

Abstract
Background
Krüppel‐like factor 4 (Klf4) is involved in a variety of cellular functions by activating or repressing the transcription of multiple genes. Results of previous studies showed that tamoxifen‐inducible global deletion of the Klf4 gene in mice accelerated neointimal formation following vascular injury, in part via enhanced proliferation of smooth muscle cells (SMCs). Because Klf4 is also expressed in non‐SMCs including endothelial cells (ECs), we determined if Tie2 promoter‐dependent deletion of Klf4 in ECs and hematopoietic cells affected injury‐induced neointimal formation.
Methods and Results
Klf4 conditional knockout (cKO) mice were generated by breeding Tie2‐Cre mice and Klf4 floxed mice, and their phenotype was analyzed after carotid ligation injury. Results showed that injury‐induced repression of SMC differentiation markers was unaffected by Tie2 promoter‐dependent Klf4 deletion. However, of interest, neointimal formation was significantly enhanced in Klf4‐cKO mice 21 days following carotid injury. Moreover, Klf4‐cKO mice exhibited an augmented proliferation rate, enhanced accumulation of macrophages and T lymphocytes, and elevated expression of cell adhesion molecules including vascular cell adhesion molecule–1 (Vcam1) and E‐selectin in injured arteries. Mechanistic analyses in cultured ECs revealed that Klf4 inhibited tumor necrosis factor‐α–induced expression of Vcam1 through blocking the binding of nuclear factor‐κB to the Vcam1 promoter.
Conclusions
These results provide evidence that Klf4 in non‐SMCs such as ECs regulates neointimal formation by repressing arterial inflammation following vascular injury.
Keywords: cell adhesion molecules, endothelium, krüppel‐like factors, smooth muscle, vascular injury
Introduction
Vascular proliferative diseases, such as atherosclerosis and restenosis after percutaneous coronary interventions, are the major source of death in westernized societies. They are now recognized as an inflammatory disorder involving multiple cell types including smooth muscle cells (SMCs), endothelial cells (ECs), and monocytes/macrophages, and lymphocytes.1 Identification of molecular factors and mechanisms controlling the progression of vascular diseases in each cell type is of considerable interest.
Krüppel‐like factor 4 (Klf4) is a zinc finger transcription factor involved in a variety of cellular functions by activating or repressing the transcriptional activity of multiple genes. For example, conventional Klf4‐knockout mice exhibited a defect in the acquisition of skin barrier function.2 They were born at the expected Mendelian ratio but died within 15 hours after birth because of rapid loss of body fluid. They also displayed a 90% decrease in the number of goblet cells in the colon, indicating the requirement of Klf4 for colonic epithelial cell differentiation.3 Loss of Klf4 in mouse stomach resulted in increased proliferation and altered differentiation of the gastric epithelia.4 Tissue‐specific deletion of Klf4 in the eye caused abnormal corneal epithelium and lack of goblet cells in the conjunctiva.5 In addition, Klf4 has been shown to regulate monocyte/macrophage differentiation.6–8 As such, Klf4 is implicated in a diverse array of cellular processes in multiple cell types.
Klf4 also plays a key role in the regulation of gene transcription in the cardiovascular system. We have shown that Klf4 represses SMC proliferation and also suppresses expression of multiple SMC differentiation markers, such as SM α‐actin and SM22α, in cultured SMCs and in animal models in vivo.9–11 Indeed, although Klf4 is not normally expressed in differentiated SMCs in vivo, it is transiently induced in phenotypically modulated SMCs after vascular injury.9,11 We showed that tamoxifen‐inducible deletion of the Klf4 gene in the whole bodies of mice resulted in the attenuation of downregulation of SMC differentiation markers and the accelerated formation of neointima following carotid ligation injury.11 We demonstrated that enhanced neointimal formation in Klf4‐deficient mice was caused in part by the reduced induction of p21WAF1/Cip1, a cell‐cycle inhibitor, in SMCs.11 On the other hand, Klf4 is constitutively expressed in vascular ECs. Hamik et al12 showed that overexpression of Klf4 increased expression of anti‐inflammatory and antithrombotic factors, including endothelial nitric oxide synthase and thrombomodulin, whereas knockdown of Klf4 led to enhancement of tumor necrosis factor–α (TNF‐α)‐induced expression of vascular cell adhesion molecule–1 (Vcam1) and tissue factor in cultured human umbilical vein ECs (HUVECs). Klf4 has also been shown to play an important role in the maintenance of endothelial barrier function, in that Klf4‐regulated VE‐cadherin expression and knockdown of Klf4 augmented lipopolysaccharide‐induced lung injury and pulmonary edema in mice.13 In addition, results of recent studies showed that endothelial Klf4 protected against atherothrombosis in Apoe−/−‐background mice.14 Moreover, myeloid Klf4 deficiency has been shown to augment atherosclerotic plaque formation in Apoe‐knockout mice.15 As such, results of the preceding studies provided evidence that Klf4 is a critical factor regulating the development of multiple vascular diseases in various cell types.
Given the multiple cellular functions of Klf4, it is of significant interest to determine if Klf4 in non‐SMCs controls neointimal formation in response to vascular injury. Although the results of our previous studies showed that Klf4 deletion in SMCs cell‐autonomously increased the SMC proliferation rate and enhanced neointimal formation in tamoxifen‐inducible conditional Klf4‐knockout mice,11 it is possible that Klf4 in non‐SMCs, such as ECs, also contributes to the phenotype. To address this question, we derived conditional Klf4‐deficient mice in ECs and hematopoietic cells by breeding Tie2‐Cre mice16 with mice carrying a loxP allele of Klf4 (Klf4loxP mice)3 and analyzed their phenotype following vascular injury.
Methods
Generation of Klf4 Conditional Knockout Mice
Animal protocols were approved by Keio University Animal Care and Use Committee. Klf4loxP/loxP mice, kindly provided by Dr Klaus H. Kaestner,3 were bred with Tie2‐Cre+/− mice16 to generate Tie2‐Cre+/−/Klf4loxP/+ mice. Tie2‐Cre+/−/Klf4loxP/+ mice were then bred with Klf4loxP/loxP mice to generate Tie2‐Cre+/−/Klf4loxP/loxP (Klf4 conditional knockout [Klf4‐cKO]) mice and Tie2‐Cre−/−/Klf4loxP/loxP (control) mice. Both mice were on a C57BL/6J background, and littermates were used for all comparisons. Genotyping was performed by polymerase chain reaction (PCR) as described previously.11 Blood pressure and heart rate were measured by the tail‐cuff method (BP‐98E; Softron, Tokyo, Japan).
Carotid Ligation Injury Model
Carotid artery ligation was performed as described previously.11,17 The right carotid artery was completely ligated just proximal to the carotid bifurcation. The left carotid artery served as an uninjured control. The right and left carotid arteries were harvested 3, 7, 14, and 21 days after injury, fixed in 4% paraformaldehyde, and embedded into OCT compound. The arteries were also harvested for real‐time reverse transcription (RT)‐PCR 7 days after injury.
