phospho‐STAT3 indicated that the IL‐6 signaling pathway was functionally active. IL‐6 deficiency in mgR/mgR mice delayed progression of ascending aorta dilation at late stages and reduced elastolytic events that resulted in better preservation of the elastin and collagen content of the ascending aortic wall but did not affect extent of rupture. Finally, we showed that IL‐6 deficiency decreased MMP‐9 activity, suggesting a mechanism in which IL‐6 regulated TAA development in MFS by modulating MMP expression in the aortic wall.
Methods
Animal Care and Use
C57BL/6J wild‐type (+/+) and IL‐6−/− mice on the same genetic background were obtained from the Jackson Laboratory. Heterozygous mutant mice (Fbn1mgR/+; mgR/+) were obtained from Johns Hopkins University and were bred to generate homozygous mutant mice (Fbn1mgR/mgR; mgR/mgR) and wild‐type littermates (Fbn1+/+; +/+). To create IL‐6−/−•Fbn1mgR/mgR double knockout (DKO) mice, Fbn1mgR/+ were bred with IL‐6‐null mice and offspring bred. For histological analysis of aortic tissues, mice were euthanized at 12 weeks of age. This time point was chosen based on our preliminary studies in which aneurysmal changes were prominent and inflammatory events were evident. All animal experiments were approved by the University of Texas Medical Branch (UTMB) Institutional Animal Care and Use Committee. Mice were housed in the UTMB Animal Resource Center in accordance with the NIH Guidelines for the Care and Use of Animals in Research.
Echosonography
mgR/mgR, mgR/+, +/+, and DKO littermates were imaged weekly with noninvasive transthoracic ultrasound echosonographic techniques. Serial ultrasound imaging provided visualization of the progression of vessel dilatation. Mice were sedated with 1% to 3% inhaled isoflurane delivered via nose cone and were positioned supine and imaged using Vevo 770 ultrasound machine for small animals (Visualsonics) equipped with a 707B transducer. Ascending aortas were imaged and diastolic diameter measured by 2 masked investigators in both transverse and longitudinal axis views with B‐mode and M‐mode. Aortic diameters of wild‐type (+/+) and mgR/mgR mutants were first measured in vivo at 12 weeks (wks) of age at the level of ascending aorta, supra‐aortic ridge, and sinus of Valsalva. Subsequent measurements were obtained in triplicate at the level of the ascending aorta.
Computerized Tomography Imaging
A small animal Inveon™ micro‐CT scanner (Siemens Preclinical Solutions) was used. Computerized tomography (CT) imaging parameters were set as follows: voltage 80 kV, current 500 μA, exposure time 850 ms per projection, 520 equidistant steps covering 360 degrees of rotation. Isotropic resolution of the reconstructed images was 0.107 mm. Animals were imaged in the prone position while lying on a radiotransparent bed. Anesthesia was required to prevent motion artifacts, and was achieved using isoflurane via a nose cone. Mice were given supplemental oxygen during scanning. Scan time was ≈15 minutes per mouse.
Aortic Explant and Cytokine Analysis
Following euthanasia and transcardial perfusion with PBS, the entire aorta was dissected and cleaned using stereoscope magnification. Thoracic aorta segments were placed in 0.5 mL DMEM medium containing ×1 ITS and 0.1% BSA and incubated in a tissue culture hood at 37°C for 4 hours as described.11 Ex vivo explant culture medium was frozen at −80°C until assayed for cytokines and chemokines using a multiplex, bead‐based ELISA kit (Lincoplex/Millipore mouse cytokine panel) according to the manufacturer's instructions. Cytokine concentrations were determined using recombinant standards.
Immunohistochemistry and Elastin Histological Staining
Formalin‐fixed, paraffin‐embedded sections from ascending aortas were rehydrated using serial concentrations of ethanol. For histological staining, 5 μm sections were stained with modified Verhoeff Van Gieson elastin stain kit (Sigma‐Aldrich) per manufacturer instructions. Light microscopy was performed using ×20 lens and quantification of elastin breaks was measured in triplicate by a masked investigator. For immunohistochemistry (IHC), antigen retrieval was performed when necessary with antigen unmasking solution (Vector Laboratories). Paraffin‐embedded sections were blocked using 0.1% Triton‐X, 5% normal serum of the species producing the secondary antibodies and incubated with rabbit anti‐IL‐6 (1:600, Abcam), rabbit anti‐MCP‐1 (1:100, Abcam), rabbit anti‐MMP‐9 (1:500, Sigma), or rat anti‐F4/80 (1:500, Abcam). 7 μm OCT‐embedded frozen sections of ascending aortas were blocked and stained with rat anti‐macrophage (1:100, MOMA‐2; Abcam) or rabbit phospho‐STAT3‐Tyr705 (D3A7, 1:100, Cell Signaling). For p‐STAT3 staining, sections pretreated with phospho‐STAT3‐Tyr705 blocking peptide (Cell Signaling) were used as negative controls. Biotinylated goat anti‐rat or goat anti‐rabbit antibodies (Vector Labs) were used as secondary antibodies, followed by staining with ABC and DAB kits (Vector Labs) per manufacturer's instructions. Secondary antibody‐only controls were used to determine staining specificity.
Quantitative Real‐Time PCR
Ascending aortas were freshly isolated and frozen at −80°C for later use. Individual ascending aortas were pulverized in liquid nitrogen inside plastic pouches (Kapak SealPAK), further homogenized on ice in glass Dounce homogenizers and RNA extracted with TriReagent (Sigma‐Aldrich) according to the manufacturer's instructions. RNA was quantified by Nanodrop (Thermo Scientific) and samples were included when the 260/280 nm ratio was >1.6. Five micrograms RNA was reverse transcribed using Superscript III (Invitrogen) according to manufacturer's instructions. Real‐time PCR reactions were performed in triplicate using 1 μL of resulting cDNA per 20 μL reaction volume containing iQ SYBR Green Supermix (Bio‐Rad). Primers were purchased from SABioscience for mouse ccl2/MCP‐1 (PPM03151F) and mouse il6 (PPM03015A). Mouse Socs3 primers were synthesized: sense: 5′‐CCGCGGGCACCTTTC‐3′; anti‐sense: 5′‐TTGACGCTCAACGTGAAGAAGT‐3′. PCR was performed on the CFX96 system (Bio‐Rad) according to preset protocol. Data were normalized to the internal control Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), and expressed as fold change calculated by the ΔΔCt method.
