

Abstract
Background
Pulmonary arterial hypertension remains a devastating disease without a cure. The major complication of this disease is the abnormal growth of vascular cells, resulting in pulmonary vascular remodeling. Thus, agents, which affect the remodeled vessels by killing unwanted cells, should improve treatment strategies. The present study reports that antitumor drugs selectively kill vascular cells in remodeled pulmonary vessels in rat models of pulmonary hypertension.
Methods and Results
After developing pulmonary vascular remodeling in chronic hypoxia or chronic hypoxia/SU‐5416 models, rats were injected with antitumor drugs including proteasome inhibitors (bortezomib and MG‐132) and daunorubicin. Within 1 to 3 days, these agents reduced the media and intima thickness of remodeled pulmonary vascular walls, but not the thickness of normal pulmonary vessels. These drugs also promoted apoptotic and autophagic death of vascular cells in the remodeled vessels, but not in normal vessels. We provide evidence that the upregulation of annexin A1, leading to GATA4‐dependent downregulation of Bcl‐xL, is a mechanism for specific apoptotic killing, and for the role of parkin in defining specificity of autophagic killing of remodeled vascular cells. The reversal of pulmonary vascular remodeling increased the capacity of vasodilators to reduce pulmonary arterial pressure.
Conclusions
These results suggest that antitumor drugs can specifically kill cells in remodeled pulmonary vascular walls and may be useful for improving the efficacy of current therapeutic strategies to treat pulmonary arterial hypertension.
Keywords: apoptosis, autophagy, blood pressure, pulmonary hypertension, vascular remodeling
Introduction
Pulmonary arterial hypertension (PAH) is a fatal disease without a cure.1–2 Because it is a progressive disease, by the time patients are diagnosed, pulmonary arterial walls have often already thickened as a result of complex pathologic features. Increased resistance in the pulmonary circulation strains the right ventricle (RV), leading to right‐sided heart failure and death. Before the existence of any approved drugs for treating this disease, the NIH registry has determined that the median duration of survival of PAH patients after diagnosis is 2.8 years with the 3‐year survival being 48%.3 Since then, 3 classes of therapeutic agents have been approved for the treatment of PAH. However, though these agents improve the quality of life for patients, their influence on survival may be minimal. Even with currently available therapies, the prognosis remains poor with 3‐year survival of PAH patients being reported as 58% to 75%.4–6 Thus, the development of improved therapeutic strategies is needed for the treatment of this disease.
The primary function of these approved drugs is to promote vasodilation. However, because the growth of vascular cells is also critical to the pathogenesis of PAH, agents that eliminate excess vascular cells may have therapeutic potential, acting to reduce the thickness of pulmonary vascular walls and thereby helping to decrease vascular resistance.7 In this regard, antitumor drugs with the ability to kill cells that have been considered for cancer chemotherapy may also be useful in the treatment of PAH.8
Proteasome inhibitors are promising therapeutic agents for the treatment of cancer. Bortezomib (MG‐341 or PS‐341) was approved by the FDA in 2003 for the treatment of multiple myeloma.9 MG‐132 was developed earlier and has been most widely used in research studies to inhibit proteasomes.10 These proteasome inhibitors have been shown to inhibit pulmonary artery smooth muscle cell (PASMC) proliferation and may be useful for treating PAH.11–12 Anthracyclines derived from Streptomyces bacteria comprise a class of chemotherapeutic agents that have been used to treat cancer since the 1960s. Daunorubicin (DNR) was the first anthracycline to be developed and has shown to be effective against acute leukemia.13–15 One mechanism of the action of anthracyclines is to promote the apoptosis of cancer cells.16–17 We have previously shown that anthracyclines can promote apoptosis of cultured PASMCs.8
The pathology of PAH is complex and involves abnormal growth of multiple types of cells in the pulmonary circulation.18 The reduction of these cells may reduce pulmonary vascular resistance, thereby reducing the strain on the RV. The chronic hypoxia model of pulmonary hypertension in rats has been well utilized for the study of pulmonary vascular remodeling. The recently developed SUGEN/hypoxia model of pulmonary hypertension exhibits pathologic features similar to human PAH.19–20
In the present study, these animal models demonstrate that the administration of antitumor agents in rats with pulmonary hypertension reduces the pulmonary vascular wall thickness of the media and the intima within 1 to 3 days. The death of pulmonary vascular cells and the reduction of pulmonary vascular wall thickness only occur in the pulmonary arteries of animals with pulmonary hypertension, but not in the vessels of normal, control animals. We investigated the mechanisms and the consequences for the susceptibility of remodeled pulmonary vessels to drug‐induced cell death.
Methods
Animal Treatment
Male Sprague‐Dawley rats (≈250 g) were subjected to chronic hypoxia in a chamber (30′′ wide×20′′ deep×20′′ high) regulated by an OxyCycler Oxygen Profile Controller (Model A84XOV; BioSpherix) that was set to maintain 10% O2 with an influx of nitrogen gas, located in the animal care facility at Georgetown University Medical Center.21 Ventilation to the outside of the chamber was adjusted to remove CO2, such that its level did not exceed 5000 ppm. Control animals were subjected to ambient 21% O2 (normoxia) in another chamber. Animals were fed normal rat chow during the treatment.
For the chronic hypoxia model of pulmonary hypertension, rats were subjected to hypoxia (10% O2) for 2 weeks.21 For the SUGEN/hypoxia model,19–20 rats were subcutaneously injected with SU‐5416 (20 mg/kg body weight), maintained in hypoxia for 3 weeks, then in normoxia for 5 weeks.
After pulmonary hypertension and pulmonary vascular remodeling are developed, rats were then injected intraperitoneally with antitumor drugs including DNR (Sigma‐Aldrich; 2 or 5 mg/kg body weight), bortezomib (Selleck Chemicals; 1 mg/kg body weight), and MG‐132 (Selleck; 10 mg/kg body weight). Vehicles used were equal amounts of saline for DNR, and equal amounts of DMSO in saline for MG‐132 and bortezomib. Animals were placed back in hypoxia or normoxia for an additional 1 to 3 days before lungs and hearts were harvested.
At the end of the experiments, some rats were anesthetized with intraperitoneal injections of xylazine (10 mg/kg) and ketamine (100 mg/kg). They were then intubated and mechanically ventilated with a volume‐controlled Inspira Advanced Safety Ventilator (Harvard Apparatus). Rats were maintained on a heat pad and the temperature was kept at 37°C using a TR‐200 Temperature Controller connected to a rectal probe (Fine Scientific Tools). After a thoracotomy through the third left intercostal space, a Millar catheter (1.4 F) was inserted to the RV. RV pressure signals were recorded using PowerLab with Chart 5 software (ADInstruments).
The Georgetown University Animal Care and Use Committee approved all animal experiments, and the investigation conforms to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Histological Measurements
Tissues were immersed in buffered 10% paraformaldehyde at room temperature, and were embedded in paraffin. Paraffin‐embedded tissues were cut and mounted on glass slides. Tissue sections were stained with hematoxylin and eosin (H&E). Slides were blinded and analyzed microscopically for pulmonary arterial wall thickness and vessel radius. Six to eight vessels were analyzed per animal, and 6 values for thickness and 2 values for radius measured for each vessel. The percentage of wall thickness values defined as wall thickness divided by vessel radius were determined using IP Lab Software (Scanalytics Inc). Tissue sections were also evaluated for the occurrence of apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore).
Immunohistochemistry (IHC) was performed using a rabbit monoclonal antibody for cleaved caspase‐3 (Asp175) (Cell Signaling Technology) or a goat polyclonal antibody for Bcl‐xL (Santa Cruz Biotechnology).
Western Blot Analysis
Extrapulmonary arteries (left and right main branches) and intrapulmonary arteries (first‐order branch) were surgically dissected, and connective tissues were gently removed in ice‐cold PBS under a dissecting microscope. Tissues were minced and homogenized, and protein gel electrophoresis samples were prepared as previously described.21 For western blotting, equal protein amounts of samples were electrophoresed through a reducing SDS polyacrylamide gel and electroblotted onto a membrane. The membrane was blocked and incubated with antibodies for cleaved caspase‐3, LC3B, parkin (Cell Signaling Technology), Bcl‐xL, annexin A1, GATA4, and p62 (Syd Labs, Inc), and levels of proteins were detected using horseradish peroxidase‐linked secondary antibodies and an Enhanced Chemiluminescence System (Amersham Life Science).
Cell Culture Experiments
Human PASMCs were purchased from ScienCell Research Laboratories and Cell Applications, Inc and were cultured in accordance with the manufacturers' instructions in 5% CO2 at 37°C. Cells in passages 3 to 7 were used. PASMCs were differentiated using the Differentiation Medium from Cell Applications in accordance with the manufacturer's instructions. For siRNA knock‐down, cells were transfected with siRNA Transfection Reagent and gene silencing siRNAs along with control with scrambled sequence from Santa Cruz Biotechnology. Cells were used for experiments 2 days after transfection. Transfection and reporter assays were performed as previously described.8
Statistical Analysis
Means and standard errors were calculated. Comparisons between 2 groups were analyzed by a 2‐tailed Student t test, and comparisons between 3 or more groups were analyzed by 2‐way analysis of variance (ANOVA) with a Student‐Newman‐Keuls post‐hoc test using the GraphPad Prism (GraphPad Software, Inc), in accordance with the Kolmogorov‐Smirnov test for normality. In some cases, the non‐parametric Mann‐Whitney test was used for the comparison between 2 groups, and rank transformation was used for the comparison between 3 or more groups. P<0.05 was considered to be significant.