Morphometric Analysis
Cross‐sections of carotid arteries (6 μm) were prepared from 1.0 mm proximal to the ligature to the aortic arch. Morphometric analysis was performed using 3 sections per artery. These sections were located at ≈2.0 mm proximal to the ligature, and each section was 300 μm apart (ie, 1700, 2000, and 2300 μm proximal to the ligature). Sections were subjected to Verhoeff–van Gieson elastin staining. The areas of the intima, the media, and the lumen were measured by Image‐Pro Plus software (Media Cybernetics, Silver Spring, MD). Six mice for each genotype and for each time were analyzed.
Immunohistochemistry
Immunohistochemistry was performed with antibodies for Klf4,10 SM α‐actin (1A4; Sigma, St. Louis, MO), SM22α (ab14106; Abcam, Cambridge, MA), Ki67 (sc7846; Santa Cruz Biotechnology, Santa Cruz, CA), CD3ε (sc1127; Santa Cruz Biotechnology), Mac2 (M3/38; Acris Antibodies, Herford, Germany), Vcam1 (sc1504; Santa Cruz Biotechnology), and E‐selectin (sc14011; Santa Cruz Biotechnology). Staining for Klf4, Ki67, CD3ε, Mac2, Vcam1, and E‐selectin was visualized by diaminobenzidine, and sections were counterstained by hematoxylin.11,17 Staining for SM α‐actin and SM22α was visualized by a Vector Red Alkaline Phosphatase Substrate kit (Vector Laboratories, Burlingame, CA), and sections were counterstained by hematoxylin.11,17–18
Real‐Time RT‐PCR
Total RNA prepared from the aorta, carotid artery, blood, and cultured ECs was used for real‐time RT‐PCR. Primer and probe sequences for mouse SM α‐actin, SM22α, Klf4, Vcam1, and 18S rRNA were described previously.9,17,19–20 Primer sequences for mouse E‐selectin and human Vcam1 were as follows: E‐selectin‐F, 5′‐CCAGAATGGCGTCATGGA‐3′; E‐selectin‐R, 5′‐TAAAGCCCTCATTGCATTGA‐3′; hVcam1‐F, 5′‐GGTGGGACACAAATAAGGGTTTTGG‐3′; and hVcam1‐R, 5′‐CTTGCAATTCTTTACAGCCTGCC‐3′.
Cell Culture
HUVECs (Japanese Collection of Research Bioresources, Osaka, Japan) were cultured in MCDB107 medium supplemented with endothelial cell growth supplement (Sigma) and 10% fetal bovine serum (Life Technologies, Carlsbad, CA). Human aortic ECs (Lonza, Allendale, NJ) were cultured according to the manufacturer's instructions. One day after plating at 20 000 cells/cm2, DNA plasmids or siRNAs21 were transfected into ECs using X‐tremeGENE HP DNA transfection reagent (Roche, Indianapolis, IN) or Lipofectamine RNAiMAX (Life Technologies), respectively. On the next day, ECs were treated with 10 ng/mL human TNF‐α (R&D Systems, Minneapolis, MN) for an additional 24 hours.
Plasmid Constructs
Expression plasmids for Klf4 and p65 were described previously.9,17 The human Vcam1 (−1716/+119) promoter‐luciferase construct and the Vcam1 (−288/+119) promoter‐luciferase construct in pGL3‐basic vector (Promega, Madison, WI) were made by PCR amplification of the DNA fragments. The 5xGAL4 binding site–containing luciferase construct (pG5‐luc) and the expression plasmid for GAL4 were purchased from Promega. The expression plasmid for GAL4‐p65 fusion protein was constructed by inserting p65 cDNA into the carboxyl terminus of GAL4.
Western Blotting, Immunofluorescence Studies, and Coimmunoprecipitation Assays
Western blotting, immunofluorescence studies, and co‐immunoprecipitation assays were performed as described previously.10,17,19–20 Antibodies used were as follows: Vcam1, p65 (F6; Santa Cruz Biotechnology), phospho‐p65 at serine 536 (93H1; Cell Signaling Technology), VE‐cadherin (sc28644; Santa Cruz Biotechnology), GAPDH (6C5; Millipore, Billerica, MA), and FLAG (F7425; Sigma).
Quantitative Chromatin Immunoprecipitation Assays
Quantitative chromatin immunoprecipitation assays were performed using anti‐p65 antibody or anti‐Klf4 antibody as previously described.11,17,20 Real‐time PCR was performed to amplify the promoter region of the human Vcam1 gene and the human E‐selectin gene. Primer sequences were as follows: Vcam1‐proF, 5′‐CAGGACAGAGAGAGGAGCT‐3′; Vcam1‐proR, 5′‐AAGGGTCTTGTTGCAGAGG‐3′; E‐selectin‐proF, 5′‐CAAGAGACAGAGTTTCTGACATCAT‐3′; and E‐selectin‐proR, 5′‐TTTATAGGAGGGATTGCTTCCTGTG‐3′.
Statistical Analyses
Data are presented as mean±SEM. Statistical analyses were done by SigmaPlot/SigmaStat9 (Systat Software Inc, San Jose, CA). After confirming that the data passed the normality test for parametric analyses, the unpaired t test (Figure 1A through 1F and Table), 1‐way factorial ANOVA (Figure 6A, 6B, and 7E), 2‐way factorial ANOVA (Figure 2B, 2C, 3C, 3D, 5B, 5D, 8A, and 8B), and 3‐way factorial ANOVA (Figure 3B, 3E, 3F, and 4D through 4F) were performed with a post hoc Fisher protected least‐significant‐difference test. P values <0.05 were considered significant.
Table 1.
White Blood Cell Counts From Klf4‐cKO Mice and Control Mice
| Control | Klf4‐cKO | |
|---|---|---|
| Total white blood cells, /μL | 9150±263 | 9000±258 |
| Neutrophils, /μL | 3138±203 | 3123±282 |
| Lymphocytes, /μL | 5900±219 | 5764±356 |
| Monocytes, /μL | 113±20 | 113±24 |
Data are presented as mean±SEM.
Results
Endothelial Klf4 Was Selectively Deleted in Klf4‐cKO Mice
To determine the role of Klf4 in non‐SMCs for neointimal formation following vascular injury, we generated Klf4‐cKO mice by breeding Tie2‐Cre mice and Klf4loxP mice. Although Tie2‐Cre transgenic mice were originally developed as a genetic tool for the analyses of EC‐specific gene targeting,16 the recombination has also been shown to occur in some hematopoietic cells.22 Klf4‐cKO mice were born at the expected Mendelian ratio, and they grew up normally to adulthood. Body weight, systolic and diastolic blood pressure, and heart rate at 10 weeks of age were similar in Klf4‐cKO mice and control mice (Figure 1A through 1C). Expression of Klf4 in the aorta was significantly decreased in Klf4‐cKO mice (Figure 1D), whereas Klf4 expression in the colon was unaltered (Figure 1E), suggesting that Klf4 was selectively deleted in ECs. Expression of Klf4 was not significantly decreased in the blood (Figure 1F), whereas partial recombination occurred in Klf4‐cKO mice (Figure 1G). Klf4 expression in circulating blood seemed to be low compared with other tissues, and this may be why the difference in Klf4 expression was undetectable between Klf4‐cKO mice and control mice. Klf4‐cKO mice and control mice at 10 to 11 weeks of age received carotid ligation injury. In uninjured arteries, Klf4 was expressed in ECs but not in SMCs of control mice, whereas it was undetectable in either ECs or SMCs of Klf4‐cKO mice (Figure 1H). Three days after injury, Klf4 was induced in SMCs in both groups of mice, but endothelial Klf4 was only detectable in control mice (Figure 1H). These results indicate that endothelial Klf4 was efficaciously deleted in Klf4‐cKO mice, and that injury‐induced expression of Klf4 in SMCs is unaffected by Klf4 deletion in ECs and hematopoietic cells.
