

Abstract
Background
Atherothrombosis is associated with platelet hyperactivity. Hypertriglyceridemia and insulin resistance (IR) are features of polycystic ovary syndrome (PCOS). The effect of induced hypertriglyceridemia on IR and platelet function was examined in young women with PCOS.
Methods and Results
Following overnight fasting, 13 PCOS and 12 healthy women were infused with saline or 20% intralipid for 5 hours on separate days. Insulin sensitivity was measured using a hyperinsulinemic euglycaemic clamp in the final 2 hours of each infusion. Platelet responses to adenosine diphosphate (ADP) and prostacyclin (PGI2) were measured by flow cytometric analysis of platelet fibrinogen binding and P‐selectin expression using whole blood taken during each infusion (at 2 hours) and at the end of each clamp. Lipid infusion increased triglycerides and reduced insulin sensitivity in both controls (median, interquartile range ) (5.25 [3.3, 6.48] versus 2.60 [0.88, 3.88] mg kg−1 min−1, P<0.001) and PCOS (3.15 [2.94, 3.85] versus 1.06 [0.72, 1.43] mg kg−1 min−1, P<0.001). Platelet activation by ADP was enhanced and ability to suppress platelet activation by PGI2 diminished during lipid infusion in both groups when compared to saline. Importantly, insulin infusion decreased lipid‐induced platelet hyperactivity by decreasing their response to 1 μmol/L ADP (78.7% [67.9, 82.3] versus 62.8% [51.8, 73.3], P=0.02) and increasing sensitivity to 0.01 μmol/L PGI2 (67.6% [39.5, 83.8] versus 40.9% [23.8, 60.9], P=0.01) in controls, but not in PCOS.
Conclusion
Acute hypertriglyceridemia induced IR, and increased platelet activation in both groups that was not reversed by insulin in PCOS subjects compared to controls. This suggests that platelet hyperactivity induced by acute hypertriglyceridemia and IR could contribute athero‐thrombotic risk.
Clinical Trial Registration
URL: www.isrctn.org. Unique Identifier: ISRCTN42448814.
Keywords: hypertriglyceridemia, insulin resistance, intralipid, PCOS, platelet activation
Introduction
Insulin resistance (IR), defined as resistance to insulin‐stimulated glucose uptake, is a common precursor to both diabetes and associated vascular disease.(2002)–(2007) IR is a multisystem disorder that is associated with multiple metabolic and cellular alterations including atherogenic dyslipidemia, glucose intolerance, and a prothrombotic state. This triad of factors predisposes patients with IR to accelerated atherothrombosis.(1998)–(1996) Dyslipidemia is very common in women with polycystic ovary syndrome (PCOS),(1985) a disorder in which IR occurs in 95% of obese cases.(2004) Consequently, subjects with PCOS are susceptible to impaired glucose tolerance (IGT), metabolic syndrome (MBS), and type 2 diabetes mellitus (T2DM)(1999)–(2007) that are associated with premature cardiovascular (CVD) related mortality.(2004)–(1999)
Postprandial triglyceride levels are strongly and positively correlated to incidence of coronary artery disease diagnosed by coronary angiography.(1992) Lowering plasma triglyceride concentrations by fibrates in patients with either isolated hypertriglyceridemia or atherogenic metabolic dyslipidemia significantly reduces CVD events.(2010) However, the nature of the link between hypertriglyceridemia and CVD is poorly defined. Hypertriglyceridemia may play an indirect role in the causation of atherosclerosis(1998) being a central component of the “atherogenic dyslipidemia triad” together with reduced levels of high‐density lipoprotein cholesterol (HDL‐c) and a preponderance of small dense low‐density lipoprotein cholesterol (LDL‐c).(1999) It is also possible that hypertriglyceridemia may have a direct effect on atherosclerosis as it has been proven that triglycerides are present in the form of triglyceride‐rich lipoproteins in the aorta,(1991) in atherosclerotic lesions,(1994) and in the foam cells formed by macrophages(2005) in previous studies.
The procoagulant state associated with IR is thought, at least in part, to be associated with platelet hyperactivity.(1990) In healthy blood vessels, platelet activation is counterbalanced by inhibitory signalling cascades that are activated by endothelial‐derived nitric oxide (NO) and prostacyclin (PGI2), which modulate excessive activation,(1977) although there is potential for platelet resistance to these key inhibitory factors in some disease states. The effective compliance of platelet function at all times is critical since, in addition to their classical role in acute thrombosis, platelets are involved in the initiation of atheroma, modulation of inflammatory responses, and contribute to endothelial dysfunction.(2002) Platelets can adhere to intact activated endothelium in the absence of exposed extracellular matrix proteins.(1997) These adherent platelets may play a critical role in atherogenesis by the secretion of the chemokines CCL5 (RANTES), CXCL4 (platelet factor 4), and interleukin‐1.(2000)
Women with PCOS are phenotypically similar to those with metabolic syndrome and diabetes as 80% of them are centrally obese, two thirds have low HDL‐c, and one third have hypertriglyceridemia.(2006)–(2005) Platelet hyperactivity was observed in lean women with PCOS(2003) and in subjects with T2DM.(1995) In the present study we examine the potential link between dyslipidemia, IR, and platelet hyperactivity. Since familial hypertriglyceridemia shows increased platelet activation in response to adenosine diphosphate (ADP) and collagen,(1986) and given that IR also shows this response in diabetes,(1990) we hypothesized that experimentally enhanced hypertriglyceridemia, using intralipid, will affect both IR and platelet function in subjects with PCOS compared to control subjects.
Materials and Methods
Study Subjects
Thirteen PCOS patients and 12 healthy women gave their written informed consent to participate in this study that was approved by the local Research Ethics Committee. PCOS patients were recruited from the endocrine clinics and healthy volunteers were recruited through advertisements at the local University and the hospital intranet. PCOS was diagnosed by the presence of 2 of 3 criteria, oligomenorrhoea, clinical and/or biochemical hyperandrogenism, and polycystic ovaries on ultrasound after exclusion of other endocrine causes of hyperandrogenism according to the Rotterdam diagnostic criteria.(2004) All subjects were nonsmokers, took no regular medications, and had no concurrent illness. All the subjects were asked not to use drugs containing acetylsalicylic acid during the week preceding the experiments. Subjects with impaired glucose regulation on an oral glucose tolerance test at screening were excluded.