Results
Antitumor Drugs Reduce the Thickness of Pulmonary Vascular Walls in Rats With Pulmonary Hypertension Without Affecting Normal Vessels
In the chronic hypoxia model, animals exhibit increased pulmonary arterial pressure, RV hypertrophy, and increased pulmonary vascular wall thickness.21 After the pulmonary vascular remodeling was established by the 2‐week treatment with sustained hypoxia, MG‐132 was administered, and animals were then placed back in the hypoxic environment. Three days after the administration of MG‐132, we found that pulmonary vascular wall thickness was significantly reduced in arterioles and small arteries (Figure 1A) and in large arteries (Figure 2A) in animals with pulmonary hypertension. In contrast, MG‐132 had no effects on pulmonary vascular wall thickness of normal control rats (Figures 1A and 2A).
Figure 1.
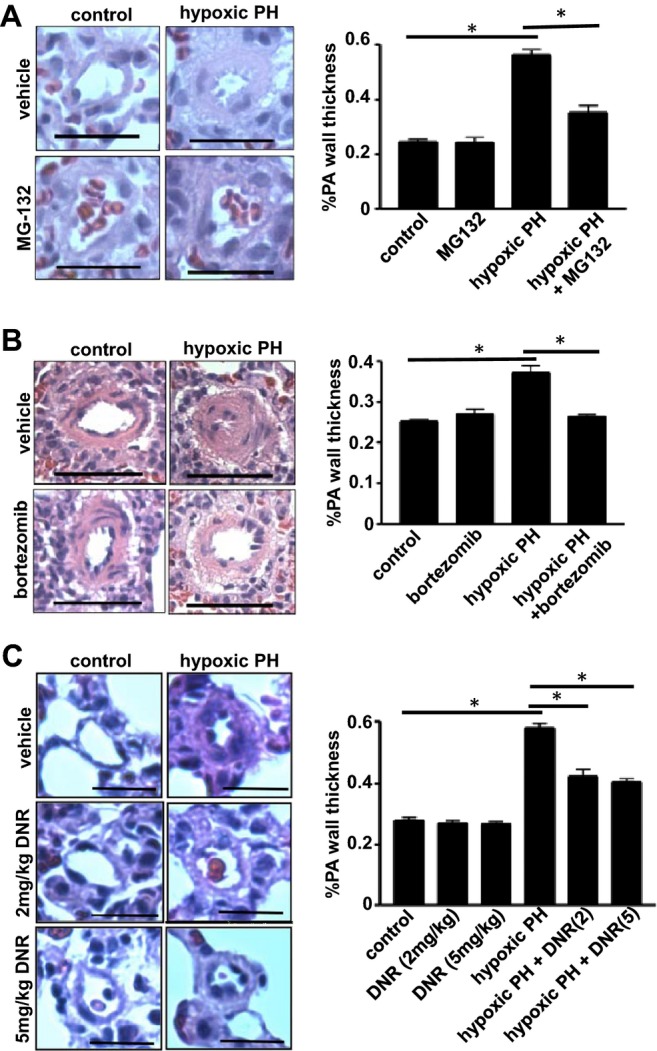
Antitumor agents reduce the wall thickness of remodeled pulmonary arterioles and small pulmonary arteries in the chronic hypoxia model of pulmonary hypertension (PH). Rats were exposed to hypoxia for 2 weeks, and were then injected with (A) 10 mg/kg body weight MG‐132 (n=4), (B) 1 mg/kg body weight bortezomib (n=3), or (C) 2 or 5 mg/kg body weight DNR (n=6). Rats were then placed back in hypoxic environment. Three days after the injection, lungs were harvested, immersed in buffered 10% paraformaldehyde, and embedded in paraffin for H&E staining. Bar graphs represent means±SEM of % pulmonary arterial (PA) wall thickness of pulmonary arterioles/small pulmonary arteries with diameters ranging from 36.4 to 97.6 μm (mean diameter 68.1 μm) as determined by analyzing H&E stained slides. *Values significantly different from each other at P<0.05. Scale bars, 50 μm. DNR indicates daunorubicin; H&E, hematoxylin and eosin.
Figure 2.
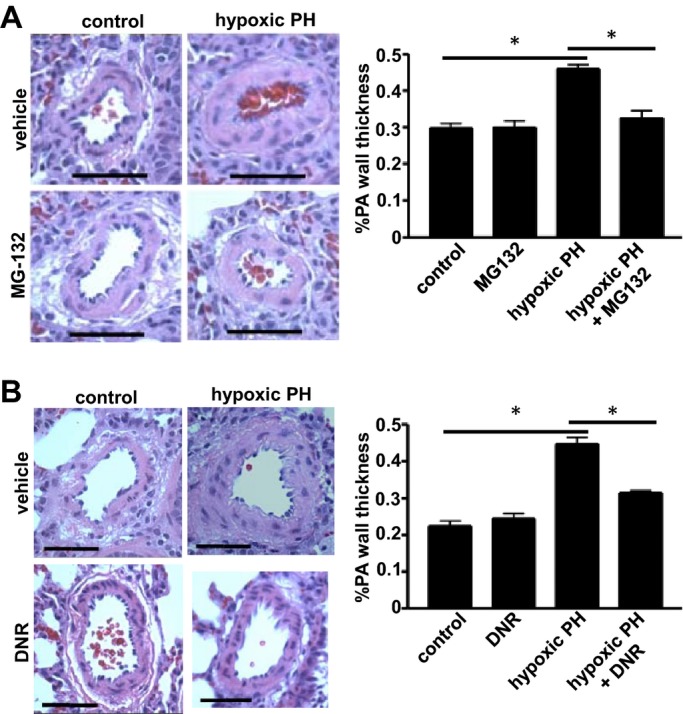
Antitumor agents reduce the wall thickness of remodeled large pulmonary arteries (PA). Rats were exposed to hypoxia (10% O2) for 2 weeks, and were then injected with (A) MG‐132 at 10 mg/kg body weight (n=4) or (B) DNR at 5 mg/kg (n=6). Rats were then placed back in hypoxia. Three days after the injection, lungs were harvested for H&E staining. Bar graphs represent means±SEM of % PA wall thickness of large PA with diameters ranging from 122.2 to 377.4 μm (mean diameter 224.4 μm). *Values significantly different from each other at P<0.05. Scale bars, 100 μm. DNR indicates daunorubicin; H&E, hematoxylin and eosin; PH, pulmonary hypertension.
Bortezomib is a proteasome inhibitor that has been approved for the treatment of multiple myeloma.9 The administration of bortezomib to intact rats with pulmonary hypertension induced by chronic hypoxia similarly resulted in reversal of pulmonary vascular remodeling without affecting the normal pulmonary vessels (Figure 1B).
DNR is effective in promoting the apoptosis of PASMCs.8 DNR was also found to reduce the wall thickness in arterioles and small arteries (Figure 1C) and large arteries (Figure 2B) of animals with pulmonary hypertension. The effect of DNR to reverse pulmonary vascular remodeling was apparent as early as 1 day after the administration (Figure 3). While DNR reduced the thickness of pulmonary vascular walls in animals with pulmonary hypertension, this agent had no effect on the pulmonary vascular wall thickness of normal rats.
Figure 3.
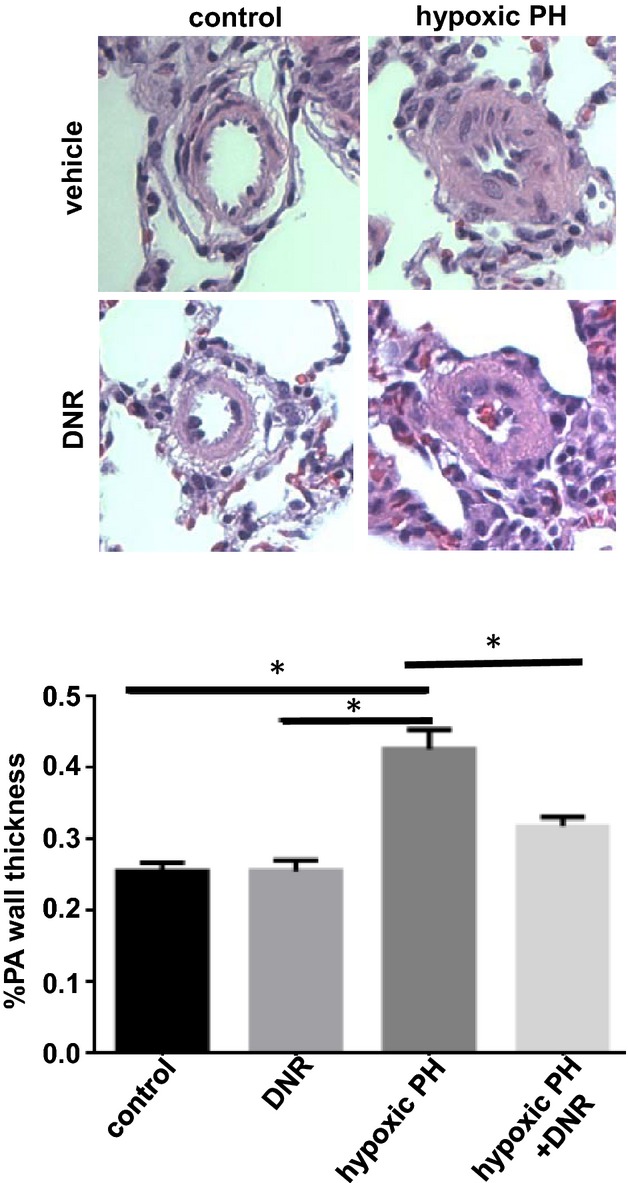
Reduction of the thickness of pulmonary arteries (PA) occurs as early as 1 day after the DNR administration. Rats were exposed to hypoxia (10% O2) for 2 weeks, and then injected with DNR at 5 mg/kg body weight. Rats were then placed back in hypoxia. One day after the injection, lungs were harvested for H&E staining. The bar graph represents means±SEM (n=4) of % PA wall thickness of pulmonary arterioles/small PA. *Values significantly different from each other at P<0.05. DNR indicates daunorubicin; H&E, hematoxylin and eosin; PH, pulmonary hypertension.