Figure 1.
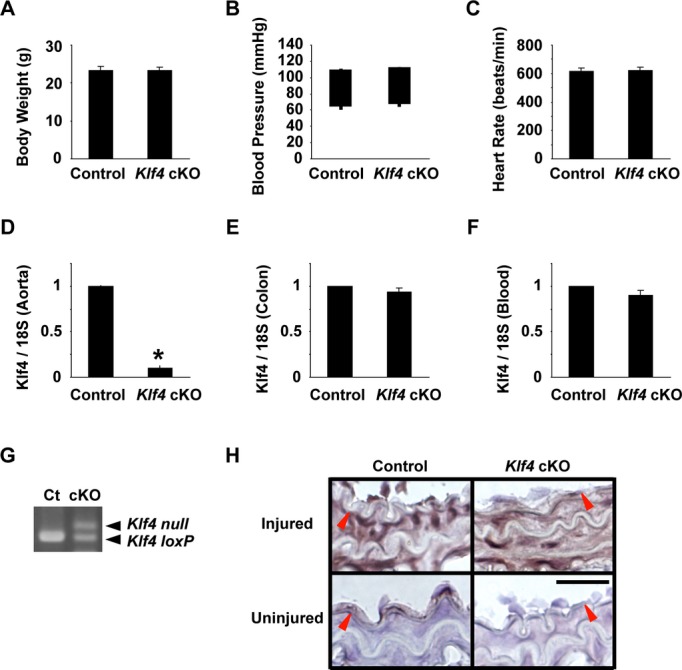
Klf4‐cKO mice grew up normally to the adulthood. A through C, Body weight (A), systolic and diastolic blood pressure (B), and heart rate (C) of Klf4‐cKO mice and control mice were measured at 11 weeks of age (n=12 for each genotype). B, The tops and bottoms of the squares indicate systolic and diastolic blood pressure, respectively. D through F, Expression of Klf4 in the aorta (D), colon (E), and blood (F) was determined by real‐time RT‐PCR in Klf4‐KO mice and control mice (n=4 for each genotype). *P<0.05 compared with control mice. G, Recombination of the Klf4loxP allele was examined by PCR in the blood of Klf4‐cKO mice (cKO) and control mice (Ct). H, Klf4 expression was examined by immunohistochemistry in the carotid arteries of Klf4‐cKO mice and control mice 3 days after injury. Klf4 expression was visualized by diaminobenzidine, and sections were counterstained with hematoxylin. Representative pictures are shown from 6 mice analyzed per genotype. Bar: 50 μm. Red arrowheads indicate the IEL. cKO indicates conditional knockout; IEL, internal elastic lamina; RT‐PCR, reverse‐transcription polymerase chain reaction.
Injury‐Induced Downregulation of SMC Differentiation Markers Was Unaffected in Klf4‐cKO Mice
Results of our previous studies showed that downregulation of SMC differentiation markers at the early times after injury was attenuated in tamoxifen‐inducible conditional Klf4‐deficient mice.11 We examined if Klf4 in ECs and hematopoietic cells contributes to injury‐induced downregulation of SMC differentiation markers. As shown in Figure 2A, expression of SM α‐actin and SM22α was markedly decreased in the medial layer of carotid arteries in both Klf4‐cKO mice and control mice on day 3 and day 7 after injury. Expression of SM α‐actin and SM22α at mRNA levels was also decreased significantly in injured arteries in both mice (Figure 2B and 2C). These results suggest that Klf4 in ECs and hematopoietic cells does not play a role in the downregulation of SMC differentiation markers following vascular injury.
Figure 2.
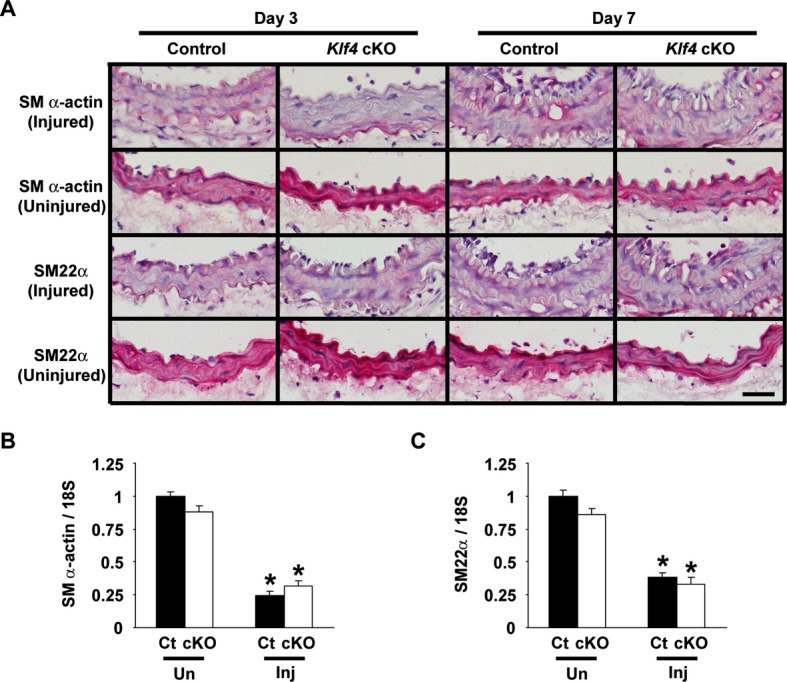
Expression of SMC differentiation markers was decreased in Klf4‐cKO mice after vascular injury. A, Expression of SM α‐actin and SM22α was examined by immunohistochemistry in the carotid arteries of Klf4‐cKO mice and control mice on day 3 and day 7 after ligation injury. Expression of SM α‐actin and SM22α was visualized by Vector Red alkaline phosphatase, and sections were counterstained with hematoxylin. Representative pictures are shown from 6 mice analyzed per genotype and from each time. Bar: 50 μm. B and C, Expression of SM α‐actin (B) and SM22α (C) was determined by real‐time RT‐PCR in the injured (Inj) and uninjured (Un) carotid arteries of Klf4‐cKO mice and control mice on day 7 after injury (n=5 per each genotype). *P<0.05 compared with uninjured arteries. cKO indicates conditional knockout; Ct, control; RT‐PCR, reverse‐transcription polymerase chain reaction; SMC, smooth muscle cell.