Second Harmonic Generation and Multiphoton Autofluorescence Microscopy
The aortic arch of freshly isolated intact aorta was imaged using the nonlinear optical microscopy techniques of multiphoton microscopy and second harmonic generation (SHG) microscopy with contrast based on intrinsic signals from the tissue.25 These include autofluorescence from elastin and the cytoplasm of cells in multiphoton autofluorescence microscopy (MPAM) and the frequency‐doubled SHG signal that arises from fibrillar collagen.26–27 Used together, high‐resolution images through the aortic adventitia into the media can be obtained for assessment of depth‐resolved structure.28 MPAM/SHG evaluation was done with a customized Zeiss 410 confocal laser scanning inverted microscope modified for multiphoton excitation and detection along nondescanned optics.29 Briefly, illumination was from a femtosecond titanium sapphire laser (Tsunami, SpectralPhysics) having a 5W frequency‐doubled Nd:YVO pump laser, and routed into the scanhead and through the sample objective using optics for ultrafast laser propagation. The system operated with a typical pulse width of 140 fs prior to the objective (40×1.2 N.A. water). Excitation for autofluorescence was 780 nm and for SHG was 840 nm. An epi‐configuration was used for collection of emitted light and detected using a cooled PMT placed in a nondescanned configuration (R6060, Hamamatsu). Fluorescence emission in the spectral region of 450 to 650 nm was collected for detection of broadband autofluorescence from the aorta. SHG was collected using a 420±14 nm bandpass filter in the nondescanned detector path. Thus, MPAM and SHG image stacks were taken sequentially. The intact aorta was placed on a 35 mm imaging dish having a #1.5 coverslip bottom (Matek) and immersed in phosphate buffered saline. Image stacks were obtained beginning above the adventitial surface and moving into the aortic wall using a step size of 1 μm to depths >150 μm. The objective provided a field of view of 320×320 μm. Image reconstructions of micrograph stacks were constructed using Metamorph (Molecular Devices). Analysis of features found in the elastin lamellae was performed on Metamorph using the measurement tools to calculate diameter of holes and represented in μm. SHG signal content was quantified by first thresholding SHG images to determine the regions positive for fibrillar collagen. The same thresholding parameters were used for the full stack and between samples. The percent area positive for collagen according to the threshold region was determined relative to the full field. A value per group was obtained by averaging this area between samples of a group.
Gelatin Zymography
Aortic proteins from 12‐week wild‐type +/+, mgR/+, mgR/mgR and DKO littermates were extracted as previously described.30–31 Protein concentrations were standardized with Bio‐Rad protein assay. Equal amounts (25 μg) of aortic proteins were electrophoresed on 10% gelatin zymogram gels (Invitrogen) as previously described.31 Molecular sizes were determined using protein standards (Invitrogen). Gels were scanned and shown in black and white for densitometry analysis by ImageJ.
Data Analysis
Data are reported as ±SEM. Differences between 2 groups were analyzed by Student t test (2‐tail, assuming unequal variances), and included comparison of diameter changes in aortic arch, mRNA analysis, cytokine secretion, hole diameter, and percent SHG threshold area. One‐way ANOVA was performed when comparing multiple groups, and included assessing change in ascending aortic diameter, elastin breaks per field, and MMP quantification. This was followed by Tukey or Bonferroni post hoc tests to determine individual pair‐wise significance. Before application of ANOVA, data sets were determined to be normally distributed (assessed by Shapiro‐Wilk normality test) and passed the equal variance test as determined by Graphpad Prism 6 or SigmaPlot. Kaplan‐Meier survival curves for different genotypes were plotted and significance analyzed with Mantel‐Cox test. In all cases, P<0.05 was considered significant.
Results
Aortic Imaging
As previously reported,20 mgR/mgR homozygous Fbn‐1 mutants developed spontaneous thoracic aortic aneurysms at 0 to 3 months of age (Figure 1A). We performed transthoracic echosonography to evaluate the development and growth of TAAs. Aortic diameters of wild‐type (+/+) and mgR/mgR mutants were first measured in vivo at 12 weeks of age at the level of ascending aorta, supra‐aortic ridge, and sinus of Valsalva. Differences between +/+ and mgR/mgR littermates were only significant at the level of the ascending aorta (Figure 1B), and this aortic segment was used in our subsequent experiments (Figure 1C).
Figure 1.
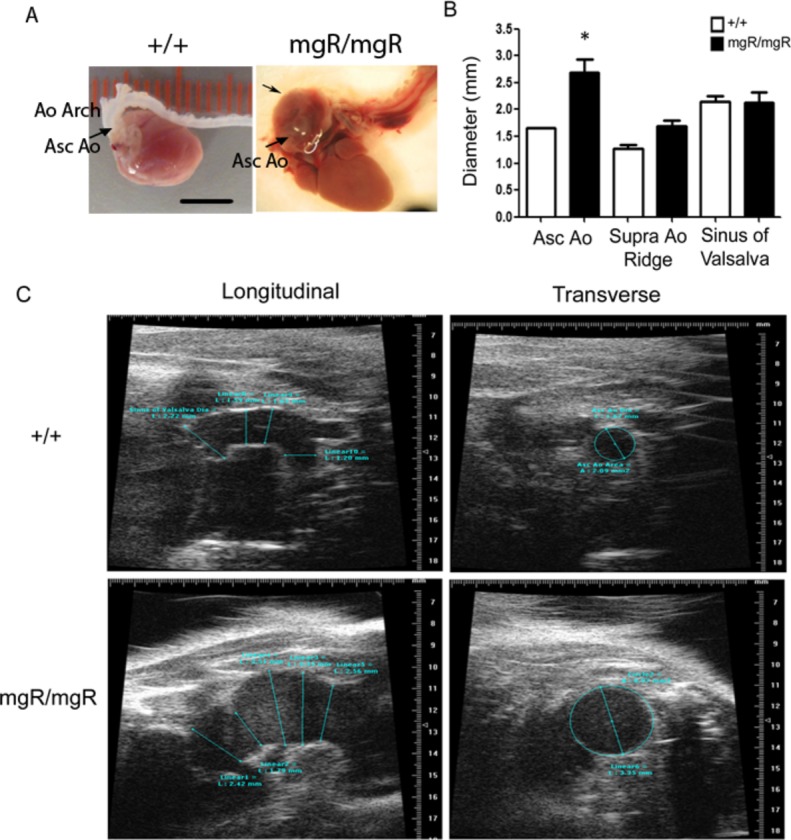
Characterization of vascular pathology of homozygous mgR mice. Age‐matched homozygous (mgR/mgR) and wild‐type (++) littermates were identified by genotyping and used for echosonography imaging of the thoracic aorta (TAA) at the levels of the ascending aorta (Asc Ao), the supra‐aortic ridge (Supra Ao Ridge) and the sinus of Valsalva. A, Representative anatomical images of aortas from a 12‐week‐old wild‐type mouse and a mgR/mgR mouse with TAA formation. Ao Arch, aortic arch; Asc Ao, ascending aorta; unalabeled arrow, TAA in ascending aorta. Scale bar is 5 mm. B, Diameters of thoracic aortas in mgR/mgR (n=9) and wild‐type (++) littermates (n=8) were measured in vivo at the level of ascending aorta, supra‐aortic ridge and the sinus of Valsalva. *P<0.05. C, Representative echosonography images of the aortic arch in age‐matched wild‐type +/+ and mgR/mgR littermates. Left panel: Echo transducer was placed longitudinally to acquire images at different levels of aortic arch. Right panel: transverse images of ascending aorta.