SU‐5416 injection plus hypoxia has been shown to generate pathologic lesions resembling human idiopathic PAH.19–20 Accordingly, rats were injected with SU‐5416, and then placed in hypoxia for 3 weeks. Subsequently, animals were maintained in normoxia for 5 weeks to develop severe PAH with RV systolic pressure of ≈70 mm Hg as well as increased intimal and medial thickening as visualized by hematoxylin and eosin (H&E) staining (Figure 4). Rats were then injected with MG‐132. Three days after the injection, pulmonary vascular remodeling of both the intima and the media was found to be reversed (Figure 4). In contrast, MG‐132 had no effects on pulmonary vascular wall thickness in control rats (Figure 4). Effects of MG‐132 to reverse pulmonary vascular remodeling persisted at least 2 weeks after the administration (Figure 5).
Figure 4.
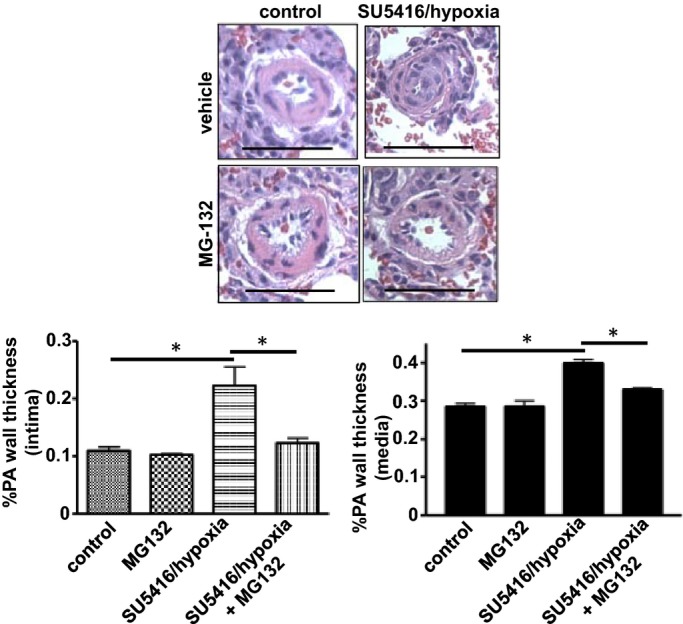
Reduction of pulmonary intimal and medial thickness in the SU‐5416/hypoxia model of pulmonary hypertension. Rats were injected with SU‐5416, placed in hypoxia for 3 weeks, and then maintained in normoxia for 5 weeks. Rats were then injected with 10 mg/kg body weight MG‐132. Three days after the injection, lungs were harvested, immersed in buffered 10% paraformaldehyde, and embedded in paraffin for H&E staining. Bar graphs represent means±SEM of intima and media thickness of pulmonary arterioles/small pulmonary arteries (PA) with diameters ranging from 36.4 to 97.6 μm (mean diameter 68.1 μm), as determined by analyzing H&E stained slides. *Values significantly different from each other at P<0.05 (n=4). Scale bars, 50 μm. H&E indicates hematoxylin and eosin.
Figure 5.
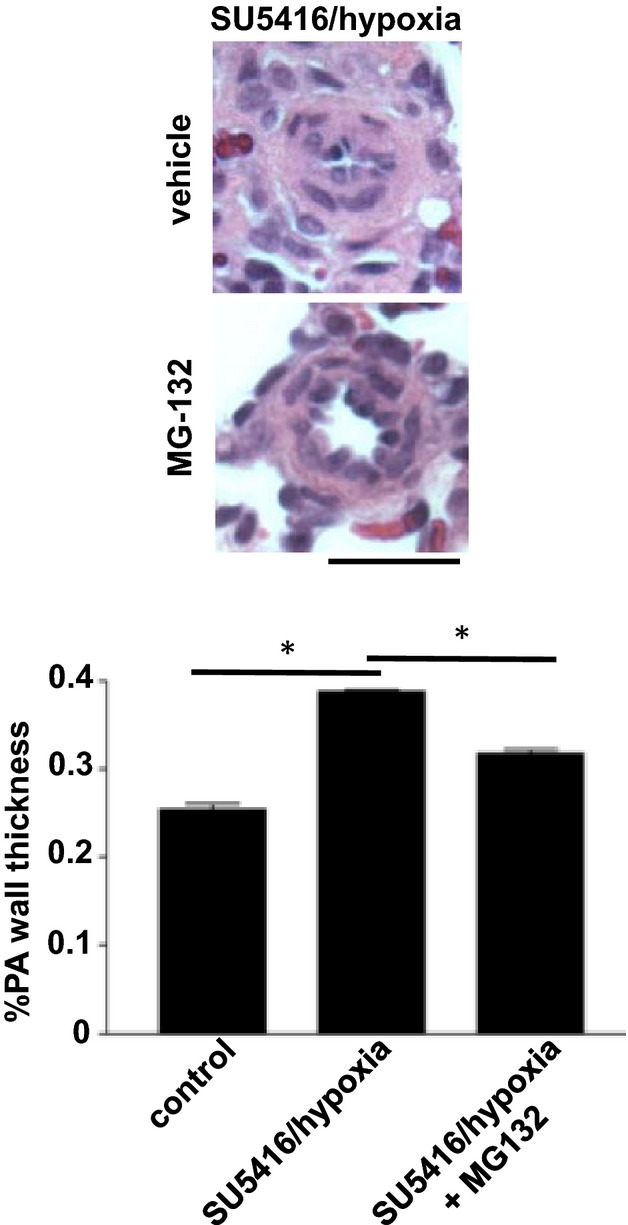
Reduction of pulmonary arterial (PA) wall thickness in the SU‐5416/hypoxia model of pulmonary hypertension after 2 weeks of MG‐132 treatment. Rats were injected with SU‐5416, placed in hypoxia for 3 weeks, and then maintained in normoxia for 5 weeks. Rats were then injected with 10 mg/kg body weight MG‐132. Two weeks after the injection, lungs were harvested, immersed in buffered 10% paraformaldehyde, and embedded in paraffin for H&E staining. The bar graph represents means±SEM of PA wall thickness of pulmonary arterioles/small pulmonary arteries (PA) with diameters ranging from 36.4 to 97.6 μm (mean diameter 68.1 μm), as determined by analyzing H&E stained slides. *Values significantly different from each other at P<0.05 (n=4). Scale bars, 50 μm. H&E indicates hematoxylin and eosin.
Antitumor Drugs Promote Apoptosis in the Remodeled Pulmonary Vessels, but not in Normal Pulmonary Vessels
To study the mechanism of how these antitumor agents may reverse pulmonary vascular remodeling, the occurrence of apoptosis was monitored. We found, using TUNEL assay, that MG‐132 indeed promoted apoptosis in the pulmonary vascular wall of intact rats with pulmonary hypertension, but not in normal rats (Figure 6A), 3 days after the administration. Similarly, DNR was found to promote apoptosis as monitored by TUNEL assay (Figure 6B) as well as immunohistochemistry (Figure S1A) and immunoblotting (Figure S1B) for cleaved caspase‐3 formation. The cleaved caspase‐3 expression was detectable as early as 1 day after DNR administration (Figure 6C).
Figure 6.