Neointimal Formation Was Enhanced in Klf4‐cKO Mice Following Vascular Injury
Morphometric analyses were performed in carotid arteries 7, 14, and 21 days following injury (Figure 3). Although the neointimal area did not differ between Klf4‐cKO mice and control mice 7 and 14 days after injury, it was significantly larger in Klf4‐cKO mice (23132±806 μm2) than in control mice (14375±614 μm2) 21 days after injury (Figure 3A and 3C). Medial areas of injured vessels were increased compared with uninjured vessels in both Klf4‐cKO mice and control mice, but did not significantly differ from one another (Figure 3B). The lumen areas were gradually decreased following injury in both groups of mice, and they were significantly smaller in Klf4‐cKO mice than in control mice on day 21 (Figure 3D). The areas within the external elastic lamina were unaltered in response to vascular injury in both groups of mice (Figure 3E). The areas within the internal elastic lamina were decreased after injury because of the expansion of the medial areas in both groups of mice (Figure 3F).
Figure 3.
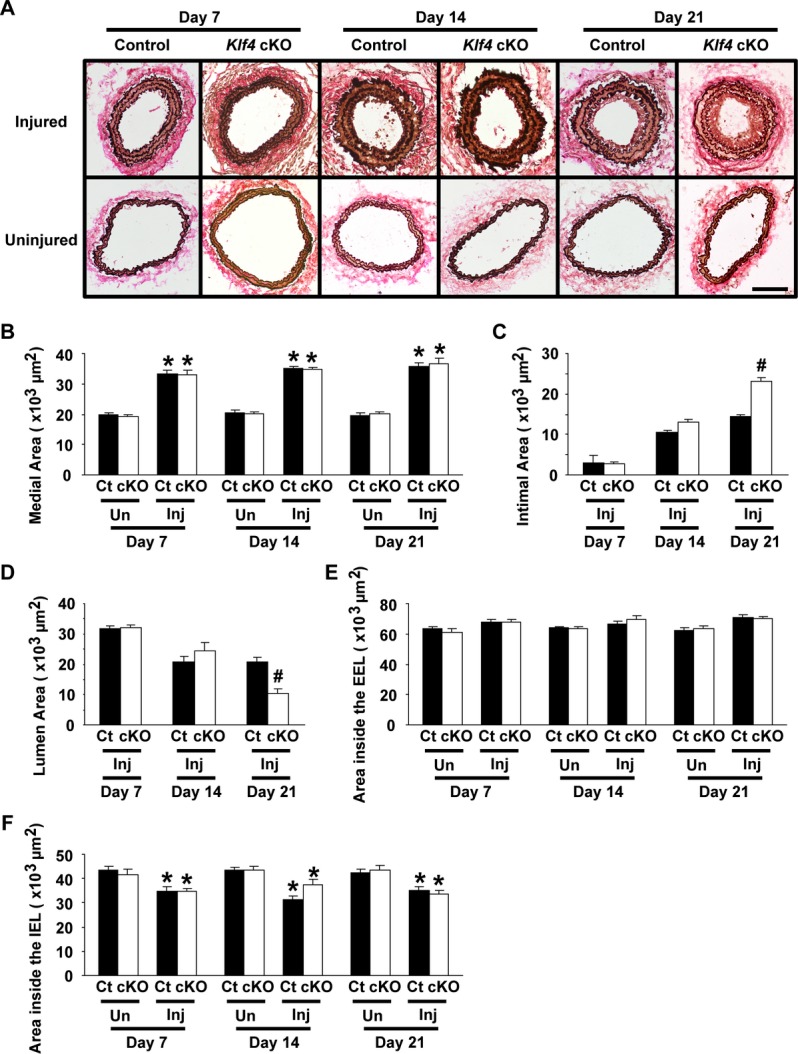
Injury‐induced neointimal formation was enhanced in Klf4‐cKO mice. Verhoeff‐van Gieson staining (A) was performed in the injured and uninjured carotid arteries of EC‐specific Klf4‐knockout mice and control mice 7, 14, and 21 days after injury. Areas of the media (B), the intima (C), and the lumen (D), as well as the areas within the EEL (E) and the IEL (F), were quantified (n=6 per genotype and each time). Bar: 100 μm. *P<0.05 compared with uninjured arteries; #P<0.05 compared with corresponding control mice. cKO indicates conditional knockout; Ct, control; EC, endothelial cell; EEL, external elastic lamina; IEL, internal elastic lamina; Inj, injured; Un, uninjured.
The proliferation rate was determined in the vessels after injury. Ki67 staining revealed that injured carotid arteries of Klf4‐cKO mice exhibited significantly increased proliferation compared with control mice at day 14 (17.1±1.1% in Klf4‐cKO mice versus 10.5±0.7% in control mice) and day 21 (16.3±0.8% in Klf4‐cKO mice versus 9.2±0.5% in control mice) after injury, although the proliferation rate in injured vessels of both mice was increased compared with uninjured vessels at any points examined (Figure 4A and 4D). Accumulation of macrophages and T lymphocytes was then examined by immunohistochemistry. Results of Mac2 staining showed that macrophage accumulation in the injured arteries of Klf4‐cKO mice (15.5±1.2% at day 7, 15.2±0.7% at day 14, and 13.2±1.3% at day 21) was augmented compared with control mice (7.4±0.9% at day 7, 7.9±0.7% at day 14, and 5.1±0.6% at day 21) (Figure 4B and 4E). Moreover, CD3ε staining revealed that accumulation of T lymphocytes was also enhanced in Klf4‐cKO mice (5.7±0.5% at day 7, 6.0±0.5% at day 14, and 3.5±0.5% at day 21) compared with control mice (2.1±0.7% at day 7, 3.3±0.4% at day 14, and 1.3±0.3% at day 21) (Figure 4C and 4F). White blood cell counts were performed to examine whether deletion of the Klf4 gene in hematopoietic cells affected the enhanced accumulation of macrophages and T lymphocytes in Klf4‐cKO mice. Results showed that the number of neutrophils, lymphocytes, and monocytes in the blood was similar between Klf4‐cKO mice and control mice (Table). In addition, expression of Klf4 mRNA in the blood did not differ between these mice, as shown in Figure 1F. These results suggest that increased infiltration of macrophages and T lymphocytes in Klf4‐cKO mice is probably a result of loss of Klf4 in ECs, although we cannot deny the possibility that Klf4 in cells derived from hematopoietic cells also affects the phenotype.
Figure 4.

Accumulation of macrophages and T lymphocytes was enhanced in the injured arteries of Klf4‐cKO mice. Left: Representative pictures of injured carotid arteries for Ki67 staining on day 14 (A), Mac2 staining on day 14 (B), and CD3ε staining on day 7 (C) after ligation injury in Klf4‐cKO mice and control mice are shown. Staining for Ki67, Mac2, and CD3ε was visualized by diaminobenzidine, and sections were counterstained with hematoxylin. Bars: 50 μm. Red and blue arrowheads indicate IEL and EEL, respectively. Right: The ratios of Ki67‐positive cells (D), Mac2‐positive cells (E), and CD3ε‐positive cells (F) in the injured and uninjured carotid arteries were calculated in Klf4‐cKO mice and control mice (n=6 per genotype and each time). *P<0.05 compared with uninjured arteries; #P<0.05 compared with corresponding control mice. cKO indicates conditional knockout; Ct, control; EEL, external elastic lamina; IEL, internal elastic lamina; Inj, injured; Un, uninjured.