Using histological staining in mgR mutants, we observed extensive loss of elastin in the media at this age, exhibiting both breaking and thinning of elastin fibers (Figure 2A). We confirmed the degree of elastolysis by MPAM. In wild‐type controls, elastin proteins formed smooth sheets in the aortic media, whereas in the mgR mutants, numerous holes as large as 50 μm were evident, suggesting substantial elastin degeneration (Figure 2B, top panel). To study the complete structural components of the ascending aortic wall, we also combined MPAM and SHG microscopy for imaging of ex vivo aortic samples. This approach has been used to detect collagen content in different animal models,32–33 and allowed us to assess ≈150 μm of the aortic wall, starting at the adventitial surface, passing through the adventitia and entering the medial layer. SHG images showed that wild‐type mice had dense, curly, and thick collagen fibers, while the mgR mutants maintained relatively straightened, dispersed, and thin collagen fibers (Figure 2B, middle panel). We interpreted this observation as a dramatic loss of fibrillar collagen in the adventitia of mgR/mgR mice. Moreover, we observed a large number of autofluorescent infiltrating cells in the border between media and adventitia (Figure 2B, bottom panel), suggesting cellular recruitment into the ascending aortic wall at this age. The cell type could not be identified with these imaging techniques.
Figure 2.

Aortic elastin and collagen abnormalities in mgR homozygous mutants. Aortas from age‐matched wild‐type (n=6) and mgR/mgR (n=6) littermates were examined for ECM components. A, Histological staining of elastin (shown in purple), collagen (shown in pink) and SMCs (shown in yellow) in sections of ascending aorta. Arrow: breaks in elastin fibers. Scale bar is 100 μm. B, SHG and MPAM images of wild‐type (n=4) and age‐matched aneurysmal mgR/mgR mice (n=6). Top panel: Elastin sheets form a smooth, curving layer in aortic media in +/+ and holes were formed in elastin layers in mgR/mgR mice. Middle panel: Collagen fibers were arranged in dense, curving structures in the aortic adventitia in wild‐type +/+ mice, but were widely dispersed in the adventitia and adventitia‐media border of mgR/mgR mice. Bottom panel: Combined images of collagen and elastin at the adventitia‐media border. Collagen is shown in green and elastin is shown in red. Arrows: holes formed in elastin layers. Cell infiltration is also pronounced at the adventitia‐media border (autofluorescent red dots). Representative images are shown. Images were shown in 320×320 μm. ECM indicates extracellular matrix; MPAM, multiphoton autofluorescence microscopy; SHG, second harmonic generation; SMC, smooth muscle cell.
IL‐6 and MCP‐1 are Elevated in mgR/mgR Mice
In order to study the contribution of IL‐6 signaling in the development of TAA in this mouse model of MFS, we evaluated the expression and secretion of cytokines in the ascending aorta. We observed a significant 2.4‐fold increase in IL‐6 mRNA expression (P<0.05), as well as an increase in the IL‐6 downstream target, suppressor of cytokine signaling 3 (SOCS3, P<0.01), suggesting the IL‐6 signaling pathway was functionally active in mgR/mgR mice (Figure 3A). Additionally, MCP‐1 mRNA expression also was elevated in mgR/mgR mice. MCP‐1 is a chemotactic cytokine whose expression is controlled by an IL‐6 amplification loop11 (Figure 3A, P<0.05). Using an established aortic explant culture system,11 we confirmed that secretion of IL‐6, MCP‐1, and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) was enhanced by 2‐ to 3‐fold in the mutant ascending aortas versus wild‐type aortas (Figure 3B, P<0.05). IHC staining of ascending aortic sections showed that IL‐6 and MCP‐1 expression was mainly localized in the aortic media (Figure 3C). Further, consistent with cytokine and chemokine activation, MOMA‐2 immunostaining showed increased presence of macrophages at the media‐adventitia border in mgR/mgR mutants (Figure 3C). Taken together, our observations confirm the presence of functional IL‐6‐signaling pathway and monocyte recruitment in the mgR mouse model.
Figure 3.