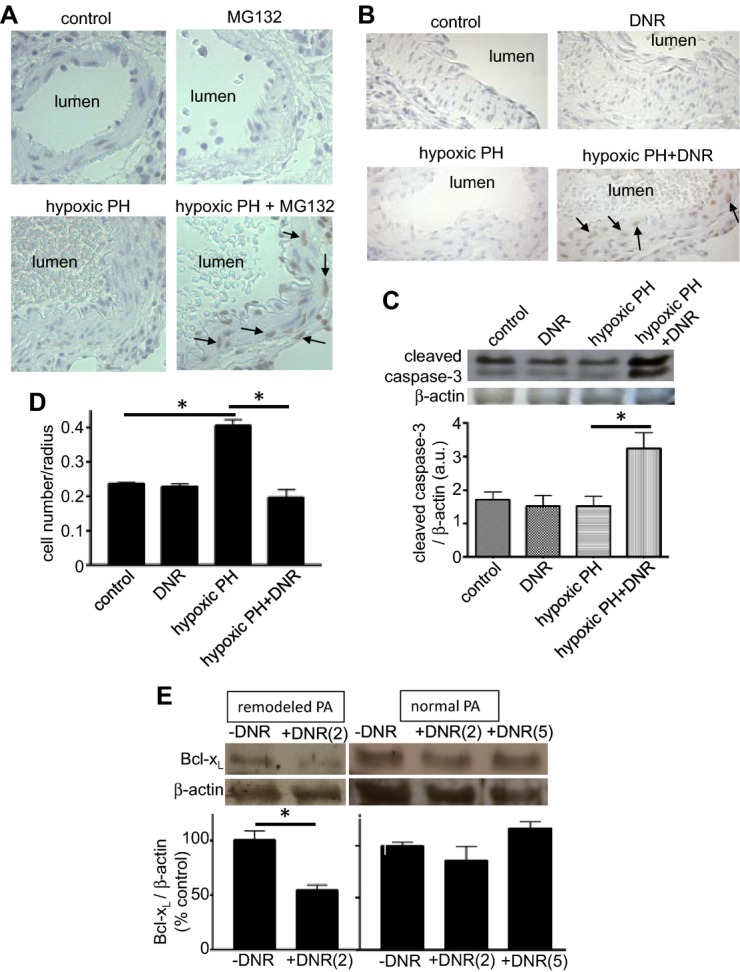
Antitumor agents promote apoptosis in remodeled pulmonary arteries without affecting normal pulmonary arteries. Rats were exposed to hypoxia for 2 weeks to promote pulmonary hypertension (PH), and were then injected with (A) MG‐132 or (B through D) DNR. Rats were then placed back in hypoxia. Three days (for A, B, and D) or 1 day (for C) after the injection, lungs were harvested. A and B, Harvested lungs were immersed in buffered 10% paraformaldehyde, and embedded in paraffin for TUNEL assay. C, Western blotting of cleaved caspase‐3 in homogenized isolated pulmonary arteries. The bar graph represents means±SEM of the ratio of cleaved caspase‐3 to β‐actin expressed in arbitrary unit (a.u.) (n=4). D, The number of cell nuclei of pulmonary arterial media was counted in H&E stained slides. The bar graph represents means±SEM (n=3). E, Rats were exposed to hypoxia for 2 weeks to develop PA remodeling, and were then injected with DNR (2 or 5 mg/kg body weight) or saline. Rats were then placed back in hypoxia. Three days after the injection, lungs were harvested. PAs were surgically isolated from rats with remodeled PA or from normal rats, homogenized, and subjected to western blotting analysis for Bcl‐xL protein expression (n=3). *Values significantly different from each other at P<0.05. DNR indicates daunorubicin; H&E, hematoxylin and eosin; PA, pulmonary arteries; TUNEL, transferase dUTP nick end labeling.
To assess if apoptosis reduced the number of cells in the vessel wall, the number of cells was counted in our histological specimen of the lungs from rats treated with and without DNR. We found that DNR significantly reduced the number of cells in remodeled pulmonary arteries (Figure 6D). Only pulmonary arteries of animals with pulmonary hypertension were susceptible to cell death induced by cancer chemotherapeutic agents; no cell deaths were detected in normal rats (Figure 6D).
Mechanism of Differential Susceptibility to Apoptosis in Remodeled and Normal Pulmonary Vessels
The reduced expression of anti‐apoptotic proteins Bcl‐xL and Bcl‐2 can serve as a mechanism for the induction of apoptosis.22 Consistently, the administration of DNR reduced the expression of Bcl‐xL and Bcl‐2 in the pulmonary arteries of rats with vascular thickening, as monitored by immunoblotting (Figure 6E) and immunohistochemistry (IHC) (Figure S2). The significant reduction of the expression of these anti‐apoptotic proteins did not occur in the pulmonary arteries of normal control rats.
We further studied the regulation of Bcl‐xL expression in cultured proliferating PASMCs to provide mechanistic insights. Bcl‐xL gene transcription is regulated by the P1/P2 region, which occurs within 802 bp upstream from the translational start site of the bcl‐x promoter as depicted in Figure 7A.23 Indeed, a truncation of 3.2 to 0.6 kb did not affect the basal expression of luciferase reporter controlled by the bcl‐x promoter24 in transfected cultured PASMCs (Figure 7B), indicating that transcription factors that regulate the P1/P2 region are active in these cells. There are 2 GATA elements within this region, which play an important role in the transcriptional control of Bcl‐xL (Figure 7A). GATA DNA‐binding activities in both distal and proximal regions can be activated by the stimulation of PASMCs (Figure 7C). While the major GATA factor in smooth muscle of the systemic circulation has been identified as GATA6,25 we have shown previously that cultured bovine PASMC express GATA4.26 Consistently, GATA4 was found to be the major GATA DNA‐binding protein in PASMCs as determined by supershifting with the GATA4 antibody. The GATA4 antibody supershifted the GATA DNA‐binding protein in PASMCs, but not in aortic SMCs (Figure 7D), demonstrating that GATA4 is a pulmonary‐specific GATA factor of SMCs. Gata4 mRNA (Figure 7E) and GATA4 protein (Figure 7F) were also detected in the rat pulmonary artery. In cultured PASMCs, the forced expression of GATA4 increased Bcl‐xL expression (Figure 7G), and the expression of dominant negative GATA4 decreased Bcl‐xL expression, but not the expression of unrelated ERK protein (Figure 7H), providing cause‐and‐effect relationships between GATA4 and Bcl‐xL regulation. In PASMCs, DNR inhibited GATA4 DNA‐binding activity (Figure 7I), consistent with the idea that DNR promotes apoptosis by reducing the levels of Bcl‐xL via suppressing its GATA4‐dependent gene transcription.
Figure 7.
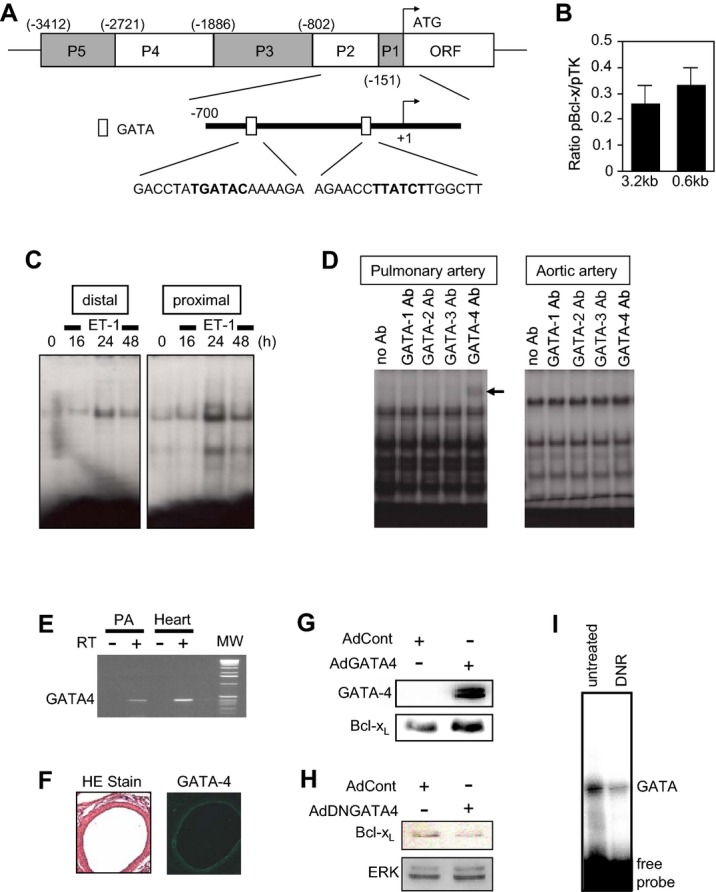
Mechanism of Bcl‐xL gene regulation in PASMCs. A, Schematics of the structure of bcl‐x gene promoter. B, PASMCs were transfected with 3.2 kb or 0.6 kb bcl‐x promoter‐controlled firefly luciferase with renilla luciferase controlled by thymidine kinase (TK) promoter as a control. Luciferase activities were monitored by dual luciferase system. C, Cells were treated with endothelin‐1 (ET‐1) for duration indicated. Nuclear extracts were subjected to band‐shift assays using 32P‐labelled oligonucleotide containing distal (−455 to −450) or proximal (−109 to −104) GATA elements within the P1/P2 region of bcl‐x promoter. D, Nuclear extracts from PASMCs and aortic SMCs were subjected to band‐shift assay with the GATA probe in the presence of antibodies (Ab) against GATA‐1, −2, −3, or −4. The arrow indicates the supershifted band by GATA4 in PASMCs. E, Total RNA was isolated from the rat pulmonary artery (PA) or the heart (as a control). GATA4 mRNA was monitored by RT‐PCR. Bands were not detected in lanes without reverse transcription (RT). F, Histological analyses of the rat lung sections with H&E stain (left panel) and immunofluorescence staining with GATA4 antibody (right panel). G, PASMCs were infected with control adenovirus (AdCont) or adenovirus expressing GATA4 (AdGATA4). Cell lysates were prepared and equal protein amounts were subjected to SDS‐PAGE and western blotting with GATA4 or Bcl‐xL antibody. H, PASMCs were infected with control adenovirus (AdCont) or adenovirus expressing dominant negative GATA4 (AdDNGATA4) for 48 hours. Cell lysates were prepared and subjected to SDS‐PAGE and western blotting with a Bcl‐xL antibody. ERK protein levels were also measured as a control. I, PASMCs were treated with DNR, and the GATA4 DNA binding activity was monitored by band‐shift assays. DNR indicates daunorubicin; H&E, hematoxylin and eosin; PAGE, polyacrylamide gel electrophoresis; PASMC, pulmonary artery smooth muscle cell; SDS, sodium dodecyl sulfate.