Taken together, these results demonstrate that Tie2 promoter‐dependent deletion of Klf4 in ECs and hematopoietic cells augmented the proliferation rate, the accumulation of macrophages and T lymphocytes, and neointimal formation in injured arteries and suggest that the enhanced formation of neointima in Klf4‐cKO mice is caused in part by the augmented recruitment of inflammatory cells, such as macrophages and T lymphocytes.
Injury‐Induced Expression of Cell Adhesion Molecules Was Enhanced in Klf4‐cKO Mice
To determine the mechanisms underlying the enhanced recruitment of inflammatory cells in Klf4‐cKO mice, expression of cell adhesion molecules such as Vcam1 and E‐selectin was examined in the vessels. As shown in Figure 5A, although endothelial expression of Vcam1 in uninjured arteries was very faint in both Klf4‐cKO mice and control mice, it was markedly augmented in injured arteries in both mice. Indeed, injury‐induced Vcam1 expression was seen in the intima and media in both mice, and it appeared to be more abundant in Klf4‐cKO mice than in control mice (Figure 5A). Quantitative analysis using real‐time RT‐PCR showed that Vcam1 expression in injured arteries was much higher in Klf4‐cKO mice (a 4.6‐fold increase versus uninjured arteries) than in control mice (a 2.7‐fold increase versus uninjured arteries), although it was increased in both mice compared with uninjured arteries (Figure 5B). Likewise, injury‐induced expression of E‐selectin in Klf4‐cKO mice was more elevated than in control mice, although E‐selectin expression in uninjured arteries was faint in both mice (Figure 5C and 5D). These results suggest that increased expression of cell adhesion molecules in injured arteries contributes to the augmented accumulation of inflammatory cells, thereby enhancing neointimal formation in Klf4‐cKO mice.
Figure 5.
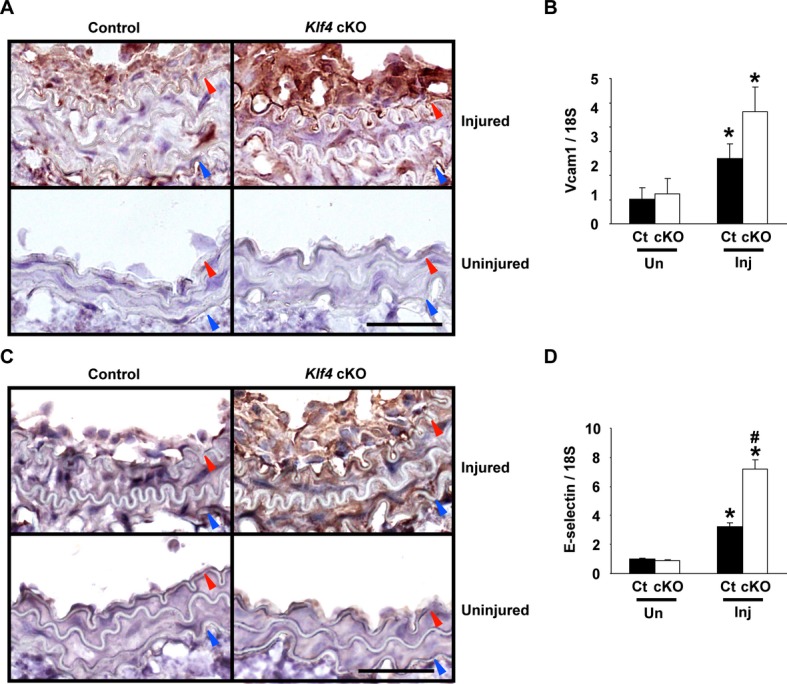
Induction of cell adhesion molecules was augmented in Klf4‐cKO mice following injury. A and C, Representative pictures for staining of Vcam1 (A) and E‐selectin (C) on day 14 after injury in Klf4‐cKO mice and control mice are shown (n=6 per genotype). Staining was visualized by diaminobenzidine, and sections were counterstained with hematoxylin. Bar: 50 μm. Red and blue arrowheads indicate IEL and EEL, respectively. B and D, Expression of Vcam1 (B) and E‐selectin (D) was determined by real‐time RT‐PCR in the carotid arteries of Klf4‐cKO mice and control mice on day 7 after ligation injury (n=5 per each genotype). *P<0.05 compared with uninjured arteries; #P<0.05 compared with corresponding control mice. cKO indicates conditional knockout; Ct, control; EEL, external elastic lamina; IEL, internal elastic lamina; Inj, injured; RT‐PCR, reverse‐transcription polymerase chain reaction; Un, uninjured; Vcam1, vascular cell adhesion molecule‐1.
Klf4 Inhibited TNF‐α‐Induced Expression of Vcam1 by Blocking the Binding of NF‐κB to the Vcam1 Promoter
Induction of cell adhesion molecules including Vcam1 and E‐selectin has been shown to be mediated by the TNF‐α–nuclear factor (NF)‐κB pathway in ECs.23 We examined if Klf4 regulates TNF‐α‐induced expression of Vcam1 in HUVECs and in human aortic ECs. As shown in Figure 6A, TNF‐α increased Vcam1 expression by 5.4‐fold in HUVECs, as determined by real‐time RT‐PCR. TNF‐α‐induced upregulation of Vcam1 was attenuated by Klf4 overexpression, whereas siRNA‐mediated knockdown of Klf4 augmented TNF‐α‐induced Vcam1 expression by 10.9‐fold in HUVECs (Figure 6A). Similar results were also obtained in human aortic ECs (Figure 6B). The human Vcam1 promoter contains 2 NF‐κB binding sites (5′‐GG(G/A)(G/A)NN(C/T)CCC‐3′), at −72/−63 bp and −57/−48 bp, as well as 1 putative consensus Klf4 binding site (5′‐(G/A)(G/A)GG(C/T)G(C/T)‐3′), at −1420/−1414 bp. To determine the role of these cis elements for the inhibitory effect of Klf4, 2 Vcam1 promoter‐luciferase constructs were made: the Vcam1 (−1716/+119) promoter‐luciferase construct containing a putative Klf4 binding site and 2 NF‐κB binding sites and the Vcam1 (−288/+119) promoter‐luciferase construct, which only contains 2 NF‐κB binding sites. Results of luciferase‐reporter assays showed that TNF‐α increased the transcriptional activity of the Vcam1 (−1716/+119) promoter‐luciferase construct by 2.2‐fold, and the stimulatory effect of TNF‐α was blunted by coexpression of Klf4 (Figure 6C). Of interest, TNF‐α also increased the transcriptional activity of the Vcam1 (−288/+119) promoter‐luciferase construct, and Klf4 still inhibited the effect of TNF‐α on this shorter construct. These results suggest that the inhibitory effect of Klf4 is not mediated through the putative Klf4 binding site.
Figure 6.