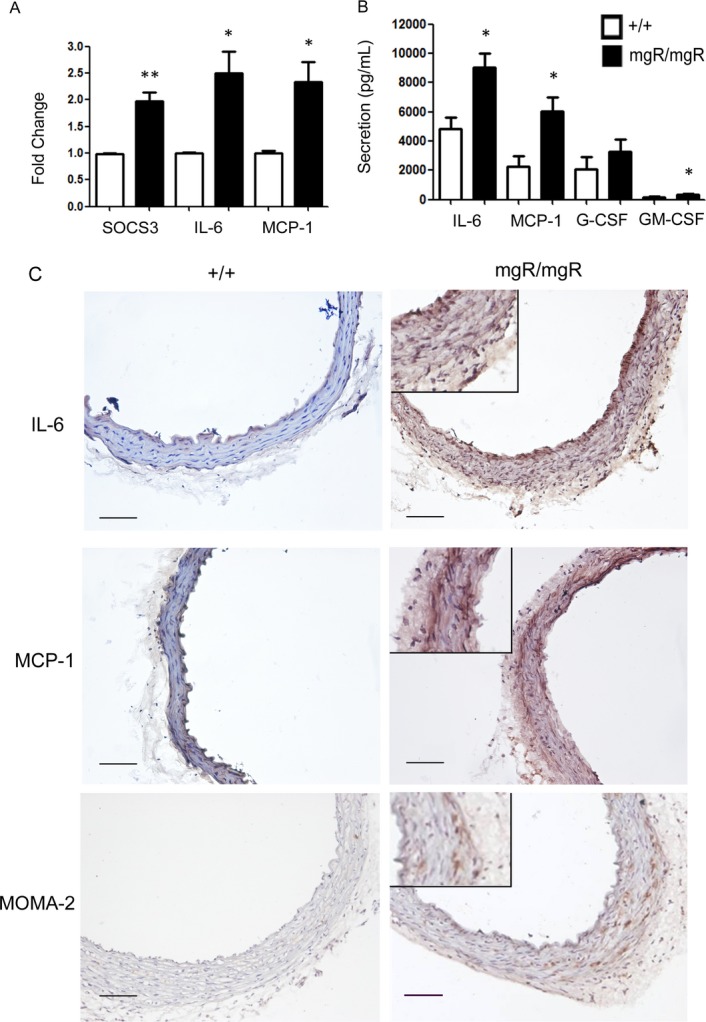
IL‐6 signaling is activated in mgR homozygous mice. A, Real‐time quantitative analysis demonstrated that gene expression levels of IL‐6, MCP‐1, and the IL‐6 downstream signaling molecule SOCS3 are significantly elevated in mgR/mgR homozygous mice (n=6) vs wild‐type controls (n=4), suggesting activation of IL‐6 signaling in the mgR mutants. *P<0.05; **P<0.01. B, Local cytokine and chemokine secretion from thoracic aortic tissues in explant culture media was measured by multiplex ELISA. Secretion of IL‐6, MCP‐1 and GM‐CSF was increased significantly. n=4 in each group. *P<0.05. **P<0.01. C, IHC staining of IL‐6, MCP‐1 and the macrophage marker MOMA‐2. Positive staining was shown in brown and counter‐staining with hematoxylin in blue. Enlarged details of the mgR/mgR sections are shown on the right. Scale bar is 100 μm. ELISA indicates enzyme‐linked immunosorbent assay; G‐CSF, granulocyte‐colony stimulating factor; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IHC, immunohistochemistry; IL, interleukin; MCP‐1, monocyte chemotactic protein‐1; SOCS3, suppressor of cytokine signaling 3.
IL‐6 Deficiency in mgR/mgR Mice Decreases Progressive Dilatation of Ascending Aorta Without Affecting Survival
In order to identify the role of IL‐6 signaling in the formation and progression of aneurysmal dilatation, we bred mgR/mgR mice with IL‐6‐null mice to generate Fbn1mgR/mgR•IL‐6−/− double mutant mouse line (DKO). We first examined the activation of IL‐6 signaling in the aortic tissues. With IHC staining of phospho‐STAT3‐Y705, an IL‐6 downstream signaling molecule, we confirmed the inactivation of STAT3 phosphorylation in the DKO mice induced by IL‐6 (Figure 4A), indicating that phospho‐STAT3 is driven predominately by IL‐6 and not other members of the IL‐6 superfamily of cytokines. We followed the progression of aortic dilation weekly using in vivo ultrasound imaging (Figure 4B). In a manner similar to that of mgR/mgR mutants, DKO mice developed spontaneous TAAs at 1 to 3 months of age but the enlargement of these aneurysms was significantly less from 14 weeks of age onward (P<0.05, Figure 4D). DKOs exhibited a similar rate of aneurysmal dilation at earlier stage of life (eg, 5 to 12 weeks). To our surprise, DKO mice did not show improved survival, with median survival of 14 weeks in DKO mice and 11.5 weeks in mgR/mgR mutants (Figure 4C). These findings indicate that IL‐6 deficiency significantly decreased aortic dilatation but had no effect on aortic rupture.
Figure 4.
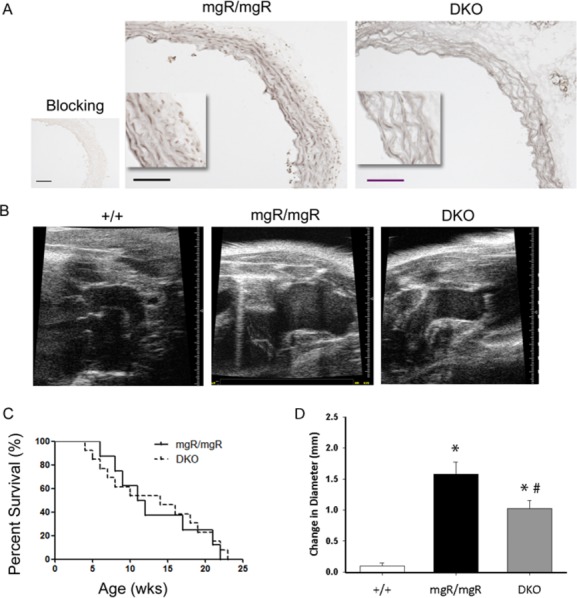
Characterization of aortic pathology of mgR/mgR•IL‐6−/− double knockout (DKO) mice. A, IHC staining of phospho‐STAT3‐Y705. Positive staining was shown in grey. Aortic sections treated with p‐STAT3 blocking peptide were shown on the left as negative control. n=6 mice in mgR/mgR and n=4 mice in DKO group. Scale bar is 100 μm. B, Representative images of echosonography of age‐matched wild‐type, mgR homozygotes, and DKO mice. C, Survival curves of mgR/mgR (n=21) and DKO (n=17) mice. D, Diameters of ascending aorta were measured weekly in vivo by transthoracic echosonography from week 5 to week 18 in wild‐type (+/+), mgR/mgR and DKO mice. Change in diameter was determined with week 5 measurements taken as the baseline, which was subtracted from the final measurement recorded during week 18. The difference, or the change in diameter, was then assessed for significance using ANOVA. n=10 in each group. *P<0.005 vs wild‐type, #P<0.05 vs mgR/mgR. ANOVA indicates analysis of variance; IHC, immunohistochemistry; IL, interleukin; p‐STAT3, phosphorylation of signal transducer and activator and transcription 3.