Study Design
Following a 12‐hour overnight fast, all participants underwent a 5‐hour saline infusion with insulin sensitivity assessed by a hyperinsulinemic euglycaemic clamp in the final 2 hours. One week later, this was repeated replacing saline with intralipid (20% soybean oil, 1.2% egg yolk phospholipids, and 2.2% glycerol; Kabi Fresenius Pharmacia). Briefly, an 18‐gauge intravenous cannula was inserted into an antecubital vein for administration of test infusions and a retrograde cannula was inserted into the dorsal hand vein on the contralateral hand. The contralateral hand was heated (60°C) to arterialize venous blood for the measurement of blood glucose. After fasted blood samples were taken, either normal saline 1.5 mL/minute or 20% intralipid 1.5 mL/minute along with unfractionated heparin sodium 0.3 units/kg/minute were infused for 5 hours. At 180 minutes, a 2‐hour hyperinsulinemic euglycemic clamp was commenced using intravenous soluble insulin (Humulin S, Eli Lilly and Co.) at a rate of 80 mU/m2 body surface area/minute for the first 20 minutes to suppress the hepatic glucose production followed by a constant rate of 40 mU/m2 surface area/minute for 100 minutes. Plasma glucose was clamped at 5.0 mmol/L with a variable infusion rate of 20% dextrose. Dextrose infusion rate was adjusted based on arterialized blood glucose measurements undertaken every 5 minutes.
Whole blood for platelet study was taken at 2 hours after commencing saline or lipid infusion (before the insulin clamp was initiated) and at 5 hours when the infusions ended. Briefly, the first 2 mL of blood were discarded in order to avoid artefactual platelet activation and the second 4.5 mL were taken into a syringe filled with 0.5 mL of 3.8% trisodium citrate and used for platelet analysis. Blood samples for biochemical measurements were taken at baseline and hourly for 5 hours during the study. At each sampling point, blood samples were centrifuged for at 1500g for 15 minutes and supernatant plasma was alloquited and stored at −80°C until analysis.
Flow Cytometric Analysis of Platelet Activation
Platelet immunostaining was performed in a manner designed to minimize sample manipulation and artefactual ex vivo activation. Briefly, immunostaining was completed within 5 minutes of venepuncture and flow cytometric analysis carried out within 2 hours of fixation. Citrated whole blood was immediately added to assay tubes containing 4‐(2‐hydroxyethyl)‐piperazine‐1‐ethanesulfonic acid (HEPES) buffer (NaCl 150 mmol/L, KCl 5 mmol/L, MgSO4 1 mmol/L, HEPES 10 mmol/L, pH adjusted to 7.4 using 1 mol/L HCl) and a saturating concentration of appropriate antibodies. Fluorescein isothiocyanate‐ (FITC)‐conjugated anti‐CD42b monoclonal antibody, activation‐independent platelet specific antibody, was used to detect platelets, FITC rabbit antifibrinogen antibody for assessment of platelet fibrinogen binding and R‐Phycoerythrin (PE)‐conjugated anti‐CD62P monoclonal antibody (Becton Dickinson) for measurement of P‐selectin, a marker of platelet granules release. ADP (0.1 to 10 μmol/L; Sigma) was then added to study platelets sensitivity to activation and then incubated for 10 minutes. The samples were then fixed with 500 μL of 0.2% formaldehyde. For the assessment of platelet inhibition, PGI2 (0.01 to 0.1 μmol/L) or 8‐(4‐chlorophenylthio)‐N6‐phenyladenosine‐3′, 5′‐cyclic monphosphate (8‐CPT‐6‐Phe‐cAMP) (50 to 200 μmol/L) was added to blood and incubated for 1 minute before the addition of ADP (final concentration 1 μmol/L) then incubated for 10 minutes prior to fixation.
The platelet population in whole blood was identified by its characteristic forward‐ and side‐scatter profiles and by FITC‐conjugated anti‐CD42b monoclonal antibody which stained positive >95% of the gated platelet population.(2004) Platelet activation was measured as the percentage of platelets that expressed either fibrinogen binding or P‐selectin receptors in 10 000 gated platelet populations on FACS Aria flow cytometer (Becton Dickinson) using BD FACS Diva Software analytical software. Results were mean of duplicated samples.
Biochemical Analysis
Serum insulin was assayed using a competitive chemiluminescent immunoassay (Euro/DPC). Plasma glucose was measured using a Synchon LX 20 analyzer (Beckman‐Coulter). Serum testosterone was measured by high performance liquid chromatography linked to tandem mass spectrometry (Waters Corporation), and sex hormone binding globulin (SHBG) was measured by immunometric assay with fluorescence detection on the DPC Immulite 2000 analyzer. The free androgen index was obtained as the quotient 100* Testosterone/SHBG. Total cholesterol, triglycerides, and HDL‐c were measured enzymatically using a Synchon LX 20 analyzer (Beckman‐Coulter). LDL‐c was calculated using the Friedewald equation.(1972) Nonesterified fatty acids (NEFA) were analyzed using enzymatic colorimetric methods (Wako NEFA‐H2) on a Konelab20 autoanalyzer with an interassay and the coefficient of variation was 1.4%. Homeostatic model assessment of IR (HOMA‐IR) was calculated by the formula: HOMA1‐IR=fasting plasma insulin (μU/mL)×fasting plasma glucose (mmol/L)/22.5.(1985) We assumed that endogenous glucose production was more than 90% suppressed by an acute rise of insulin level with primed insulin infusion.(1986) During the experimental procedure, we used the HemoCue glucose 201+ with plasma glucose conversion to measure blood glucose at regular intervals.(2005) Rate of glucose disposal (mg kg−1 min−1) (M), a measure of insulin sensitivity, was calculated from the means of the five 20‐minute periods from 20 to 120 minutes during the clamp using the Defronzo method.(1979)
Statistical Analysis
Statistical analysis was performed using SPSS for Windows NT, version 19.0 (SPSS Inc). Wilcoxon signed ranks test was applied to skewed variables that violated the assumptions of normality when tested with the Kolmogorov–Smirnov test and paired sample t test for normally distributed data within the group. Mann–Whitney test and the independent sample t test were used respectively for comparison between the groups. Data are presented as medians (IQR) for skewed variables and mean±SD for normally distributed variables. For all analyses, a 2‐tailed P≤0.05 was considered to indicate statistical significance. The missing values were handled by casewise deletion.
Results
Baseline characteristics of PCOS and controls are shown in Table 1. Subjects with PCOS were more overweight but not significantly different compared to controls. They had higher androgen profile, waist circumference, HOMA‐IR, and low HDL‐c, which were characteristic features of PCOS status, than controls.
Table 1.