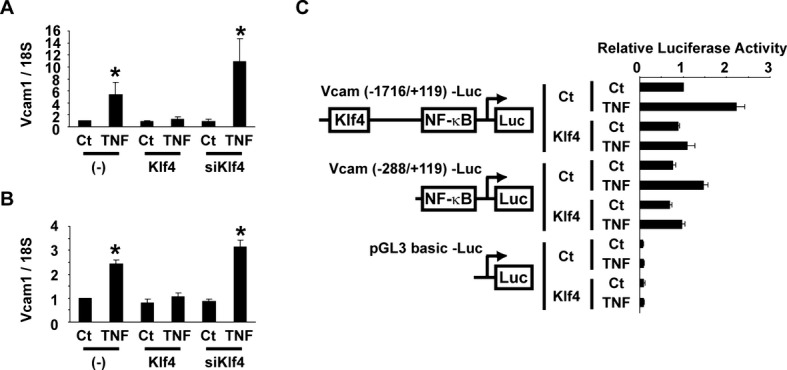
Klf4 attenuated TNF‐α‐induced expression of Vcam1 in ECs. A and B, HUVECs (A) and human aortic ECs (B) were transfected with Klf4 expression plasmid or siRNA for Klf4 (siKlf4), and then treated with TNF‐α. Expression of Vcam1 was determined by real‐time RT‐PCR (n=4). *P<0.05 compared with cells without TNF‐α treatment. C, HUVECs were transfected with the Vcam1 (−1716/+119) promoter‐luciferase construct, the Vcam1 (−288/+119) promoter‐luciferase construct, or pGL3‐basic plasmid with Klf4 expression plasmid. One day after transfection, HUVECs were treated with TNF‐α for an additional 24 hours. Luciferase activity was measured and normalized to protein content (n=4). Ct indicates control; ECs, endothelial cells; HUVECs, human umbilical vein ECs; NF‐κB, nuclear factor‐κB; RT‐PCR, reverse‐transcription polymerase chain reaction; TNF‐α, tumor necrosis factor–α; Vcam1, vascular cell adhesion molecule‐1.
To clarify the mechanism whereby Klf4 blocks the induction of Vcam1 expression by the TNF‐α–NF‐κB pathway, we then examined the phosphorylation and nuclear translocation of p65, an active component of NF‐κB, in HUVECs. Results showed that even in the presence of Klf4, both TNF‐α‐induced phosphorylation of p65 (Figure 7A) and TNF‐α‐induced nuclear translocation of p65 (Figure 7B) were unaffected, suggesting that Klf4 inhibits the activity of p65 in the nucleus. Moreover, results of cotransfection assays showed that although overexpression of p65 in the nucleus strongly increased the transcriptional activity of the Vcam1 gene in human aortic ECs, the effect was markedly attenuated by Klf4 (Figure 7C), also suggesting that Klf4 inhibits p65 in the nucleus. In addition, results of GAL4 binding assays showed that Klf4 robustly suppressed the stimulatory effect of the GAL4‐p65 fusion protein on the activity of the 5xGAL4 binding site–containing luciferase construct in human aortic ECs (Figure 7D). These results suggest that the inhibitory effect of Klf4 on p65 is not dependent on the DNA sequence of the NF‐κB binding site and/or its flanking sequence within the Vcam1 promoter. Chromatin immunoprecipitation assays also showed that Klf4 was not associated with the Vcam1 promoter in HUVECs in the presence or the absence of TNF‐α (Figure 7E). Of further interest, chromatin immunoprecipitation assays showed that Klf4 inhibited the binding of p65 to the Vcam1 promoter (Figure 8A). Indeed, the TNF‐α‐mediated increase in p65 binding to the Vcam1 promoter was attenuated in the presence of Klf4 in HUVECs, whereas the binding was enhanced by the knocking down of Klf4. TNF‐α‐induced binding of p65 to the E‐selectin promoter was also regulated by Klf4 (Figure 8B). Finally, coimmunoprecipitation assays revealed Klf4 physically bound to p65 (Figure 8C). Taken together, these results suggest that Klf4 inhibits TNF‐α‐mediated induction of Vcam1 expression by blocking the binding of p65 to the Vcam1 promoter through the physical association between Klf4 and p65.
Figure 7.

Klf4 attenuated TNF‐α‐induced expression of Vcam1 without binding to the Vcam1 promoter. A, HUVECs were transfected with FLAG‐tagged Klf4 expression plasmid, and then treated with TNF‐α. Expression of Vcam1, FLAG, phosphorylated p65 (p‐p65), p65, VE‐cadherin, and GAPDH was examined by Western blotting (n=4). B, HUVECs were transfected with the expression plasmid for FLAG‐tagged Klf4, and treated with TNF‐α. Top: Intracellular localization of endogenous p65 (green) was determined by anti‐p65 antibody. Middle: Merged images for endogenous p65 (green), FLAG‐tagged Klf4 (red), and DAPI nuclear staining (blue) are shown. Bottom: DAPI nuclear staining (blue) is shown. Bar: 50 μm. C, Human aortic ECs were transfected with the Vcam1 (−1716/+119) promoter‐luciferase construct, the Vcam1 (−288/+119) promoter‐luciferase construct, or pGL3‐basic plasmid with Klf4 expression plasmid and p65 expression plasmid. Luciferase activity was measured and normalized to protein content (n=4). D, Human aortic ECs were transfected with Klf4 expression plasmid (0, 200, or 400 ng), expression plasmid for GAL4 or GAL4‐p65 fusion protein, and 5xGAL4 binding site–containing luciferase construct. Luciferase activity was measured and normalized to protein content (n=4). E, HUVECs were treated with TNF‐α. Association of Klf4 with the Vcam1 promoter was determined by chromatin immunoprecipitation assays (n=4). Ct indicates control; DAPI, 4′, 6‐diamidino‐2‐phenylindole; ECs, endothelial cells; GAPDH; glyceraldehyde‐3‐phosphate dehydrogenase; IgG, immunoglobulin G; HUVECs, human umbilical vein ECs; TNF‐α, tumor necrosis factor–α; Vcam1, vascular cell adhesion molecule‐1.
Figure 8.
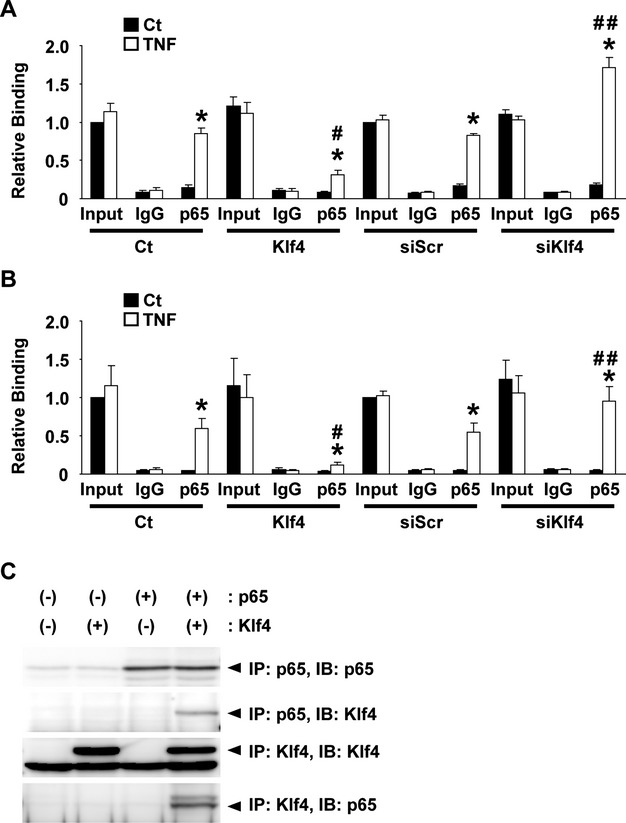
Klf4 attenuated TNF‐α‐induced expression of Vcam1 by blocking the binding of NF‐κB to the Vcam1 promoter. A and B, HUVECs were transfected with empty expression plasmid, Klf4 expression plasmid, siRNA for Klf4 (siKlf4), or siRNA for scrambled sequence (siScr) and treated with TNF‐α. Association of p65 with the Vcam1 promoter (A) and the E‐selectin promoter (B) was determined by chromatin immunoprecipitation assays (n=4). *P<0.05 compared with cells without TNF‐α treatment; #P<0.05 compared with cells without transfecting Klf4 expression plasmid; ##P<0.05 compared wutg cells without transfecting siKlf4. C, Expression plasmids for p65 and FLAG‐Klf4 were transfected into COS7 cells. Coimmunoprecipitation assays were performed with anti‐p65 antibody and anti‐FLAG antibody. Ct indicates control; HUVECs, human umbilical vein endothelial cells; IB, immunoblot; IP, immunoprecipitation; TNF‐α, tumor necrosis factor–α; Vcam1, vascular cell adhesion molecule‐1.