We have previously cloned GATA4 promoter and identified a mechanism of its regulation, in which CBF/NF‐Y‐binding to the GATA4 promoter is suppressed by annexin A1.21 To test the hypothesis that drug‐induced downregulation of Bcl‐xL is dependent on annexin A1, PASMCs were transfected with siRNA to knock‐down annexin A1. We found that knocking‐down annexin A1 inhibited DNR‐induced downregulation of Bcl‐xL (Figure 8A). Annexin A1 knock‐down also suppressed the death of PASMCs induced by MG‐132 (Figure 8B), which also downregulates Bcl‐xL (data not shown). Consistent with our previous findings that annexin A1 is susceptible to iron‐catalyzed, oxidation‐dependent proteasomal degradation,27 we found that cell death agents such as MG‐132 increased the expression of annexin A1 (Figure S3), whereas growth factors such as endothelin‐1 and chronic hypoxia decreased annexin A1 expression in an iron‐dependent fashion (Figure S4), further providing the evidence for the role of annexin A1 in cell regulation. In intact rats, the DNR administration increased the annexin A1 expression only in remodeled pulmonary arteries, but not in normal pulmonary arteries (Figure 8C).
Figure 8.
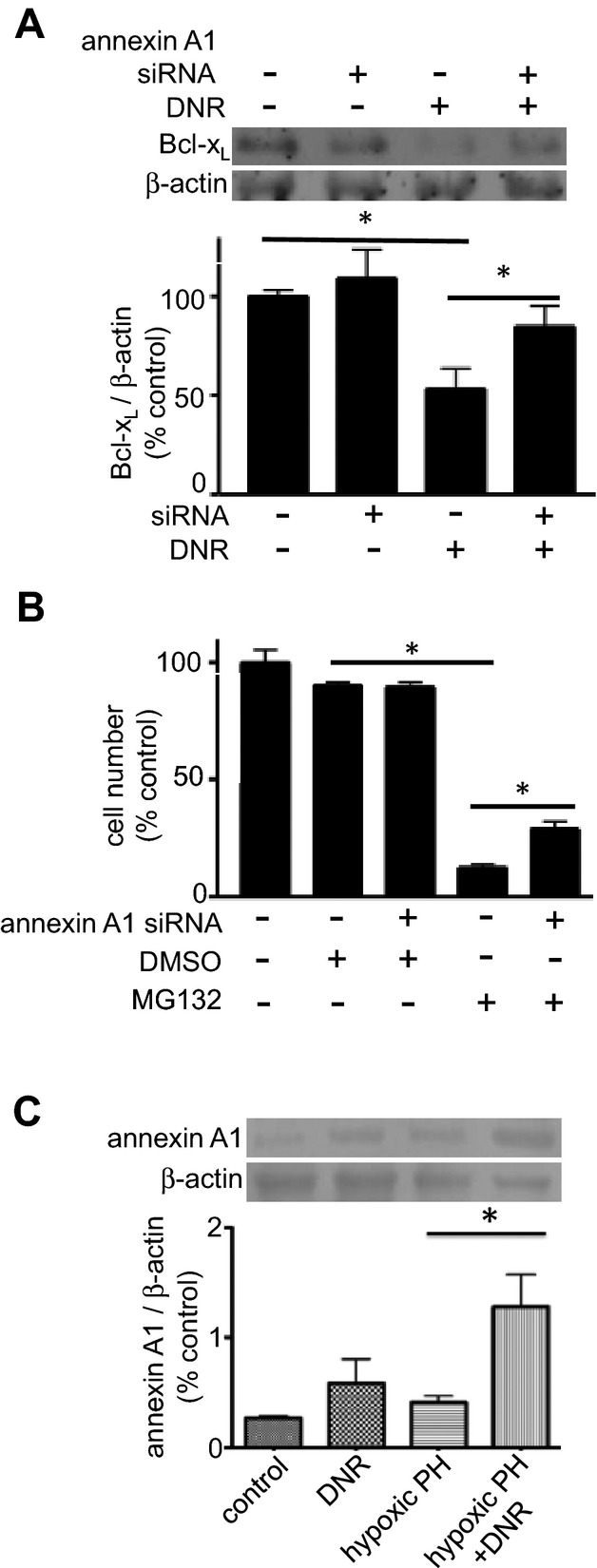
Mechanism of apoptosis in remodeled pulmonary arteries (PA). A, Cultured PASMCs were transfected with siRNA to knock‐down annexin A1. Cells were then treated with DNR (1 μmol/L) for 22 hours and Bcl‐xL protein expression was monitored by western blotting (n=6). B, The number of PASMCs was determined after siRNA knock‐down of annexin A1 and MG‐132 (0.9 μmol/L) treatment for 4 days (n=8 for all the values with the exception of the “annexin A1 siRNA+DMSO” value with n=5). C, Rats were exposed to hypoxia for 2 weeks, and then injected with DNR (5 mg/kg body weight). Rats were placed back in hypoxia. One day after the injection, pulmonary arteries were isolated, homogenized, and subjected to western blotting for annexin A1 protein expression (n=4). DMSO indicates dimethyl sulfoxide; DNR, daunorubicin; PASMC, pulmonary artery smooth muscle cell; PH, pulmonary hypertension.
Role of Autophagy in the Mechanism of Reversal of Pulmonary Vascular Remodeling
In addition to apoptosis, autophagy can also regulate programmed cell death.28 We found that DNR promoted the formation of LC3B‐II (Figure 9A) and the downregulation of p62 (Figures 9B and S5), 2 major hallmarks for the occurrence of autophagy,29 in remodeled pulmonary arteries, but not in normal pulmonary arteries. Similarly, both the formation of LC3B‐II (Figure 9C) and the downregulation of p62 (Figure 9D) occurred in response to treating cultured human PASMCs, when cells are in the proliferating phenotype, but not in the differentiated phenotype. We propose that these results represent proliferating PASMCs mimicking cells that are killed in the remodeled pulmonary vasculature, and differentiated PASMCs having a phenotype that is resistant to drug‐induced killing, as in the contractile phenotypic cells that exist in the normal pulmonary vessels. Consistent with this idea, proliferating PASMCs were found to be more susceptible to DNR‐induced killing as shown in the representative images of cultured cells (Figure S6) and in the quantitative assessment of the number of cells (Figure 9E). To provide the direct evidence for the role of the autophagic process in cell killing, siRNA was used to knock down LC3B (Figure S7). Knocking down LC3B in proliferating human PASMCs significantly suppressed DNR‐induced cell killing (Figure 9F).
Figure 9.
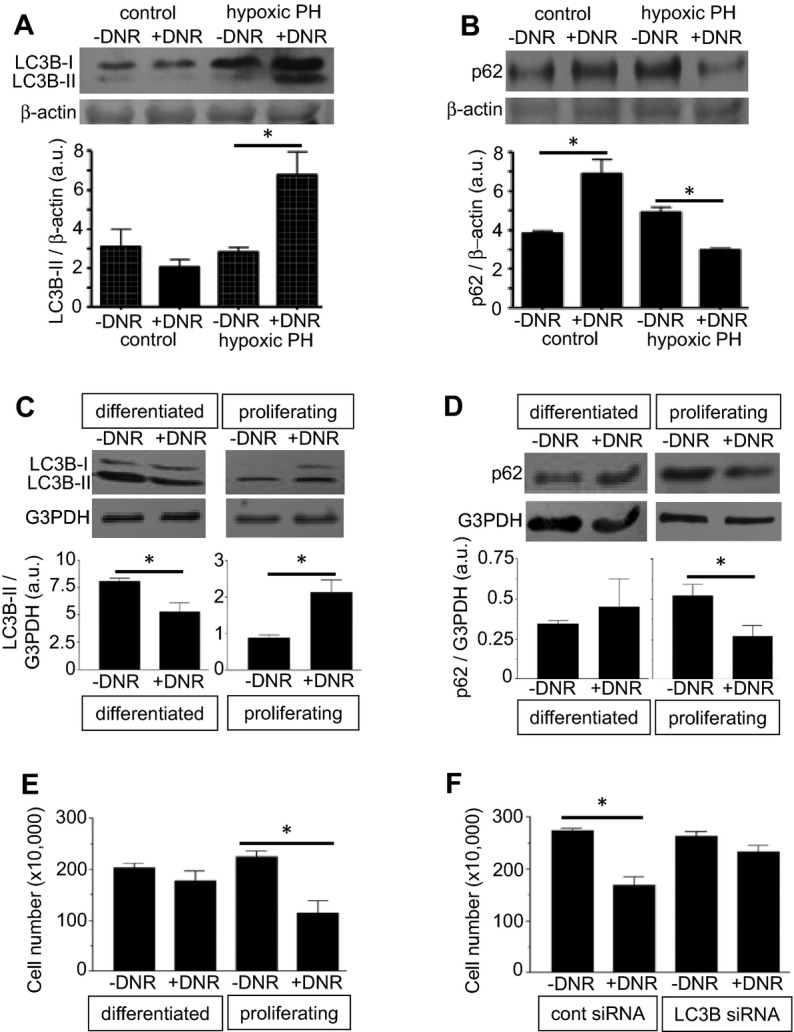
Antitumor agents promote autophagy in remodeled pulmonary arteries without affecting normal pulmonary arteries. A and B, Rats were exposed to hypoxia for 2 weeks to develop pulmonary hypertension (PH), and were then injected with DNR. Rats were then placed back in hypoxia. Three days after the injection, isolated pulmonary arteries were homogenized and LC3B (n = 4) and p62 (n=3) levels were monitored by western blotting. C and D, Differentiated and proliferating cultured human PASMCs were treated with DNR for 22 hours. Cell lysates were prepared and LC3B (n=5 for differentiated; n=4 for proliferating) and p62 (n=3 for differentiated; n=4 for proliferating) levels were monitored by western blotting. E, The cell number was determined by counting on a hemocytometer (n=4). F, Human PASMCs were transfected with control or LC3B siRNA, treated with DNR, then the cell number was determined by counting on a hemocytometer (n=4). Bar graphs represent means±SEM. *Values significantly different from each other at P<0.05. DNR indicates daunorubicin; PASMC, pulmonary artery smooth muscle cell.