Baseline Characteristics of the Participants
| Controls (n=12) | PCOS (n=13) | P Value | |
|---|---|---|---|
| Age, y | 24.1±5.8 | 28.0±6.3 | 0.13 |
| Body mass index, kg/m2 | 25.5±5.0 | 29.7±6.0 | 0.07 |
| Waist, cm | 80.9±12.5 | 96.5±17 | 0.02 |
| Waist:hip ratio | 0.79±0.07 | 0.86±0.02 | 0.04 |
| Systolic blood pressure, mm Hg | 116±9 | 118±11 | 0.56 |
| Diastolic blood pressure, mm Hg | 74±9 | 75±8 | 0.73 |
| Testosterone, nmol/L | 1.03±0.34 | 1.51±0.77 | 0.05 |
| Free androgen index | 1.6±1.0 | 6.6±3.2 | <0.001 |
| Sex hormone binding globulin, nmol/L | 75±30 | 28±19 | <0.001 |
| Alanine aminotransferase, IU/L | 17±5 | 27±13 | 0.01 |
| Total cholesterol, mmol/L | 4.7±0.79 | 4.1±0.63 | 0.14 |
| Triglycerides, mmol/L | 0.96±0.32 | 1.12±0.51 | 0.35 |
| HDL‐c, mmol/L | 1.58±0.48 | 1.2±0.37 | 0.06 |
| LDL‐c, mmol/L | 2.31±0.49 | 2.68±0.58 | 0.10 |
| Fasting plasma glucose, mmol/L | 4.69±0.51 | 4.74±0.45 | 0.08 |
| HOMA‐IR | 1.33±0.69 | 2.50±1.5 | 0.03 |
| NEFA, mmol/L | 0.55 (0.33, 0.66) | 0.54 (0.35, 0.54) | 0.23 |
Results are expressed as mean±SD, NEFA levels are expressed as median (IQR), P value ≤0.05 is significance of difference. HDL‐c indicates high‐density lipoprotein cholesterol; HOMA‐IR, homeostatic model assessment of insulin resistance; LDL‐c, low‐density lipoprotein cholesterol; NEFA, nonesterified fatty acids; PCOS, polycystic ovary syndrome.
Biochemical Changes During the Lipid Infusion
In the first instance, the study examined the effects of saline and intralipid infusion on the metabolic profile of the subjects (Table 2). Lipid infusion increased plasma triglyceride and NEFA levels, and subsequently decreased insulin‐stimulated glucose disposal in both PCOS patients (rate of glucose disposal 3.15 [2.94, 3.85] versus 1.06 [0.72, 1.43] mg kg−1 min−1, P<0.001) and controls (5.25 [3.30, 6.48] versus 2.60 [0.88, 3.88] mg kg−1 min−1, P≤0.001). In both groups, no significant difference in plasma glucose was seen after receiving 2 hours either saline or lipid infusion or, predictably, at 5 hours where glucose concentration was clamped at 5 mmol/L by design.
Table 2.
Biochemical Changes Following Intralipid Infusion
| Controls (n=12) | PCOS (n=13) | |||||
|---|---|---|---|---|---|---|
| Saline | Intralipid | P Value | Saline | Intralipid | P Value | |
| Glucose disposal, mg·kg·min | 5.25 (3.30, 6.48) | 2.6 (0.88, 3.9) | <0.001 | 3.15 (2.94, 3.9) | 1.06 (0.72, 1.43) | <0.001 |
| TG AUC 2 h, mmol/L | 1.67±0.37 | 5.98±1.8 | <0.001 | 2.21±1.37 | 7.23±2.83 | <0.001 |
| TG AUC 5 h, mmol/L | 3.78±0.86 | 22.5±7.8 | <0.001 | 5.29±2.99 | 23.9±11.4 | <0.001 |
| NEFA AUC 2 h, mmol/L | 1.13 (0.86, 1.36) | 3.2 (2.29, 4.0) | 0.01 | 1.1 (0.81, 12.4) | 3.30 (2.71, 4.80) | 0.01 |
| NEFA AUC 5 h, mmol/L | 2.24 (1.83, 2.86) | 12.0 (8.5, 13.5) | <0.001 | 2.26 (1.8, 2.55) | 12.4 (9.25, 1.44) | <0.001 |
Triglycerides value is expressed as means±SDs and the rest are expressed as medians (IQR). P value ≤0.05 is significance of difference. AUC indicates area under the curve; NEFA, nonesterified fatty acids; PCOS, polycystic ovary syndrome; TG, triglycerides.
Platelet Activation During the Saline Infusion
Platelet reactivity in vivo was determined by sensitivity to both endogenous activators (ADP) and inhibitors (PGI2 and 8‐CPT‐6‐Phe‐cAMP). Incubation of whole blood with ADP (0.1 to 10 μmol/L) led to a concentration‐dependent increase in platelet activation in all samples in both groups as evidenced by elevated levels of P‐selectin and fibrinogen binding. Preincubation of platelets with either PGI2 (0.01 to 0.1 μmol/L) or the direct activator of Protein Kinase A (PKA), 8‐CPT‐6‐Phe‐cAMP (50 to 200 μmol/L), resulted in concentration‐dependent decrease in ADP‐ (1 μmol/L) induced platelet activation. These results were not significantly different between controls and PCOS at baseline and after saline infusion (data not shown).
Platelet Activation During the Lipid Infusion
The effect of lipid infusion on platelet activity was examined from the blood samples taken at 2 hours of lipid infusions which as before were concomitant with insulin infusion. The lipid infusion had no effect on the basal levels of fibrinogen binding in either group. However, the propensity for ADP‐induced platelet activation was enhanced in both controls and PCOS following the infusion of lipid rather than saline (Table 3). For example, in the control group, ADP (1 μmol/L) increased fibrinogen binding to 57.9% (46.7, 68) after saline infusion, compared to 78.7% (67.9, 82.3), P=0.01, after lipid infusion. Similarly, fibrinogen binding in response to ADP was increased from 51.4% (44.4, 61.8) to 71.8% (58.7, 81), P=0.01, after lipid infusion in PCOS (Figure 1). P‐selectin expression, an alternative marker of platelet activation, showed a similar response to fibrinogen binding (Table 3). In the control group, ADP‐induced P‐selectin expression was increased from 30.5% (25, 47) to 54.1% (31.5, 63.4), P=0.02, and in PCOS from 36.2% (23.9, 47.4) to 44.9% (36, 55), P=0.02, by the lipid infusion (Figure 2).
Table 3.