IL‐6 Deficiency Causes Less ECM Degeneration in mgR/mgR Mice
We examined ECM components in the aortic wall to investigate potential IL‐6‐mediated effects that might impact vessel wall dilation. Using histological staining, we observed significantly fewer elastin fiber breaks in the DKO mice relative to mgR/mgR homozygous mice (Figure 5A, P<0.05). To evaluate structural elements of the aortic wall more accurately, we performed 3‐dimensional imaging of ex vivo ascending aortas using MPAM and SHG microscopy. These techniques allowed us to reconstruct the vessel wall to a depth of ≈150 μm starting at the adventitial surface in a 320×320 μm field of view (Figure 5B). In contrast to the large hole‐like elastin structures in mgR/mgR mice, the DKO mice developed only small gaps in the elastin sheets (Top panel, Figure 5C) that were considerably smaller in the DKO mice (Figure 5D, P<0.001). Additionally, there was improved preservation of collagen fibers in the adventitia as indicated by SHG (Figure 5C, middle panel; 5D, P<0.0001), and there were fewer autofluorescent infiltrating cells in the border between media and adventitia (Figure 5C, bottom panel).
Figure 5.
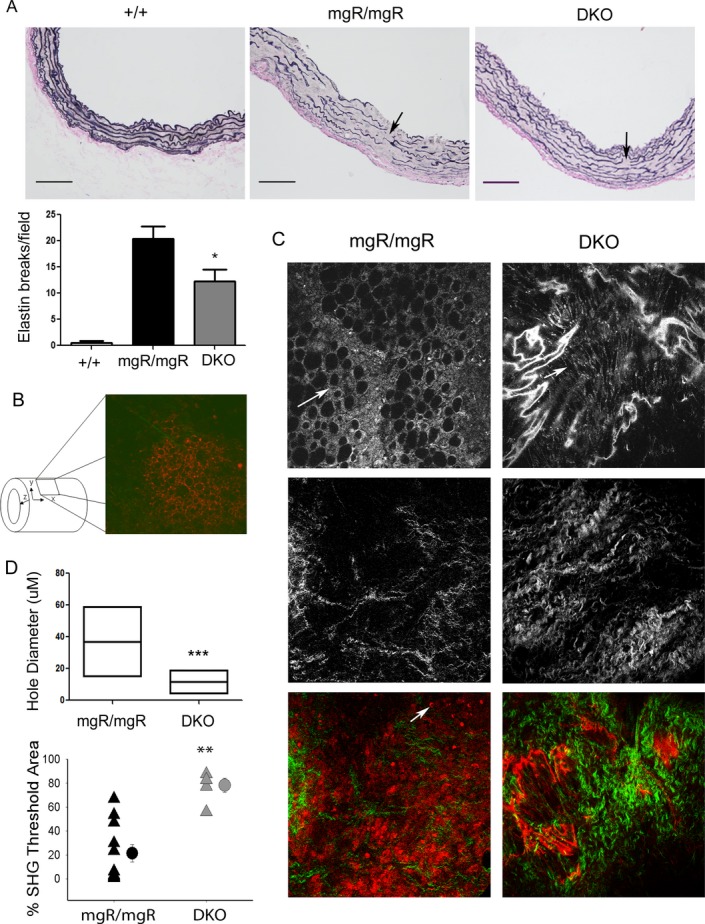
ECM degeneration in mgR/mgR and DKO mice. A, Elastin staining of ascending aortic sections of wild‐type (n=6), mgR/mgR (n=5), and DKO mice (n=6). Scale bar is 100 μm. Bottom panel: number of breaks in elastin fibers was quantified. *P<0.003 vs ++; **P<0.05 vs mgR/mgR. B, Schematic diagram of SHG/MPAM microscopy of ascending aortic samples. C, SHG and MPAM microscopic images of mgR/mgR (n=6) and age‐matched DKO (n=4) mice. Top panel: Elastin content was shown by MPAM at the wavelength of 770 μm. Round and large hole formation is evident in mgR/mgR. In DKOs, small gaps were found in elastin sheets. Arrows: Degeneration tears and holes in elastin sheets. Middle panel: Collagen fibers. Bottom panel: Combined images of collagen and elastin at the adventitia‐media border. Collagen was shown in green and elastin was shown in red. Arrow: Cell infiltration was more pronounced at the adventitia‐media border in mgR/mgR mice. Representative images are shown for each group. Images were taken in 320×320 μm. D, Sizes of holes and gaps in elastin sheets, indicators of elastin degradation, were analyzed and quantified using MetaMorph Premier S software. Hole diameter is represented as a box plot with the middle horizontal line representing the mean value, and the upper and lower lines representing the maximum and minimum for each group, respectively. n=100 in mgR/mgR and 133 in DKO group. Collagen content in aortic samples was measured as percentage of SHG signals. Triangles represent individual samples and circles represent the mean of each group. ***P<0.001; **P<0.0001 by unpaired t test. DKO indicates double knockout; ECM, extracellular matrix; MPAM, multiphoton autofluorescence microscopy; SHG, second harmonic generation.
MMP‐9 Activity is Reduced in mgR/mgR Mice With IL‐6 Deficiency
We evaluated activities of MMP‐2 and MMP‐9 in aortic extracts to test for improved ECM preservation in the DKO mice. Gelatin zymography using ascending aortic extracts from different genotypes showed significantly higher activities of both MMP‐2 and MMP‐9 in mgR/mgR mice versus wild‐type (++) mice (P<0.05 and P<0.001 for MMP‐2 and MMP‐9, respectively). Interestingly, there was a higher activity of MMP‐9, but not MMP‐2, in mgR/+ relative to wild‐type littermates (Figure 6B, P<0.02), a phenomenon not associated with an increase in vessel rupture in this group. IL‐6 deficiency in mgR homozygous mice significantly decreased MMP‐9 activity but did not significantly affect MMP‐2 activity. To determine the location of MMP‐9, we performed IHC analysis on ascending aortic sections of wild‐type, mgR/mgR, and DKO mice. We found increased immunostaining of MMP‐9 in the adventitia and media of mgR and DKO aortas, sites of significant collagen and elastin degeneration, respectively (Figure 5C). Also, there was a significant increase in F4/80+ macrophages in mgR/mgR and DKO aortas compared to wild‐type, but no difference could be discerned between mgR homozygous and DKO aortas. Since IL‐6 signaling is known to affect bone resorption, we also analyzed the skeletal structures of mgR/mgR and DKO mice via CT imaging (Figure 6D). We found kyophoscoliosis in mgR/mgR mice, as reported previously,20 but found no apparent difference between mgR/mgR and DKO mice in the small sample we assessed.
Figure 6.