Mechanism of Differential Susceptibility to Autophagy in Remodeled and Normal Pulmonary Vessels
To address the mechanism of differential susceptibilities of cells of remodeled and normal pulmonary vessels to drug‐induced killing, we tested a novel hypothesis for the involvement of parkin, which has been described as a cell survival protein.30 We found that the parkin expression is upregulated in normal pulmonary vessels, but not in remodeled vessels, in rats treated with DNR (Figures 10A and S8). Similarly, DNR upregulated parkin in differentiated human PASMCs, but not in proliferating cells (Figure 10B). These results may suggest that the normal differentiated contractile phenotype of PASMCs may be more resistant to drug‐induced cell death because of the ability to upregulate parkin that serves as a survival factor. Consistent with this concept, siRNA knock‐down of parkin in human PASMCs (Figure S9) enhanced DNR‐induced cell killing (Figure 10C). Further, results showing that siRNA knock‐down of parkin enhanced DNR‐induced autophagy (as indicated by the downregulation of p62; Figure 10D) suggest that parkin‐mediated cell survival is linked to its ability to suppress autophagy.
Figure 10.
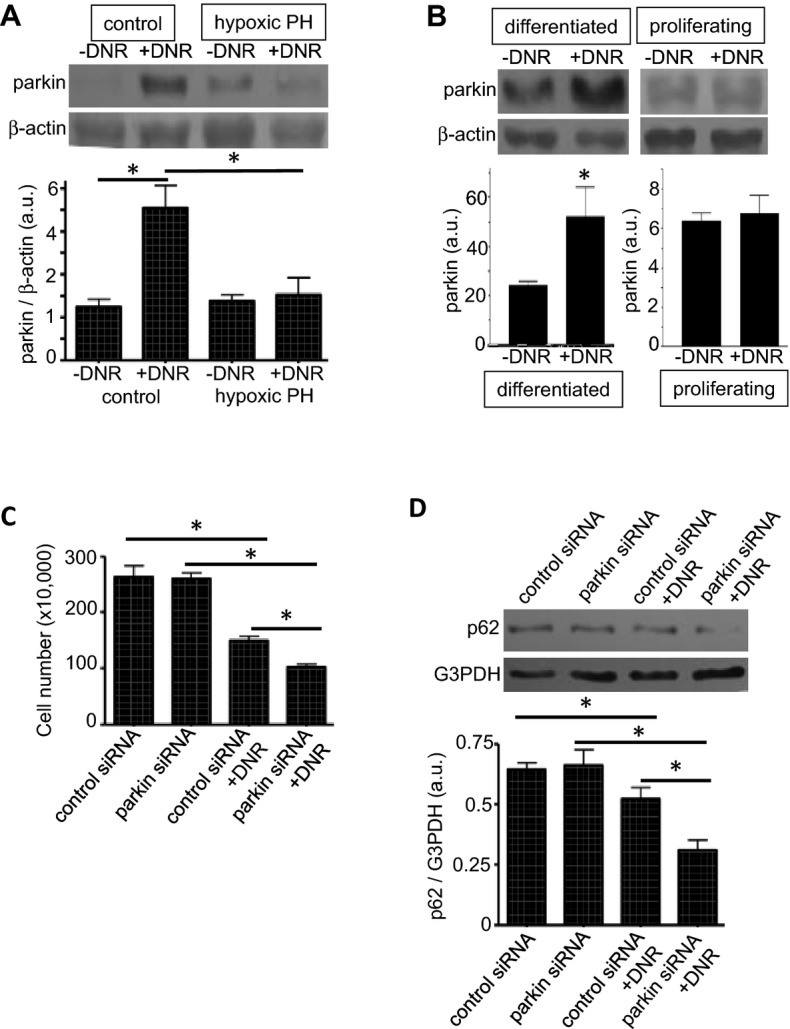
Parkin regulates drug‐induced autophagy. A, Rats were exposed to hypoxia for 2 weeks to develop pulmonary hypertension (PH), and were then injected with DNR (5 mg/kg body weight). Rats were then placed back in hypoxia. One day after the injection, isolated pulmonary arteries were homogenized, and parkin levels were monitored by western blotting (n=4). B, Differentiated and proliferating cultured human PASMCs were treated with DNR for 5 hours. Cell lysates were prepared, and parkin levels were monitored by western blotting (n=6 for differentiated; n=4 for proliferating). C, Human PASMCs were transfected with control or parkin siRNA, treated with DNR, then the cell number was determined by counting on a hemocytometer (n=3 for −DNR; n=4 for +DNR). D, Human PASMCs were transfected with control or parkin siRNA, treated with DNR, then p62 expression levels were determined by western blotting (n=4). Bar graphs represent means±SEM. *Values significantly different from each other at P<0.05. DNR indicates daunorubicin; PASMC, pulmonary artery smooth muscle cell.
Reversal of Pulmonary Vascular Remodeling Increases the Efficacy of Vasodilators to Reduce Pulmonary Arterial Pressure
Ultimately, the treatment for pulmonary hypertension should reduce the burden to the RV by decreasing pulmonary arterial and RV pressures. Using a Millar catheter, we measured RV pressure in pulmonary hypertensive rats treated with apoptotic agents that reduced pulmonary vascular thickness. We found that neither DNR (Figure 11A), bortezomib (Figure 11B), nor MG‐132 (Figures 11C and 11D) reduced RV systolic pressure that was increased in response to treatment of rats with chronic hypoxia or with SU‐5416/hypoxia when these agents were administered after pulmonary vascular remodeling had been developed.
Figure 11.
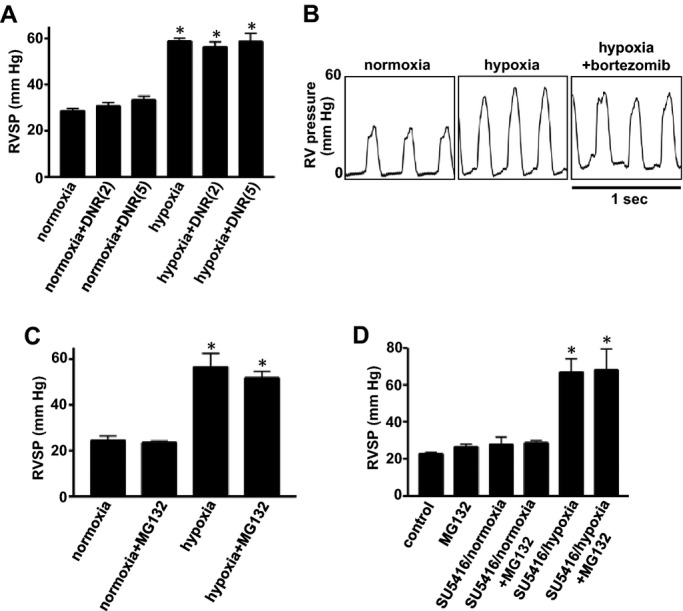
Effects of apoptotic agents on RV pressure. A through C, Rats were exposed to normoxia or hypoxia for 2 weeks, and then injected with DNR (2 and 5 mg/kg body weight; n=4 for “normoxia+DNR(5)” and “hypoxia+DNR(5)”; n=3 for others), bortezomib (1 mg/kg body weight), or MG‐132 (10 mg/kg body weight; n=4). Rats were then placed back in normoxic and hypoxic environments. Three days after the injection, rats were anesthetized and mechanically ventilated. A Millar catheter was inserted into the RV, and RV pressure was recorded. Bar graphs represent means±SEM of RV systolic pressure (RVSP). D, Rats were subjected to SU‐5416 administration and 3 weeks of hypoxia. Five weeks later after severe pulmonary hypertension was developed, rats were injected with MG‐132 (10 mg/kg body weight). RV pressure was recorded 3 days after the injection of MG‐132 (n=4). Bar graphs represent means±SEM. *P<0.05 vs normoxia. DNR indicates daunorubicin; RV, right ventricle.