Effect of 2‐Hour Intralipid Infusion on Platelets Activation
| Agents That Added | Controls (n=12) | PCOS (n=13) | ||||
|---|---|---|---|---|---|---|
| 2‐Hour Normal Saline | 2‐Hour Intralipid | P Value | 2‐Hour Normal Saline | 2‐Hour Intralipid | P Value | |
| Percentage of platelets expressed fibrinogen binding | ||||||
| Basal | 2.30 (1.93, 2.55) | 2.50 (2.34, 2.89) | 0.07 | 2.20 (2.10, 2.78) | 2.35 (2.20, 2.65) | 0.41 |
| +1 μmol/L ADP | 57.9 (46.7, 68.0) | 78.7 (67.9, 82.3) | 0.01 | 51.4 (44.4, 61.8) | 71.8 (58.7, 81.0) | 0.01 |
| +0.01 μmol/L PGI2+1 μmol/L ADP | 11.6 (8.44, 26.0) | 67.6 (39.5, 83.8) | <0.001 | 8.40 (4.90, 13.0) | 34.9 (17.1, 50.9) | <0.001 |
| +0.05 μmol/L PGI2+1 μmol/L ADP | 2.50 (1.90, 3.42) | 20.9 (4.45, 36.3) | <0.001 | 2.45 (2.08, 2.95) | 3.30 (2.75, 6.48) | 0.01 |
| +50 μmol/L 8CPT cAMP+1 μmol/L ADP | 46.4 (26.7, 58.6) | 68.5 (58.6, 72.8) | <0.001 | 35.5 (29.0, 48.5) | 50.2 (43.4, 67.0) | <0.001 |
| Percentage of platelets expressed P‐selectin | ||||||
| Basal | 1.98 (1.03,2.90) | 1.90 (1.65, 2.61) | 0.75 | 1.85 (1.28, 4.18) | 1.70 (2.50, 2.18) | 0.20 |
| +1 μmol/L ADP | 30.5 (25.2, 47.2) | 54.1 (31.5, 63.4) | 0.02 | 36.2 (23.9, 47.4) | 44.9 (35.5, 54.6) | 0.02 |
| +0.01 μmol/L PGI2+1 μmol/L ADP | 13.4 (8.35, 20.9) | 49.6 (26.4, 65.8) | <0.001 | 10.6 (8.23, 13.1) | 28.3 (13.6, 37.1) | <0.001 |
| +0.05 μmol/L PGI2+1 μmol/L ADP | 5.58 (3.24, 7.71) | 15.6 (3.15, 23.6) | 0.03 | 6.40 (4.00, 8.35) | 5.95 (4.00, 11.2) | 0.38 |
| +50 μmol/L 8CPT cAMP+1 μmol/L ADP | 30.6 (16.5, 41.0) | 45.8 (27.5, 55.9) | 0.01 | 24.4 (15.3, 36.7) | 27.9 (20.0, 48.5) | 0.05 |
Results are expressed as medians (IQR), P value ≤0.05 is significance of difference. Wilcoxon signed ranks test. ADP indicates adenosine diphosphate; cAMP, cyclic adenosine monophophate; PCOS, polycystic ovary syndrome; PGI2, prostacyclin.
Figure 1.
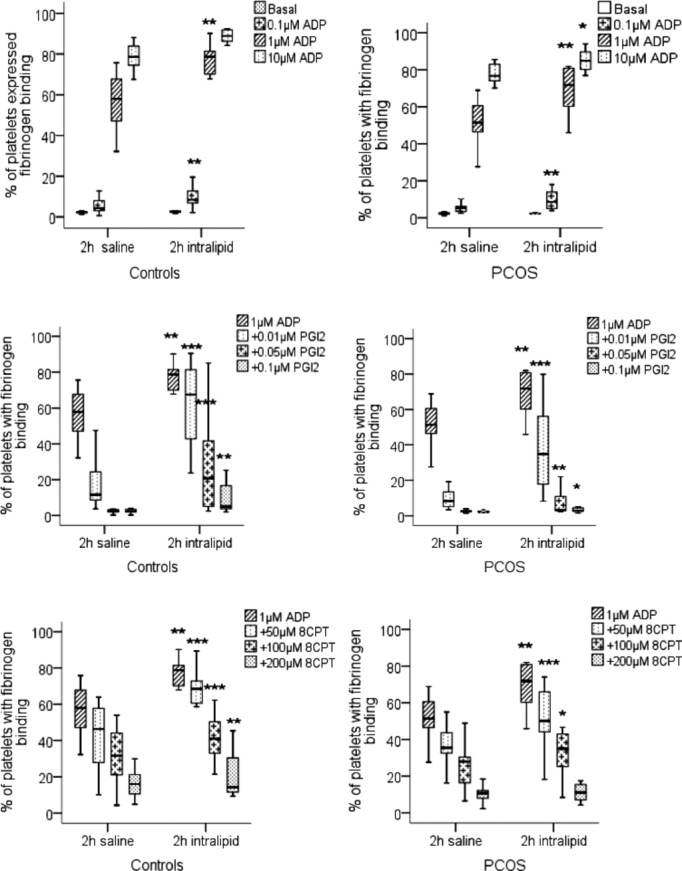
Fibrinogen binding to activated platelets in whole blood. Platelets in whole blood taken at 2 hours during saline infusion and at 2 hours during intralipid infusion from controls and women with PCOS were stimulated with ADP alone, ADP+PGI2 and ADP+8‐CPT‐6‐Phe‐cAMP (8CPT) and fibrinogen binding measured by flow cytometry. The data are expressed as percentage positive cells for fluorescence and represent median±interquartile range (box) and range (whisker). Asterisk (*P=0.05, **P=0.01, ***P<0.01) compared effect of saline to intralipid within the groups. ADP indicates adenosine diphosphate; PCOS, polycystic ovary syndrome; PGI2, prostacyclin.
Figure 2.