IL‐6 deficiency decreases MMP‐9 activity in DKO vs mgR/mgR homozygous mice but does not affect skeletal abnormality. A, Representative gelatin zymography of age‐matched wild‐type (++), mgR/+, mgR/mgR, and DKO mice. Activities of pro‐MMP‐2, MMP‐2 and MMP‐9 are shown. B, Quantification of MMP‐2 and ‐9 activity in different genotypes. n=6 for wild‐type and mgR+ and n=4 for mgR/mgR and DKO. *P<0.05 vs ++; ‡P<0.02 vs ++; #P<0.001 vs ++ and mgR/+; ##P<0.001 vs ++, mgR/+, and DKO. C, Immunohistochemistry for MMP‐9 and F4/80+ macrophages in wild‐type, mgR/mgR and DKO ascending aortas. Lumen is at the top. n=3 per group. Scale bar is 50 μm. Magnification is ×400. D, Representative CT images of mgR/mgR and DKO mice (n=2 per group). CT indicates computed tomography; DKO, double knockout; IL, interleukin; MMP‐9, matrix metalloproteinase‐9.
Discussion
Acute aortic dissections are the major cause of premature deaths in patients with MFS. One of the goals for developing treatments for MFS is to delay ascending aortic aneurysm formation and prevent aortic dissections. In this study, we demonstrated activation of IL‐6‐mediated inflammatory signaling in the medial layer of the ascending aorta, the site of greatest vessel dilation and aneurysm formation in this homozygous mgR/mgR mouse model of MFS. We confirmed recruitment of macrophages and increased activities of MMP‐2 and MMP‐9 that contribute to increased elastin and collagen degradation in this animal model. Furthermore, IL‐6 deficiency in this model improved aortic pathology by partially preserving ECM structure, reducing MMP‐9 expression, and decreasing progression of aortic dilation at later stages. Despite these beneficial effects of IL‐6 deficiency, DKO mice did not have significantly improved survival versus the hypomorphic mgR/mgR homozygous mice, suggesting that the IL‐6–MMP‐9 pathway is a contributor to aneurysm formation, but is only one of many pathways involved in the disease progression. Therefore, inhibition of multiple signaling pathways may be necessary to prevent aortic rupture and enhance survival in this mouse model of MFS.
Recent studies have identified an inflammatory component in the etiology of TAA.34 In TAA patients undergoing surgical repair, enhanced expression of cytokines, such as IL‐6 and interferon‐γ (IFN‐γ), are found in aortic tissues. These events are spatially correlated with increased monocyte/macrophage accumulation and enhanced MMP production.11 Enhanced Ang II signaling, a potent inducer of cytokines and chemokines, has been linked to MFS.6–7 In the aorta, Ang II activates NF‐κB to regulate various inflammatory molecules including proinflammatory cytokines (eg, interleukins), chemokines (MCP‐1, GM‐CSF), adhesion molecules (E‐selectin, ICAM‐1, VCAM‐1), and ECM‐degrading MMPs. A major target of Ang II is to activate IL‐6 expression by VSMCs, fibroblasts and recruited monocytes.35 In the vessel, IL‐6 promotes ROS production and macrophage recruitment and differentiation, partially through induction of MCP‐1.11,15 We observed both induction of IL‐6 and MCP‐1, and increased macrophage infiltration in the aorta of mgR/mgR mice.
Homozygous mgR mice spontaneously die of dissecting TAAs that exhibit leukocytic infiltration at advanced stages of disease progression.20 Infiltrating leukocytes may accelerate pathogenesis by producing MMPs that are capable of degrading the important structural components of the aortic wall, including elastin and collagen. Matrix degenerated products, in turn, may be able to induce macrophage chemotaxis,22,36 amplifying local inflammatory cascades. Furthermore, in patients with MFS, matrix‐degrading MMPs are elevated in aortic tissue and are associated with destruction of matrix structural macromolecules such as elastin and collagen.20,37–38 Of numerous MMPs, MMP‐2 and MMP‐9 expression correlates with the stage of aneurysmal dilation.39 They are capable of degrading elastin and collagen in the aortic wall, but are also able to catalyze activation of TGF‐β, further contributing to the disease pathogenesis.40–41 Since we observed a decrease in aortic dilation in mgR/mgR mice that were deficient in IL‐6, we further examined whether this beneficial effect was due to a decrease in activities of MMP‐2 and ‐9. Our results indicate that IL‐6 deficiency decreases only MMP‐9 activity and that MMP‐9 localizes to both the adventitial and medial layers of mgR homozygous and DKO aortas. Since the amount of MMP‐9 based on immunohistochemistry increases similarly in both mgR/mgR and DKO aortas, these findings suggest that IL‐6 plays a pivotal role in the activation of MMP‐9. In addition, localization of MMP‐9 to the adventitial and medial layer suggests that MMP‐9 contributes to both the collagen and elastin fiber degradation that was observed by SHG/MPAM microscopy. Since macrophages localized mostly to the adventitia, MMP‐9 is likely being produced by VSMCs in the media and by macrophages in the adventitia. Similar observations have been made in a bone resorption study42 and in cancer43 where IL‐6 signaling was found to regulate mRNA expression and biological activities of MMP‐2 and ‐9 through the soluble IL‐6 receptor. It remains unclear why IL‐6 deficiency did not affect MMP‐2 activity in DKO aortas. MMP‐2 is produced by medial VSMCs and adventitial fibroblasts,44 and its level is significantly elevated in TAA in MFS.20,45 MMP‐2 is capable of catalyzing the release of TGF‐β from the latency‐associated peptide (LAP) causing activation of TGF‐β. Excess TGF‐β signaling is detrimental for the aortic wall in MFS because neutralization of TGF‐β prevents medial hypertrophy and elastin fragmentation. Since MMP‐2 activity in DKO mice is similar to mgR/mgR, it is conceivable that dysregulated TGF‐β signaling remains unhindered in the DKO mice, offering another explanation for the lack of any benefit in survival.
Loss of IL‐6 had only a minor impact on the significantly increased macrophage influx observed in the hypomorphic mgR/mgR mice despite the beneficial effect on MMP activity. This finding is not surprising because IL‐6 is not a chemotactic cytokine, and suggests that other chemotactic factors, such as GxxPG‐fragment of fibrillin‐1 may contribute to monocyte recruitment. We note that similar levels of F480+ macrophage accumulation were observed between mgR/mgR and DKO mice. In spite of this finding, it is interesting that loss of IL‐6 does affect the accumulated macrophages by significantly decreasing their MMP‐9 activation.