We hypothesized that the inability of apoptotic agents to normalize the RV pressure, despite the successful regression of thickened pulmonary arterial walls, is because of vasoconstriction that may occur as an adaptation to maintain pulmonary arterial pressure. Thus, we tested whether the reduction of pulmonary vascular wall thickness resulted in an increased susceptibility to vasodilators by examining the acute responses to sodium nitroprusside (SNP). Rats with pulmonary vascular remodeling with or without DNR administration were injected with SNP while monitoring the RV pressure by a Millar catheter. Under the condition where SNP had minimal effects on RV pressure in rats with pulmonary hypertension that were injected with saline, SNP significantly reduced RV pressure in rats that were treated with DNR to reverse pulmonary vascular remodeling in a dose‐dependent manner (Figure 12). Similarly in the SU‐5416/hypoxia model, SNP‐induced reduction of RV pressure was greater in rats treated with MG‐132 (66.6±3.2 to 44.2±4.3 mm Hg) than those treated with the vehicle only (67.5±3.9 to 58.3±6.8 mm Hg).
Figure 12.
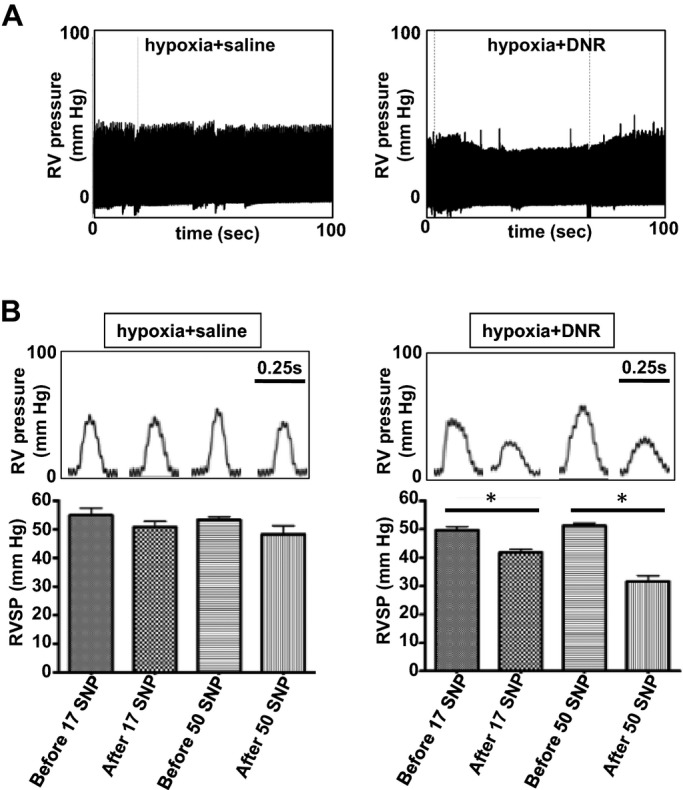
Apoptotic agents increase the susceptibility of vasodilators to reduce RV pressure. Rats were exposed to chronic hypoxia for 2 weeks, and then injected with DNR (5 mg/kg body weight). Rats were then placed back in the hypoxic environment. Three days after the injection, rats were anesthetized and mechanically ventilated. A Millar catheter was inserted into the RV apex, and RV pressure was recorded. After stabilization of hemodynamic recording for 10 minutes, SNP (17 or 50 μg/kg body weight) was slowly administered through the jugular vein in a 100 μL injection volume. A, Representative traces in which SNP was administered in animals treated with saline or DNR. B, Bar graphs represent means±SEM (n=4) of RV systolic pressure (RVSP) before and after the SNP administration. *P<0.05. DNR indicates daunorubicin; RV, right ventricle; SNP, sodium nitroprusside.
Effects of DNR on the RV Structure and Function
Effects of cardiotoxic agents like DNR on normal or hypertrophied RV are unknown. We found that DNR at 2 or 5 mg/kg body weight did not influence the RV contractility, as determined by monitoring dP/dtmax (Figure 13A) and dP/dtmin (Figure 13B) either in control animals or in animals with pulmonary hypertension. DNR did not affect the RV structure as determined by Fulton index calculating RV/(LV+S) (Figure 13C) and as indicated histologically in H&E‐stained hearts (Figure 13D). DNR did not promote apoptosis of cardiomyocyes in the hypertrophied RV in rats with pulmonary hypertension, as indicated by the expression of cleaved caspase 3 (Figure 13E).
Figure 13.

Effects of DNR on RV structure and function. Rats were exposed to normoxia or hypoxia for 2 weeks, and were then injected with DNR. Rats were then placed back in normoxic and hypoxic environments. Three days after the injection, rats were anesthetized, mechanically ventilated, a Miller catheter was inserted into the RV, and (A) dP/dtmax, and (B) dP/dtmin were monitored. C, Hearts were isolated and RV, LV and septum (S) were surgically separated and the mass ratio of RV/(LV+S) was calculated. D, Hearts were fixed and embedded in paraffin. Tissue sections were stained with H&E (n=4 for “normoxia+DNR(5)” and “hypoxia+DNR(5)”; n=3 for others). E, RV homogenates were subjected to immunoblotting using cleaved caspase‐3 antibody to monitor the occurrence of apoptosis (n=3). Bar graphs represent means±SEM. *P<0.05 vs normoxia. DNR indicates daunorubicin; H&E, hematoxylin and eosin; LV, left ventricle; RV, right ventricle.
Discussion
The major finding of this study is that apoptotic agents used to treat cancer such as proteasome inhibitors and anthracyclines reverse pulmonary vascular remodeling within 1 to 3 days after the administration in intact animals. Notably, the action of these drugs in reducing pulmonary vascular wall thickness is specific to remodeled vessels; drugs do not affect the normal pulmonary vasculature. The reversal of pulmonary vascular remodeling significantly increased the sensitivity of vasodilators to reduce RV pressure.
The antitumor drug‐induced regression of pulmonary vascular thickening in vivo appears to involve apoptosis. We found that Bcl‐xL expression in the pulmonary artery is reduced in response to apoptotic agents. Since Bcl‐xL serves as an anti‐apoptotic factor,22 the downregulation of this protein may trigger apoptosis. It is noteworthy that both the reduction of pulmonary vascular thickness and cell death in response to antitumor agents only occurred in the remodeled pulmonary vessels in animals with pulmonary hypertension, and not in pulmonary vessels from healthy control animals. These results suggest that remodeled pulmonary vessels contain cells that are more susceptible to cell death. This is therapeutically advantageous in that antitumor drugs may eliminate only unwanted vascular cells. We found that Bcl‐xL downregulation only occurred in remodeled vessels, not in normal vessels, suggesting that molecular mechanisms regulating gene transcription of this anti‐apoptotic protein define this susceptibility. In this study, we present evidence for a novel mechanism where the upregulation of annexin A1 inhibits the gene transcription of GATA4, which in turn defines the Bcl‐xL expression (Figure S10). Inhibition of GATA4 gene expression would reduce the activity of this transcription factor. The upregulation of annexin A1 by antitumor agents only occurs in pulmonary arteries of animals with pulmonary hypertension, but not in normal vessels, supporting the notion that this mechanism defines the difference in the susceptibility to apoptotic cell killing.
Autophagy has been shown to mediate both cell death and cell survival.31 We found that the drug‐induced reversal of pulmonary vascular remodeling is associated with the induction of markers for autophagy, including the upregulated expression of LC3B‐II as well as the downregulation of p62. Choi and co‐workers reported that chronic hypoxia increased autophagic signals, concluding that autophagy exerts a protective function during the pathogenesis of pulmonary hypertension.28 Our data may suggest that antitumor drugs may further promote autophagy in PASMCs, which serves as an endogenous protective mechanism against pulmonary vascular remodeling. This concept for the role of autophagy as a cell death mediator is supported by our results in cultured PASMCs showing that siRNA knockdown of LC3B inhibited drug‐induced cell death.
Like apoptosis, indications of autophagy were found to occur only in remodeled pulmonary arteries, but not in normal pulmonary vessels, in rats. SMCs undergo phenotypic switching from the differentiated contractile phenotype to the proliferating synthetic phenotype.32–33 Further experiments using cultured human PASMCs revealed that proliferating SMCs are more susceptible to drug‐induced cell death and the induction of autophagy, compared to differentiated SMCs. This is consistent with the idea that normal pulmonary arteries largely consist of differentiated SMC phenotypic cells, while remodeled vessels containing proliferating SMCs, which are more susceptible to cell death. Interestingly, our studies revealed that the mechanism of this differential susceptibility to autophagic cell death relies on the regulation by a protein called parkin, an E3 ubiquitin ligase.34 In rats, we showed that the parkin expression is upregulated in response to antitumor agents only in normal pulmonary arteries, but not in remodeled vessels. Similarly, in cultured human PASMCs, drug‐induced upregulation of parkin occurred only in differentiated, but not in proliferating cells. Parkin is a known regulator in the brain for the development of Parkinson's disease, and a recent report points to its importance in heart failure.30 To our knowledge, this is the first demonstration that parkin is expressed in pulmonary cells. In the brain and the heart, parkin is thought to mediate cell survival, and our results also show that this protein is involved as a survival factor in pulmonary vascular cells. Further work on the parkin regulation in drug‐induced cell death and the mechanism of the parkin upregulation in differentiated PASMCs should help define this clinically important biologic process, in which cells from remodeled vessels sense antitumor drugs differently.