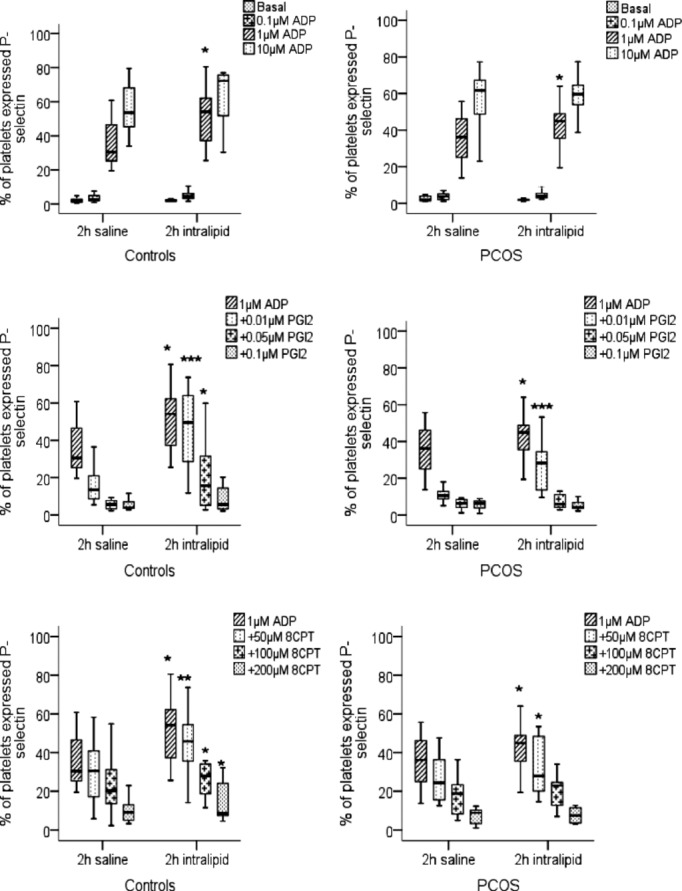
P‐selectin expression on stimulated platelets in whole blood. Whole blood taken at 2 hours during saline infusion and at 2 hours during lipid infusion from controls and women with PCOS were stimulated with ADP in the presence and absence of PGI2 or and P‐selectin expression measured by flow cytometry. The data are expressed as percentage positive cells for fluorescence and represent median±interquartile range (box) and range (whisker). Asterisk (*P=0.05, **P=0.01, ***P<0.01) compared effect of saline to lipid infusion within the group. ADP indicates adenosine diphosphate; PCOS, polycystic ovary syndrome; PGI2, prostacyclin.
To examine if this apparently enhanced platelet activation was due to increased sensitivity to agonists or diminished responsiveness to endogenous inhibitors, the ability of PGI2 (0.01 to 0.1 μmol/L) to inhibit on fibrinogen binding stimulated by ADP (1 μmol/L) was assessed. The effect of PGI2 to inhibit platelet fibrinogen binding was diminished after lipid fusion (Figure 1, Table 3). To ensure that this was not an effect of intralipid on the availability of PGI2, we repeated the experiments using 8‐CPT‐6‐Phe‐cAMP, the cell permeable cAMP analogue and a direct activator of PKA. Using 8‐CPT‐6‐Phe‐cAMP (50 μmol/L) inhibited ADP‐induced fibrinogen binding in both control and PCOS groups (Figure 1), with decreased platelet inhibition following the lipid infusion as seen for PGI2. Consistent with these data we found that the ability of both PGI2 and 8‐CPT‐6‐Phe‐cAMP to inhibit P‐selectin expression was attenuated after the lipid infusion (Figure 2, Table 3). Lipid infusions enhanced platelet hyperactivity consistently between PCOS and control groups.
Effect of Insulin Infusion on Lipid Induced Platelet Hyperactivity
The hyperinsulinemic euglycaemic clamp was performed to assess the effect of supraphysiological insulin levels on lipid‐induced increased platelet activity while maintaining plasma glucose level at 5 mmol/L in both controls and PCOS. In both groups, infusing insulin did not alter platelet activation/inhibition during the saline infusion. It is likely that platelet homeostasis is maximally optimized at the physiological milieu. However, the infusion of insulin decreased the platelet hyperactivity induced by lipid infusion in controls. Here, increased platelet fibrinogen binding in response to 1 μmol/L ADP was reduced from 78.8% (67.9, 82.3) to 62.8% (51.8, 73.3), P=0.02. Furthermore, the sensitivity of platelets to inhibition by cAMP signaling was restored. Platelet fibrinogen binding in response to ADP in the presence of PGI2 (0.01 μmol/L) was 67.6% (39.5, 83.8), but this declined to 40.9% (23.8, 60.9), P=0.01. Consistent with these data, fibrinogen binding in response to ADP in the presence of 8‐CPT‐6‐Phe‐cAMP (50 μmol/L) was 68.5% (58.6, 72.8) prior to insulin infusion and 54.0% (46.3, 67.1), P=0.02, after insulin infusion (Table 4).
Table 4.
Effect of Exogenous Insulin on Platelets Activation While Intralipid Infusion
| Agents That Added | Controls (n=12) | PCOS (n=13) | ||||
|---|---|---|---|---|---|---|
| 2‐Hour Intralipid | 5‐Hour Intralipid | P Value | 2‐Hour Intralipid | 5‐Hour Intralipid | P Value | |
| Percentage of platelets expressed fibrinogen binding | ||||||
| Basal | 2.50 (2.34, 2.89) | 2.53 (2.08, 2.96) | 1.00 | 2.35 (2.20, 2.65) | 2.35 (2.2, 2.8) | 0.92 |
| +1 μmol/L ADP | 78.7 (67.9, 82.3) | 62.8 (51.8, 73.3) | 0.02 | 71.8 (58.7, 81.0) | 68.5 (56.3, 74.3) | 0.17 |
| +0.01 μmol/L PGI2+1 μmol/L ADP | 67.6 (39.5, 83.8) | 40.9 (23.8, 60.9) | 0.01 | 34.9 (17.1, 50.9) | 31.8 (21.4, 45.4) | 0.38 |
| +0.05 μmol/L PGI2+1 μmol/L ADP | 20.9 (4.45, 36.3) | 6.78 (3.06, 26.8) | 0.05 | 3.30 (2.75, 6.48) | 4.10 (2.5, 6.1) | 0.40 |
| +50 μmol/L 8CPT cAMP+1 μmol/L ADP | 68.5 (58.6, 72.8) | 54.0 (46.3, 67.1) | 0.02 | 50.2 (43.4, 67.0) | 55.2 (48.5, 59.5) | 0.75 |
| Percentage of platelets expressed P‐selectin | ||||||
| Basal | 1.90 (1.65, 2.61) | 2.48 (1.95, 3.29) | 0.11 | 1.70 (2.50, 2.18) | 2.40 (1.78, 3.40) | 0.13 |
| +1 μmol/L ADP | 54.1 (31.5, 63.4) | 41.3 (28.0, 46.8) | 0.04 | 44.9 (35.5, 54.6) | 44.4 (37.4, 60.1) | 0.97 |
| +0.01 μmol/L PGI2+1 μmol/L ADP | 49.6 (26.4, 65.8) | 30.3 (12.5, 46.0) | 0.01 | 28.3 (13.6, 37.1) | 22.3 (18.7, 40.0) | 0.70 |
| +0.05 μmol/L PGI2+1 μmol/L ADP | 15.6 (3.15, 23.6) | 8.70 (3.65, 22.5) | 0.31 | 5.95 (4.00, 11.2) | 7.70 (5.10, 12.7) | 0.51 |
| +50 μmol/L 8CPT cAMP+1 μmol/L ADP | 45.8 (27.5, 55.9) | 35.4 (27.9, 41.0) | 0.02 | 27.9 (20.0, 48.5) | 36.7 (29.2, 45.0) | 0.13 |
Results are expressed as medians (IQR), P value ≤0.05 is significance of difference. Wilcoxon signed ranks test. ADP indicates adenosine diphosphate; cAMP, cyclic adenosine monophophate; PCOS, polycystic ovary syndrome; PGI2, prostacyclin.