Discussion
In the present study, we showed that deletion of the Klf4 gene in ECs and hematopoietic cells in mice accelerated neointimal formation following carotid ligation injury. Indeed, although injury‐induced downregulation of SMC differentiation markers was unaffected by Tie2 promoter‐dependent Klf4 deletion, knockout of Klf4 in ECs and hematopoietic cells increased the cellular proliferation rate in the injured arteries and also enhanced the recruitment of inflammatory cells such as macrophages and T lymphocytes. Enhanced recruitment of inflammatory cells was likely to be caused by the augmented induction of cell adhesion molecules such as Vcam1 and E‐selectin in Klf4‐cKO mice. The phenotype seen in Klf4‐cKO mice is probably caused by Klf4 deletion in ECs rather than in hematopoietic cells, because the number of neutrophils, lymphocytes, and macrophages did not differ between Klf4‐cKO mice and control mice and because Klf4 expression in circulating white blood cells did not differ between these mice. Moreover, we demonstrated a novel mechanism whereby Klf4 inhibits the inflammation‐related induction of Vcam1 by blocking the binding of NF‐κB to the Vcam1 promoter through the association between Klf4 and p65 in ECs. As such, results of the present study provide compelling evidence that Klf4, at least in ECs and maybe in hematopoietic precursor‐derived cells, plays an anti‐inflammatory role in the formation of neointima following vascular injury.
Previously, we generated tamoxifen‐inducible Klf4‐knockout mice and showed that downregulation of SMC differentiation markers was attenuated at early points after injury, but neointimal formation was accelerated in the mice.11 Based on the results of the present study using Klf4‐cKO mice, the attenuation of injury‐induced downregulation of SMC differentiation markers in tamoxifen‐inducible Klf4‐knockout mice is probably a result of the loss of Klf4 in SMCs, but not in ECs or circulating white blood cells. This notion is consistent with the previous findings that Klf4 was directly bound to the promoter region of SMC differentiation marker genes in injured arteries, as determined by in vivo chromatin immunoprecipitation assays.11 On the other hand, multiple mechanisms seem to be involved in the accelerated neointimal formation in tamoxifen‐inducible Klf4‐knockout mice. One of the mechanisms is the reduced induction of p21WAF1/Cip1 in the medial layer of SMCs following vascular injury, as demonstrated previously using tamoxifen‐inducible Klf4‐deficient mice.11 This mechanism is supported by multiple findings including: (1) Klf4 had no effect on the proliferation rate in p21WAF1/Cip1‐deficient cultured SMCs, whereas overexpression of Klf4 decreased cellular proliferation in wild‐type SMCs; (2) Klf4 increased the transcriptional activity of the p21WAF1/Cip1 gene through the binding of Klf4 to the promoter‐enhancer region of the p21WAF1/Cip1 gene in concert with p53; and (3) the association of Klf4 with the p21WAF1/Cip1 promoter‐enhancer was increased after injury in wild‐type mice.11 In the present study, we have added a novel mechanism whereby lack of Klf4 in ECs and/or hematopoietic precursor‐derived cells also contributes to the injury‐induced accelerated neointimal formation by enhancing the accumulation of inflammatory cells via the augmented induction of cell adhesion molecules in the injured vessels. The relationship between neointimal formation and cell adhesion molecules, as well as the accumulation of inflammatory cells has been well established in a number of previous studies.1,24–27 Indeed, deficiency of cell adhesion molecules such as Vcam1 has been shown to decrease neointimal formation,25–27 as well as to decrease the accumulation of monocytes/macrophages in atherosclerotic lesions.26 The infiltrated inflammatory cells stimulate SMC proliferation in a paracrine manner. These mechanisms are not mutually exclusive and would enhance neointimal formation additively in response to vascular injury.
Although the carotid ligation model used in the present study is frequently used for the analyses of injury‐induced neointimal formation, the relevance of this model to neointimal formation following mechanical vascular injury, such as balloon‐ and stent‐induced injury, needs attention. The carotid ligation model preserves the endothelium, whereas balloon‐ and stent‐induced mechanical injury results in endothelial denudation, platelet and fibrin deposition, and robust inflammatory cell recruitment. Further studies using multiple injury models are needed to confirm the effect of Klf4 in vascular diseases.
Results of our present study showed that Klf4 is capable of binding with p65 to inhibit the inflammation‐related induction of cell adhesion molecules in ECs. Coimmunoprecipitation assays revealed that Klf4 and p65 physically interacted with each other, although there is a possibility that the interaction observed by the overexpression of proteins is artifactual. In addition, using chromatin immunoprecipitation assays, we have shown that TNF‐α‐mediated increases in the binding of p65 to the Vcam1 promoter was attenuated in the presence of Klf4. The inhibitory action of Klf4 occurred in the nucleus, because phosphorylation as well as nuclear translocation of p65 were unaffected by overexpression of Klf4. Moreover, results of the present study showed that Klf4 inhibited p65 activity even in the absence of the NF‐κB binding site, the Klf4 binding site, and their flanking DNA sequences. These findings are highly novel, in that Klf4, a zinc‐finger‐containing transcription factor, executes its function without binding to its consensus DNA binding site. However, although these in vitro results provide a mechanical basis for injury‐induced accelerated inflammation and subsequent enhanced neointimal formation in Klf4‐cKO mice, they are different from results of previous studies showing that Klf4 did not affect DNA‐binding properties of p65, as determined by electrophoresis mobility shift assays.12 Because electrophoresis mobility shift assays could detect biochemical binding between DNA and proteins in vitro, but do not necessarily reflect the DNA–protein binding within intact chromatin, some chromatin factors might be required for the inhibitory action of Klf4 against the NF‐κB pathway.