Our SHG/MPAM observations of the appearance of holes in the elastin sheets and disruption of the collagen fibers are consistent with the increased MMP activity shown in Figure 6. The observation that the DKO mice had a significant reduction in the number of elastin breaks (quantification of Figure 5A), but that the total number of breaks remained elevated above wild‐type control mice suggests that additional mechanisms in addition to IL‐6 contribute to the structural degeneration of the aortic wall in the mgR/mgR mice. This conclusion is consistent with the MMP activity shown in Figure 6, in which IL‐6 knockout in the mgR/mgR mice resulted in significant decline in MMP‐9 activity, but both MMP‐2 and MMP‐9 remained significantly above the level observed in wild‐type mice. These findings suggest multiple pathophysiological mechanisms, in addition to IL‐6, are responsible for the aneurysm formation and morbidity in this animal model. For example, inhibition of AT1R with losartan and inhibition of MMPs with doxycyline confer protection from aortic dilation and early death in mgR/mgR mice. This beneficial effect is additive when treating with losartan plus doxycycline,46 suggesting that angiotensin II signaling, activation of downstream TGF‐β pathway, and activation of MMPs are the main drivers of TAAD in Marfan syndrome. Furthermore, our data suggest that although IL‐6 is involved in pathological remodeling of the vessel wall, it may not be a critical component of vascular signaling in Marfan syndrome. Our finding also raises the possibility that inhibiting aortic dilation after an undefined threshold has been crossed may not confer protection from dissection and rupture. This finding has broader implications clinically and implies that prevention of aortic dilation is imperative in early stages of aneurysm formation.
In summary, our data suggest that in late stage of TAA in the mgR/mgR homozygous mice, increased IL‐6 production contributes significantly to the aortic ECM degeneration and increased activity of MMP‐9, thus contributing to aneurysmal dilation of the ascending aorta. IL‐6 deficiency delayed dilation of the ascending aorta, but did not prevent rupture and did not increase survival, suggesting the involvement of other important contributors to the earlier stages of disease progression.
Sources of Funding
This work was supported by the National Institutes of Health (P50 HL083794 to Drs Brasier and Milewicz, HL70925 to Dr Brasier, DK079053 to Dr Tilton) and the Ted Nash Long Life Foundation to Dr Brasier.
Disclosures
None.
Acknowledgments
We would like to acknowledge the assistance of Dr Massoud Motamedi, Igor Patrikeev, and Jingna Wei in the Center for Biomedical Engineering. We also acknowledge Dr Heidi Spratt for professional help on statistical analysis of data.
References
- 1.Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005; 139C:4-9 [DOI] [PubMed] [Google Scholar]
- 2.Williams A, Davies S, Stuart AG, Wilson DG, Fraser AG. Medical treatment of Marfan syndrome: a time for change. Heart. 2008; 94:414-421 [DOI] [PubMed] [Google Scholar]
- 3.Yetman AT. Cardiovascular pharmacotherapy in patients with Marfan syndrome. Am J Cardiovasc Drugs. 2007; 7:117-126 [DOI] [PubMed] [Google Scholar]
- 4.Hirata K, Triposkiadis F, Sparks E, Bowen J, Wooley CF, Boudoulas H. The Marfan syndrome: abnormal aortic elastic properties. J Am Coll Cardiol. 1991; 18:57-63 [DOI] [PubMed] [Google Scholar]
- 5.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin‐1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995; 4 Spec No:1799-1809 [DOI] [PubMed] [Google Scholar]
- 6.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006; 312:117-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011; 332:361-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones KG, Brull DJ, Brown LC, Sian M, Greenhalgh RM, Humphries SE, Powell JT. Interleukin‐6 (IL‐6) and the prognosis of abdominal aortic aneurysms. Circulation. 2001; 103:2260-2265 [DOI] [PubMed] [Google Scholar]
- 9.Dawson J, Cockerill GW, Choke E, Belli AM, Loftus I, Thompson MM. Aortic aneurysms secrete interleukin‐6 into the circulation. J Vasc Surg. 2007; 45:350-356 [DOI] [PubMed] [Google Scholar]
- 10.Dawson J, Cockerill G, Choke E, Loftus I, Thompson MM. Aortic aneurysms as a source of circulating interleukin‐6. Ann N Y Acad Sci. 2006; 1085:320-323 [DOI] [PubMed] [Google Scholar]
- 11.Tieu BC, Lee C, Sun H, LeJeune W, Recinos A, III, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL‐6/MCP1 amplification loop accelerates macrophage‐mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009; 119:3637-3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison SC, Smith AJ, Jones GT, Swerdlow DI, Rampuri R, Bown MJ, Folkersen L, Baas AF, de Borst GJ, Blankensteijn JD, Price JF, van der Graaf Y, McLachlan S, Agu O, Hofman A, Uitterlinden AG, Franco‐Cereceda A, Ruigrok YM, Van't Hof FN, Powell JT, van Rij AM, Casas JP, Eriksson P, Holmes MV, Asselbergs FW, Hingorani AD, Humphries SE. Interleukin‐6 receptor pathways in abdominal aortic aneurysm. Eur Heart J. 2013; 34:3707-3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, Pekalski ML, Chen WM, Onengut‐Gumuscu S, Rich SS, Butterworth AS, Malarstig A, Danesh J, Todd JA. Functional IL6R 358Ala allele impairs classical IL‐6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013; 9:e1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brasier AR. The nuclear factor‐kappaB‐interleukin‐6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010; 86:211-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, Mantovani A, Lazzarin A, Sozzani S, Poli G. Interleukin‐6 induces monocyte chemotactic protein‐1 in peripheral blood mononuclear cells and in the U937 cell line. Blood. 1998; 91:258-265 [PubMed] [Google Scholar]
- 16.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998; 394:894-897 [DOI] [PubMed] [Google Scholar]
- 17.Ishibashi M, Egashira K, Zhao Q, Hiasa K, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K. Bone marrow‐derived monocyte chemoattractant protein‐1 receptor CCR2 is critical in angiotensin II‐induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2004; 24:e174-e178 [DOI] [PubMed] [Google Scholar]
- 18.He R, Guo DC, Estrera AL, Safi HJ, Huynh TT, Yin Z, Cao SN, Lin J, Kurian T, Buja LM, Geng YJ, Milewicz DM. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006; 131:671-678 [DOI] [PubMed] [Google Scholar]