In contrast, platelet hyperactivity in subjects with PCOS was not attenuated by insulin infusion. After insulin was provided, ADP‐induced fibrinogen binding remained elevated, 71.8% (58.7, 81) compared to 68.5% (56.3, 74.3), P=0.17, as did P‐selectin expression 44.9% (35.5, 54.6) versus 44.4% (37.4, 60.1), P=0.97. Consistent with the sustained platelet hyperactivity, platelet sensitivity to PGI2 and 8‐CPT‐6‐Phe‐cAMP (Table 4) remained compromised. Thus, elevated platelet activity induced by acute hyperlipidemia in PCOS was resistant to insulin and remained in the increased activated state throughout the lipid infusion (Figure 3).
Figure 3.
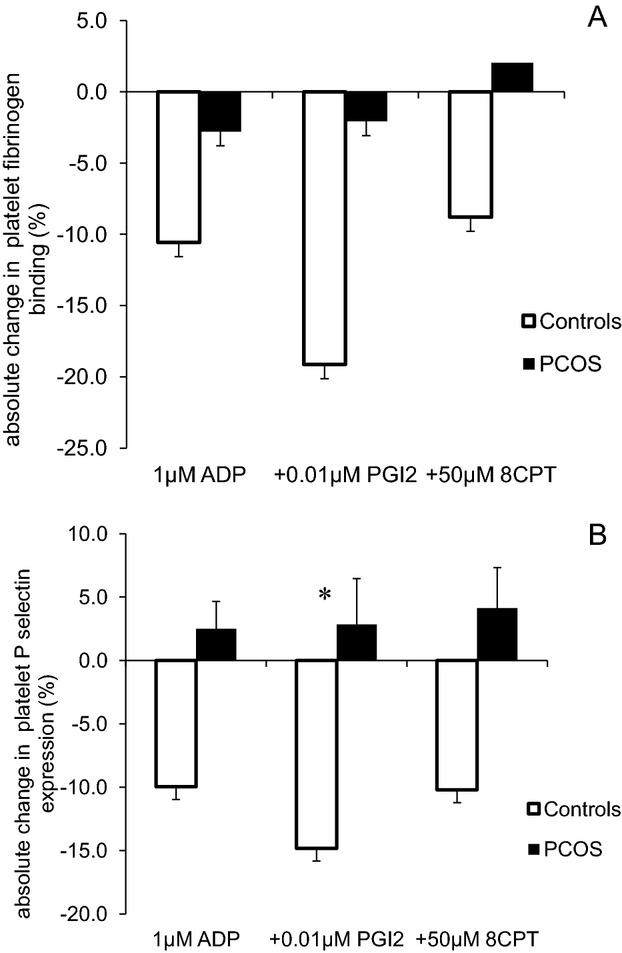
Effect of hyperinsulinaemia on intralipid induced platelet activation. Platelets in whole blood taken at 2 hours (before hyperinsulinaemic euglycaemic clamp) and at 5 hours (after the clamp) during intralipid trial were stimulated by 1 μmol/L ADP, 1 μmol/L ADP+0.01 μmol/L PGI2, and 1 μmol/L ADP+50 μmol/L 8‐CPT‐6‐Phe‐cAMP (8CPT). Absolute changes in % platelet activation were compared between controls and PCOS (A: platelet fibrinogen binding; B: platelet P‐selectin expression). Absolute change is calculated by difference of percentage of activated platelets between the samples taken before and after the clamp, data were expressed by means±SE and compared by independent sample t test between the groups, *P value ≤0.05. ADP indicates adenosine diphosphate; PCOS, polycystic ovary syndrome; PGI2, prostacyclin.
Discussion
In the present study, the lipid infusion, with an acute rise in triglyceride and NEFA levels, decreased insulin‐stimulated glucose disposal significantly in both PCOS and control subjects. Lipid infusion is well recognized to induce IR by reducing insulin‐stimulated glucose oxidation and enhancing insulin‐inhibited lipid oxidation.(1991)
The lipid infusion had no effect on baseline unstimulated platelet fibrinogen binding or P‐selectin expression in either group, suggesting that a rapid increase in plasma lipid per se did not activate platelets in vivo. Indeed, de Man et al reported that there were no differences in baseline platelet expression of P‐selectin and fibrinogen binding in patients with primary hypertriglyceridemia compared to age‐ and sex‐matched controls.(2000) Therefore, both acute and chronic rises of triglycerides do not appear to induce platelet activation at resting state.
In contrast to these baseline findings, this acute dysmetabolic state, induced hypertriglyceridemia with IR, resulted in platelets having a greater propensity for activation when challenged with ADP and a reduced sensitivity to the key physiological regulator of platelets, PGI2, not only in insulin resistant PCOS patients but also in healthy subjects. Therefore, the lipid‐induced platelet activation was not entirely related to PCOS status.
The mechanism by which acute hypertriglyceridemia influence increased platelet activation could be explained by acute lipemia priming platelets for activation by a 2‐pronged mechanism; first, platelets are more readily activated when confronted by activating stimuli, and second, platelets have reduced their sensitivity to inhibitory stimuli through cAMP‐mediated inhibition. In combination, these 2 mechanisms could exaggerate platelet responsiveness once activated, contributing to the procoagulant phenotype in dyslipidemia. Induction of hyperlipidemia, IR, and obesity, by a high fat high calorie diet, was associated with increased reactivity of platelets to ADP and collagen in Ossabaw miniature swine.(2011) Lipid infusion enhanced ADP stimulated platelet aggregation, and induced early atherosclerosis in the aorta of the laboratory rats.(1987)–(1987) Hypertriglyceridemia is associated with endothelial dysfunction and an increase in endothelial activation markers sICAM‐1, soluble E‐selectin, and von Willebrand factor,(2003) which may in turn decrease the production of endothelial NO and PGI2.(1999) Therefore, the present study suggests that the presence of high triglycerides in the circulation could act as a driver of atherosclerosis and atherothrombosis through platelet activation in PCOS as well as in controls.