In addition to the interaction between Klf4 and p65, accumulating evidence indicates that Klf4 is capable of binding with multiple proteins to carry out various functions. For example, we previously showed that Klf4 was induced following vascular injury, and it repressed SMC proliferation by increasing p21WAF1/Cip1 expression via the recruitment of p53 to the promoter‐enhancer region of the p21WAF1/Cip1 gene.11 Zhang et al28 showed the physical interaction between Klf4 and p53. We also showed that Klf4 bound with Elk‐1 and that they cooperatively repressed the transcription of SMC differentiation marker genes in response to oxidized phospholipids in SMCs.10 Moreover, Klf4 has been shown to contribute to high phosphate‐induced phenotypic switching of SMCs into osteogenic cells,21 and Michikami et al29 demonstrated that Klf4 physically associated with Runx2, a key transcription factor for osteogenesis. As such, the previous studies and our present study strongly support a model wherein Klf4, which is activated in response to multiple stress signals, executes multiple functions by changing partner proteins.
In summary, we provide evidence that Klf4 in ECs and/or hematopoietic precursor‐derived cells regulates neointimal formation following vascular injury. Klf4 in multiple cell types including SMCs and ECs plays anti‐inflammatory and antiproliferative roles. To dissociate the function of Klf4 in ECs from hematopoietic precursor‐derived cells, further studies are needed to use VE‐cadherin‐Cre mice instead of Tie2‐Cre mice, and/or the bone marrow transplantation approach.
Sources of Funding
This study was supported by a grant‐in‐aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T.Y. and M.H.) and by The Mochida Memorial Foundation for Medical and Pharmaceutical Research (to T.Y.).
Disclosures
None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002; 420:868-874 [DOI] [PubMed] [Google Scholar]
- 2.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999; 22:356-360 [DOI] [PubMed] [Google Scholar]
- 3.Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, Kaestner KH. The zinc‐finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. 2002; 129:2619-2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005; 128:935-945 [DOI] [PubMed] [Google Scholar]
- 5.Swamynathan SK, Katz JP, Kaestner KH, Ashery‐Padan R, Crawford MA, Piatigorsky J. Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol Cell Biol. 2007; 27:182-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feinberg MW, Wara AK, Cao Z, Lebedeva MA, Rosenbauer F, Iwasaki H, Hirai H, Katz JP, Haspel RL, Gray S, Akashi K, Segre J, Kaestner KH, Tenen DG, Jain MK. The Kruppel‐like factor KLF4 is a critical regulator of monocyte differentiation. EMBO J. 2007; 26:4138-4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alder JK, Georgantas RW, III, Hildreth RL, Kaplan IM, Morisot S, Yu X, McDevitt M, Civin CI. Kruppel‐like factor 4 is essential for inflammatory monocyte differentiation in vivo. J Immunol. 2008; 180:5645-5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, Flask CA, Kim J, Doreian BW, Lu KQ, Kaestner KH, Hamik A, Clément K, Jain MK. Krüppel‐like factor 4 regulates macrophage polarization. J Clin Invest. 2011; 121:2736-2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem. 2005; 280:9719-9727 [DOI] [PubMed] [Google Scholar]
- 10.Yoshida T, Gan Q, Owens GK. Krüppel‐like factor 4, Elk‐1, and histone deacetylases cooperatively suppress smooth muscle cell differentiation markers in response to oxidized phospholipids. Am J Physiol Cell Physiol. 2008; 295:C1175-C1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshida T, Kaestner KH, Owens GK. Conditional deletion of Krüppel‐like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ Res. 2008; 102:1548-1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerszten RE, Edelman ER, Jain MK. Kruppel‐like factor 4 regulates endothelial inflammation. J Biol Chem. 2007; 282:13769-13779 [DOI] [PubMed] [Google Scholar]
- 13.Cowan CE, Kohler EE, Dugan TA, Mirza MK, Malik AB, Wary KK. Krüppel‐like factor‐4 transcriptionally regulates VE‐cadherin expression and endothelial barrier function. Circ Res. 2010; 107:959-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, Feaver RE, Gao H, Desai A, Schmaier A, Gerson SL, Wang Y, Atkins GB, Blackman BR, Simon DI, Jain MK. Endothelial Kruppel‐like factor 4 protects against atherothrombosis in mice. J Clin Invest. 2012; 122:4727-4731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma N, Lu Y, Zhou G, Liao X, Kapil P, Anand P, Mahabeleshwar GH, Stamler JS, Jain MK. Myeloid Krüppel‐like factor 4 deficiency augments atherogenesis in apoE−/− mice—brief report. Arterioscler Thromb Vasc Biol. 2012; 32:2836-2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2‐Cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo. Dev Biol. 2001; 230:230-242 [DOI] [PubMed] [Google Scholar]
- 17.Yoshida T, Yamashita M, Horimai C, Hayashi M. Smooth muscle‐selective inhibition of nuclear factor‐κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. J Am Heart Assoc. 2013; 2:e000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida T, Yamashita M, Horimai C, Hayashi M. High glucose concentration does not modulate the formation of arterial medial calcification in experimental uremic rats. J Vasc Res. 2013; 50:512-520 [DOI] [PubMed] [Google Scholar]
- 19.Yoshida T, Sinha S, Dandré F, Wamhoff BR, Hoofnagle MH, Kremer BE, Wang DZ, Olson EN, Owens GK. Myocardin is a key regulator of CArG‐dependent transcription of multiple smooth muscle marker genes. Circ Res. 2003; 92:856-864 [DOI] [PubMed] [Google Scholar]
- 20.Shang Y, Yoshida T, Amendt BA, Martin JF, Owens GK. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J Cell Biol. 2008; 181:461-473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida T, Yamashita M, Hayashi M. Krüppel‐like factor 4 contributes to high phosphate‐induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem. 2012; 287:25706-25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Constien R, Forde A, Liliensiek B, Gröne HJ, Nawroth P, Hämmerling G, Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001; 30:36-44 [DOI] [PubMed] [Google Scholar]
- 23.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF‐κB and cytokine‐inducible enhancers. FASEB J. 1995; 9:899-909 [PubMed] [Google Scholar]
- 24.Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991; 251:788-791 [DOI] [PubMed] [Google Scholar]
- 25.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez‐Ramos JC, Connelly PW, Milstone DS. A major role for VCAM‐1, but not ICAM‐1, in early atherosclerosis. J Clin Invest. 2001; 107:1255-1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oguchi S, Dimayuga P, Zhu J, Chyu KY, Yano J, Shah PK, Nilsson J, Cercek B. Monoclonal antibody against vascular cell adhesion molecule‐1 inhibits neointimal formation after periadventitial carotid artery injury in genetically hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2000; 20:1729-1736 [DOI] [PubMed] [Google Scholar]
- 27.Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. The combined role of P‐ and E‐selectins in atherosclerosis. J Clin Invest. 1998; 102:145-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, Biggs JR, Kraft AS, Yang VW. The gut‐enriched Krüppel‐like factor (Krüppel‐like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000; 275:18391-18398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michikami I, Fukushi T, Tanaka M, Egusa H, Maeda Y, Ooshima T, Wakisaka S, Abe M. Krüppel‐like factor 4 regulates membranous and endochondral ossification. Exp Cell Res. 2012; 318:311-325 [DOI] [PubMed] [Google Scholar]