- 19.He R, Guo DC, Sun W, Papke CL, Duraisamy S, Estrera AL, Safi HJ, Ahn C, Buja LM, Arnett FC, Zhang J, Geng YJ, Milewicz DM. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg. 2008; 136:922-929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin‐1. Proc Natl Acad Sci USA. 1999; 96:3819-3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radonic T, de Witte P, Groenink M, de Waard V, Lutter R, van Eijik M, Jansen M, Timmermans J, Kempers M, Scholte AJ, Hilhorst‐Hofstee Y, van den Berg MP, van Tintelen JP, Pals G, Baars MJ, Mulder BJ, Zwinderman AH. Inflammation aggravates disease severity in Marfan syndrome patients. PLoS One. 2012; 7:e32963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo G, Booms P, Halushka M, Dietz HC, Ney A, Stricker S, Hecht J, Mundlos S, Robinson PN. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG‐containing fibrillin‐1 fragment. Circulation. 2006; 114:1855-1862 [DOI] [PubMed] [Google Scholar]
- 23.Booms P, Ney A, Barthel F, Moroy G, Counsell D, Gille C, Guo G, Pregla R, Mundlos S, Alix AJ, Robinson PN. A fibrillin‐1‐fragment containing the elastin‐binding‐protein GxxPG consensus sequence upregulates matrix metalloproteinase‐1: biochemical and computational analysis. J Mol Cell Cardiol. 2006; 40:234-246 [DOI] [PubMed] [Google Scholar]
- 24.Booms P, Pregla R, Ney A, Barthel F, Reinhardt DP, Pletschacher A, Mundlos S, Robinson PN. RGD‐containing fibrillin‐1 fragments upregulate matrix metalloproteinase expression in cell culture: a potential factor in the pathogenesis of the Marfan syndrome. Hum Genet. 2005; 116:51-61 [DOI] [PubMed] [Google Scholar]
- 25.Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton‐excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA. 2003; 100:7075-7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campagnola PJ, Loew LM. Second‐harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol. 2003; 21:1356-1360 [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc. 2012; 7:654-669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulesteix T, Pena AM, Pages N, Godeau G, Sauviat MP, Beaurepaire E, Schanne‐Klein MC. Micrometer scale ex vivo multiphoton imaging of unstained arterial wall structure. Cytometry A. 2006; 69:20-26 [DOI] [PubMed] [Google Scholar]
- 29.Edward K, Qiu S, Resto V, McCammon S, Vargas G. In vivo layer‐resolved characterization of oral dysplasia via nonlinear optical micro‐spectroscopy. Biomed Opt Express. 2012; 3:1579-1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis V, Persidskaia R, Baca‐Regen L, Itoh Y, Nagase H, Persidsky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase‐2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998; 18:1625-1633 [DOI] [PubMed] [Google Scholar]
- 31.Hu X, Beeton C. Detection of functional matrix metalloproteinases by zymography. J Vis Exp. 2010; 4510.3791/2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vargas G, Shilagard T, Ho KH, McCammon S. Multiphoton autofluorescence microscopy and second harmonic generation microscopy of oral epithelial neoplasms. Conf Proc IEEE Eng Med Biol Soc. 2009; 2009:6311-6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor‐stromal interface facilitates local invasion. BMC Med. 2006; 4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ejiri J, Inoue N, Tsukube T, Munezane T, Hino Y, Kobayashi S, Hirata K, Kawashima S, Imajoh‐Ohmi S, Hayashi Y, Yokozaki H, Okita Y, Yokoyama M. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res. 2003; 59:988-996 [DOI] [PubMed] [Google Scholar]
- 35.Han Y, Runge MS, Brasier AR. Angiotensin II induces interleukin‐6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor‐kappa B transcription factors. Circ Res. 1999; 84:695-703 [DOI] [PubMed] [Google Scholar]
- 36.Guo G, Gehle P, Doelken S, Martin‐Ventura JL, von Kodolitsch Y, Hetzer R, Robinson PN. Induction of macrophage chemotaxis by aortic extracts from patients with Marfan syndrome is related to elastin binding protein. PLoS One. 2011; 6:e20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikonomidis JS, Jones JA, Barbour JR, Stroud RE, Clark LL, Kaplan BS, Zeeshan A, Bavaria JE, Gorman JH, III, Spinale FG, Gorman RC. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006; 114:I365-I370 [DOI] [PubMed] [Google Scholar]
- 38.Segura AM, Luna RE, Horiba K, Stetler‐Stevenson WG, McAllister HA, Jr, Willerson JT, Ferrans VJ. Immunohistochemistry of matrix metalloproteinases and their inhibitors in thoracic aortic aneurysms and aortic valves of patients with Marfan's syndrome. Circulation. 1998; 98:II331-II337 [PubMed] [Google Scholar]
- 39.Petersen E, Wagberg F, Angquist KA. Proteolysis of the abdominal aortic aneurysm wall and the association with rupture. Eur J Vasc Endovasc Surg. 2002; 23:153-157 [DOI] [PubMed] [Google Scholar]
- 40.Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta‐binding protein. J Cell Biol. 2006; 175:111-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, Monticone R, Lakatta EG. Matrix metalloproteinase 2 activation of transforming growth factor‐beta1 (TGF‐beta1) and TGF‐beta1‐type II receptor signaling within the aged arterial wall. Arterioscler Thromb Vasc Biol. 2006; 26:1503-1509 [DOI] [PubMed] [Google Scholar]
- 42.Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, Kamoi K, Suda T. Regulation of matrix metalloproteinases (MMP‐2, ‐3, ‐9, and ‐13) by interleukin‐1 and interleukin‐6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology. 1998; 139:1338-1345 [DOI] [PubMed] [Google Scholar]
- 43.Kossakowska AE, Edwards DR, Prusinkiewicz C, Zhang MC, Guo D, Urbanski SJ, Grogan T, Marquez LA, Janowska‐Wieczorek A. Interleukin‐6 regulation of matrix metalloproteinase (MMP‐2 and MMP‐9) and tissue inhibitor of metalloproteinase (TIMP‐1) expression in malignant non‐Hodgkin's lymphomas. Blood. 1999; 94:2080-2089 [PubMed] [Google Scholar]
- 44.Rizas KD, Ippagunta N, Tilson MD., III Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009; 17:201-210 [DOI] [PubMed] [Google Scholar]
- 45.Nataatmadja M, West J, West M. Overexpression of transforming growth factor‐beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aortic aneurysm. Circulation. 2006; 114:I371-I377 [DOI] [PubMed] [Google Scholar]
- 46.Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP‐2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res. 2012; 110:e92-e101 [DOI] [PMC free article] [PubMed] [Google Scholar]