This study also reported for the first time that the infusion of insulin during this hyperlipidemic state is able to rectify changes in platelet reactivity. This protective effect of insulin was only observed in control subjects, but a similar amount of insulin made no improvement in PCOS and resulted in prolonged lipid‐induced platelet activation. This could be due to PCOS status or an increased requirement of insulin dose in insulin resistant PCOS than controls to reverse this lipid‐induced IR and increased platelet activation. Trovati et al reported that the dose of insulin required to induce platelet antiaggregatory effect in patients with T2DM is 8 times higher than controls.(1995)
While reporting reduced platelet sensitivity to PGI2 during lipid infusion, this study also demonstrated that platelet inhibition through direct activation of PKA using the cell permeable activator, 8‐CPT‐6‐Phe‐cAMP, was also compromised. The mechanisms underlying the compromised PKA activity in induced insulin resistant state is unclear and requires further investigation, but suggest the IR associated with acute lipidemia supresses activity of the whole cAMP signalling pathway and that the diminished platelet response to PGI2 was likely due to a direct effect of lipid on the platelet rather than high lipid interference with the PGI2 receptor on the platelet surface.
In this study, women with PCOS were slightly older and more overweight than controls. These differences did not seem to have an impact on platelet activation because there was no difference in platelet activation at resting state, with activation with ADP, and with inhibition with PGI2 and 8‐CPT‐6‐Phe‐cAMP, at baseline (during saline infusion) between PCOS and control groups. However, effect of obesity cannot be excluded as a possible reason why PCOS failed to improve lipid‐induced platelet hyperactivity by insulin infusion.
We have examined the platelet activation and inhibition using 3 different concentrations of ADP, PGI2 and 8‐CPT‐6‐Phe‐cAMP to ensure the concentration‐dependent changes. Within group comparisons, the maximal effect of lipid infusion on platelet activation/inhibition pathways has been seen at the optimal activation dose, the dose that did not minimally or maximally stimulate platelets, of 1 μM ADP and at a lower inhibitory concentration of PGI2 and 8‐CPT‐6‐Phe‐cAMP. However, with this sample size, this study could not exclude the negative findings being due to Type II errors, especially since many expected differences, with or without lipid infusions in both groups, only just managed or just failed to reach statistical significance.
In summary, acute metabolic derangement with hypertriglyceridemia and IR induced by lipid infusion enhanced platelet activation by enhanced platelet response to ADP and increased resistance to inhibitory effect of PGI2, in both PCOS and controls. IR in PCOS appeared to deprive the apparent cardioprotective effects of insulin on platelet activation during acute lipemia that has been seen in controls. Therefore, platelet hyperactivity appears related to acute hypertriglyceridemia and IR, and thus could be a surrogate marker of athero‐thrombotic risk, especially in PCOS patients with underlying IR.
Acknowledgments
We gratefully acknowledge Hasan Kahal for his assistance in platelet work, and Paul Afolabi and John M. Jackson from Southampton NIHR Biomedical Research Centre, University Hospital Southampton NHS Foundation Trust for analyzing the NEFA samples.
Sources of Funding
The Diabetes Endowment Fund.
Disclosures
None.
References
- Bonora E, Formentini G, Calcaterra F, Lombardi S, Marini F, Zenari L, Saggiani F, Poli M, Perbellini S, Raffaelli A, Cacciatori V, Santi L, Targher G, Bonadonna R, Muggeo M. HOMA‐estimated insulin resistance is an independent predictor of cardiovascular disease in type 2 diabetic subjects: prospective data from the Verona Diabetes Complications Study. Diabetes Care. 2002; 25:1135-1141 [DOI] [PubMed] [Google Scholar]
- Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Meigs JB, Bonadonna RC, Muggeo M. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in Caucasian subjects from the general population: the Bruneck study. Diabetes Care. 2007; 30:318-324 [DOI] [PubMed] [Google Scholar]
- Austin MA. Plasma triglyceride as a risk factor for cardiovascular disease. Can J Cardiol. 1998; 14suppl B:14B-17B [PubMed] [Google Scholar]
- Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high‐density lipoprotein cholesterol level: a meta‐analysis of population‐based prospective studies. J Cardiovasc Risk. 1996; 3:213-219 [PubMed] [Google Scholar]
- Wild RA, Painter PC, Coulson PB, Carruth KB, Ranney GB. Lipoprotein lipid concentrations and cardiovascular risk in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1985; 61:946-951 [DOI] [PubMed] [Google Scholar]
- Carmina E, Lobo RA. Use of fasting blood to assess the prevalence of insulin resistance in women with polycystic ovary syndrome. Fertil Steril. 2004; 82:661-665 [DOI] [PubMed] [Google Scholar]
- Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999; 84:165-169 [DOI] [PubMed] [Google Scholar]
- Barber TM, McCarthy MI, Franks S, Wass JA. Metabolic syndrome in polycystic ovary syndrome. Endokrynol Pol. 2007; 58:34-41 [PubMed] [Google Scholar]
- Ford ES. The metabolic syndrome and mortality from cardiovascular disease and all‐causes: findings from the National Health and Nutrition Examination Survey II Mortality Study. Atherosclerosis. 2004; 173:309-314 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Eguchi H, Manaka H, Igarashi K, Kato T, Sekikawa A. Impaired glucose tolerance is a risk factor for cardiovascular disease, but not impaired fasting glucose. The Funagata Diabetes Study. Diabetes Care. 1999; 22:920-924 [DOI] [PubMed] [Google Scholar]
- Patsch JR, Miesenbock G, Hopferwieser T, Muhlberger V, Knapp E, Dunn JK, Gotto AM, Jr, Patsch W. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler Thromb. 1992; 12:1336-1345 [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Fisman EZ. “If it ain't broke, don't fix it”: a commentary on the positive‐negative results of the ACCORD Lipid study. Cardiovasc Diabetol. 2010; 9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am J Cardiol. 1998; 81:18B-25B [DOI] [PubMed] [Google Scholar]
- Grundy SM, Pasternak R, Greenland P, Smith S, Jr, Fuster V. AHA/ACC scientific statement: assessment of cardiovascular risk by use of multiple‐risk‐factor assessment equations: a statement for healthcare professionals from the American Heart Association and the American College of Cardiology. J Am Coll Cardiol. 1999; 34:1348-1359 [DOI] [PubMed] [Google Scholar]
- Shaikh M, Wootton R, Nordestgaard BG, Baskerville P, Lumley JS, La Ville AE, Quiney J, Lewis B. Quantitative studies of transfer in vivo of low density, SF 12‐60, and SF 60‐400 lipoproteins between plasma and arterial intima in humans. Arterioscler Thromb. 1991; 11:569-577 [DOI] [PubMed] [Google Scholar]
- Rapp JH, Lespine A, Hamilton RL, Colyvas N, Chaumeton AH, Tweedie‐Hardman J, Kotite L, Kunitake ST, Havel RJ, Kane JP. Triglyceride‐rich lipoproteins isolated by selected‐affinity anti‐apolipoprotein B immunosorption from human atherosclerotic plaque. Arterioscler Thromb. 1994; 14:1767-1774 [DOI] [PubMed] [Google Scholar]
- Kawakami A, Tani M, Chiba T, Yui K, Shinozaki S, Nakajima K, Tanaka A, Shimokado K, Yoshida M. Pitavastatin inhibits remnant lipoprotein‐induced macrophage foam cell formation through apoB48 receptor‐dependent mechanism. Arterioscler Thromb Vasc Biol. 2005; 25:424-429 [DOI] [PubMed] [Google Scholar]
- Tschoepe D, Roesen P, Kaufmann L, Schauseil S, Kehrel B, Ostermann H, Gries FA. Evidence for abnormal platelet glycoprotein expression in diabetes mellitus. Eur J Clin Invest. 1990; 20:166-170 [DOI] [PubMed] [Google Scholar]
- Tateson JE, Moncada S, Vane JR. Effects of prostacyclin (Pgx) on cyclic AMP concentrations in human platelets. Prostaglandins. 1977; 13:389-397 [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002; 8:1227-1234 [DOI] [PubMed] [Google Scholar]
- Gawaz M, Neumann FJ, Dickfeld T, Reininger A, Adelsberger H, Gebhardt A, Schomig A. Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation. 1997; 96:1809-1818 [DOI] [PubMed] [Google Scholar]
- Gawaz M, Brand K, Dickfeld T, Pogatsa‐Murray G, Page S, Bogner C, Koch W, Schomig A, Neumann F. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin‐1‐dependent mechanism. Implications for atherogenesis. Atherosclerosis. 2000; 148:75-85 [DOI] [PubMed] [Google Scholar]
- Ehrmann DA, Liljenquist DR, Kasza K, Azziz R, Legro RS, Ghazzi MN. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006; 91:48-53 [DOI] [PubMed] [Google Scholar]
- Apridonidze T, Essah PA, Iuorno MJ, Nestler JE. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005; 90:1929-1935 [DOI] [PubMed] [Google Scholar]
- Dereli D, Ozgen G, Buyukkececi F, Guney E, Yilmaz C. Platelet dysfunction in lean women with polycystic ovary syndrome and association with insulin sensitivity. J Clin Endocrinol Metab. 2003; 88:2263-2268 [DOI] [PubMed] [Google Scholar]
- Trovati M, Mularoni EM, Burzacca S, Ponziani MC, Massucco P, Mattiello L, Piretto V, Cavalot F, Anfossi G. Impaired insulin‐induced platelet antiaggregating effect in obesity and in obese NIDDM patients. Diabetes. 1995; 44:1318-1322 [DOI] [PubMed] [Google Scholar]
- Riess H, Merk W, Falkner C, Hiller E. Increased in vitro platelet aggregation in hypertriglyceridemias. Thromb Res. 1986; 41:281-289 [DOI] [PubMed] [Google Scholar]
- Rotterdam ESHRE/ASRM‐Sponsored PCOS Consensus Workshop Group Revised 2003 consensus on diagnostic criteria and long‐term health risks related to polycystic ovary syndrome. Fertil Steril. 2004; 81:19-25 [DOI] [PubMed] [Google Scholar]
- Riba R, Nicolaou A, Troxler M, Homer‐Vaniasinkam S, Naseem KM. Altered platelet reactivity in peripheral vascular disease complicated with elevated plasma homocysteine levels. Atherosclerosis. 2004; 175:69-75 [DOI] [PubMed] [Google Scholar]
- Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972; 18:499-502 [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985; 28:412-419 [DOI] [PubMed] [Google Scholar]
- Zuniga‐Guajardo S, Jimenez J, Angel A, Zinman B. Effects of massive obesity on insulin sensitivity and insulin clearance and the metabolic response to insulin as assessed by the euglycemic clamp technique. Metabolism. 1986; 35:278-282 [DOI] [PubMed] [Google Scholar]
- Stork ADM, Kemperman H, Erkelens DW, Veneman TF. Comparison of the accuracy of the hemocue glucose analyzer with the yellow springs instrument glucose oxidase analyzer, particularly in hypoglycemia. Eur J Endocrinol. 2005; 153:275-281 [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979; 237:E214-E223 [DOI] [PubMed] [Google Scholar]
- Boden G, Jadali F, White J, Liang Y, Mozzoli M, Chen X, Coleman E, Smith C. Effects of fat on insulin‐stimulated carbohydrate metabolism in normal men. J Clin Invest. 1991; 88:960-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Man FH, Nieuwland R, van der Laarse A, Romijn F, Smelt AH, Gevers Leuven JA, Sturk A. Activated platelets in patients with severe hypertriglyceridemia: effects of triglyceride‐lowering therapy. Atherosclerosis. 2000; 152:407-414 [DOI] [PubMed] [Google Scholar]
- Kreutz RP, Alloosh M, Mansour K, Neeb Z, Kreutz Y, Flockhart DA, Sturek M. Morbid obesity and metabolic syndrome in Ossabaw miniature swine are associated with increased platelet reactivity. Diabetes Metab Syndr Obes. 2011; 4:99-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladino CF, Fox RL, Yeh Q, Karpowicz F, Feffer SE, Jonas EA. Platelet aggregability in rats with early atherosclerotic changes induced by parenterally‐administered lipid emulsions. Atherosclerosis. 1987; 66:19-28 [DOI] [PubMed] [Google Scholar]
- Saladino CF, Klein RA, Jonas EA. Induction of early atherosclerosis in rats using parenterally‐administered lipid emulsions. Artery. 1987; 14:304-315 [PubMed] [Google Scholar]
- Lundman P, Eriksson MJ, Silveira A, Hansson LO, Pernow J, Ericsson CG, Hamsten A, Tornvall P. Relation of hypertriglyceridemia to plasma concentrations of biochemical markers of inflammation and endothelial activation (C‐reactive protein, interleukin‐6, soluble adhesion molecules, von Willebrand factor, and endothelin‐1). Am J Cardiol. 2003; 91:1128-1131 [DOI] [PubMed] [Google Scholar]
- Shimokawa H. Primary endothelial dysfunction: atherosclerosis. J Mol Cell Cardiol. 1999; 31:23-37 [DOI] [PubMed] [Google Scholar]