Our observation that the reversal of pulmonary vascular remodeling alone was not accompanied by normalization of right ventricular pressure has critical importance. We observed this event by 3 different apoptotic agents and in 2 different models of pulmonary hypertension. Similar results have been reported in the studies of the effects of rosiglitazone in hypoxia‐induced pulmonary arterial remodeling.35 The elucidation of this mechanism may be the key to developing effective therapeutic strategies to treat PAH. We hypothesized that reduced pulmonary vascular wall thickness may have triggered rebound pulmonary vasoconstriction in order to maintain the given pulmonary arterial pressure as an adaptive mechanism. This hypothesis is indeed supported by our observations that the administration of vasodilators in combination with cancer chemotherapeutic agents effectively reversed pulmonary vascular remodeling and pulmonary arterial pressure. These results suggest exciting combination therapies with available vasodilators and antitumor agents that are capable of promoting apoptosis of remodeled vascular cells, for the treatment of PAH. Types, doses, and timing of vasodilators, however, still need to be identified in order to obtain successful therapeutic strategies. Further work is also needed to select the most efficacious apoptotic agents as well as the strategies for administration, which may allow for long‐lasting beneficial effects.
In summary, the present study demonstrates that antitumor drugs such as proteasome inhibitors and anthracyclines are capable of reducing the thickness of pulmonary vascular walls in rat models of pulmonary hypertension. The mechanism of the regression appears to involve the promotion of apoptotic as well as autophagic cell death. We identified that the differential susceptibility to apoptosis in remodeled and normal vessels is regulated by the annexin A1/GATA4 pathway, while that to autophagic cell death is defined by parkin serving as a survival factor in normal vessels. These antitumor drugs that are capable of reducing pulmonary vascular wall thickness effectively increase the efficacy of vasodilators. These results suggest that the combination of apoptotic agents and vasodilators may be promising for treating patients with PAH, who often have already developed pulmonary vascular remodeling at the time of diagnosis.
Sources of Funding
This work was supported in part by National Institutes of Health (R01HL72844 and R01HL97514) and the American Heart Association Mid‐Atlantic Affiliate to Suzuki.
Disclosures
None.
References
- 1.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007; 30:104-109 [DOI] [PubMed] [Google Scholar]
- 2.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez‐Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau GESC Committee for Practice Guidelines (CPG) Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009; 30:2493-2537 [DOI] [PubMed] [Google Scholar]
- 3.D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Kernis JT, Levy PS, Pietra GG, Reid LM, Reeves JT, Rich S, Vreim CE, Williams GW, Wu M. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991; 115:343-349 [DOI] [PubMed] [Google Scholar]
- 4.Benza RL, Miller DP, Frost A, Barst RJ, Krichman AM, McGoon MD. Analysis of the lung allocation score estimation of risk of death in patients with pulmonary arterial hypertension using data from the REVEAL Registry. Transplantation. 2010; 90:298-305 [DOI] [PubMed] [Google Scholar]
- 5.Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau GFrench Pulmonary Arterial Hypertension Network Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010; 36:549-555 [DOI] [PubMed] [Google Scholar]
- 6.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg‐Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010; 35:1079-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Archer SL, Michelakis ED. An evidence‐based approach to the management of pulmonary arterial hypertension. Curr Opin Cardiol. 2006; 21:385-392 [DOI] [PubMed] [Google Scholar]
- 8.Suzuki YJ, Nagase H, Wong CM, Kumar SV, Jain V, Park AM, Day RM. Regulation of Bcl‐xL expression in lung vascular smooth muscle. Am J Respir Cell Mol Biol. 2007; 36:678-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003; 8:508-513 [DOI] [PubMed] [Google Scholar]
- 10.Giuliano M, D'Anneo A, De Blasio A, Vento R, Tesoriere G. Apoptosis meets proteasome, an invaluable therapeutic target of anticancer drugs. Ital J Biochem. 2003; 52:112-121 [PubMed] [Google Scholar]
- 11.Li M, Dong X, Liu Y, Sun X, Li Z, He J. Inhibition of ubiquitin proteasome function suppresses proliferation of pulmonary artery smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol. 2011; 384:517-523 [DOI] [PubMed] [Google Scholar]
- 12.Kim SY, Lee JH, Huh JW, Kim HJ, Park MK, Ro JY, Oh YM, Lee SD, Lee YS. Bortezomib alleviates experimental pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2012; 47:698-708 [DOI] [PubMed] [Google Scholar]
- 13.Dimarco A, Gaetani M, Orezzi P, Scarpinato BM, Silvestrini R, Soldati M, Dasdia T, Valentin L. ‘Daunomycin’, a new antibiotic of the rhodomycin group. Nature. 1964; 201:706-707 [DOI] [PubMed] [Google Scholar]
- 14.Holton CP, Vietti TJ, Nora AH, Donaldson MH, Stuckey WJ, Jr, Watkins WL, Lane DM. Clinical study of daunomycin and prednisone for induction of remission in children with advanced leukemia. N Engl J Med. 1969; 280:171-174 [DOI] [PubMed] [Google Scholar]
- 15.Boiron M, Weil M, Jacquillat C, Tanzer J, Levy D, Sultan C, Bernard J. Daunorubicin in the treatment of acute myelocytic leukaemia. Lancet. 1969; 1:330-333 [DOI] [PubMed] [Google Scholar]
- 16.Ling YH, Priebe W, Perez‐Soler R. Apoptosis induced by anthracycline antibiotics in P388 parent and multidrug‐resistant cells. Cancer Res. 1993; 53:1845-1852 [PubMed] [Google Scholar]
- 17.Zaleskis G, Berleth E, Verstovsek S, Ehrke MJ, Mihich E. Doxorubicin‐induced DNA degradation in murine thymocytes. Mol Pharmacol. 1994; 46:901-908 [PubMed] [Google Scholar]
- 18.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012; 186:261-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taraseviciene‐Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death‐dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001; 15:427-438 [DOI] [PubMed] [Google Scholar]
- 20.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010; 121:2747-2754 [DOI] [PubMed] [Google Scholar]
- 21.Park AM, Wong CM, Jelinkova L, Liu L, Nagase H, Suzuki YJ. Pulmonary hypertension‐induced GATA4 activation in the right ventricle. Hypertension. 2010; 56:1145-1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boise LH, González‐García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. bcl‐x, a bcl‐2‐related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993; 74:597-608 [DOI] [PubMed] [Google Scholar]
- 23.Pecci A, Viegas LR, Baranao JL, Beato M. Promoter choice influences alternative splicing and determines the balance of isoforms expressed from the mouse bcl‐X gene. J Biol Chem. 2001; 276:21062-21069 [DOI] [PubMed] [Google Scholar]
- 24.Grillot DA, Gonzalez‐Garcia M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Nunez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl‐x gene. J Immunol. 1997; 158:4750-4757 [PubMed] [Google Scholar]
- 25.Suzuki E, Evans T, Lowry J, Truong L, Bell DW, Testa JR, Walsh K. The human GATA‐6 gene: structure, chromosomal location, and regulation of expression by tissue‐specific and mitogen‐responsive signals. Genomics. 1996; 38:283-290 [DOI] [PubMed] [Google Scholar]
- 26.Suzuki YJ, Day RM, Tan CC, Sandven TH, Liang Q, Molkentin JD, Fanburg BL. Activation of GATA‐4 by serotonin in pulmonary artery smooth muscle cells. J Biol Chem. 2003; 278:17525-17531 [DOI] [PubMed] [Google Scholar]
- 27.Wong CM, Cheema AK, Zhang L, Suzuki YJ. Protein carbonylation as a novel mechanism in redox signaling. Circ Res. 2008; 102:310-318 [DOI] [PubMed] [Google Scholar]
- 28.Lee SJ, Smith A, Guo L, Alastalo TP, Li M, Sawada H, Liu X, Chen ZH, Ifedigbo E, Jin Y, Feghali‐Bostwick C, Ryter SW, Kim HP, Rabinovitch M, Choi AM. Autophagic protein LC3B confers resistance against hypoxia‐induced pulmonary hypertension. Am J Respir Crit Care Med. 2011; 183:649-658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007; 3:542-545 [DOI] [PubMed] [Google Scholar]
- 30.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN‐inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011; 108:9572-9577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132:27-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beamish JA, He P, Kottke‐Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. 2010; 16:467-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001; 90:358-368 [DOI] [PubMed] [Google Scholar]
- 34.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008; 183:795-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crossno JT, Jr, Garat CV, Reusch JEB, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia‐induced pulmonary arterial remodeling. Am J Physiol Lung Cell Mol Physiol. 2007; 292:L885-L897 [DOI] [PubMed] [Google Scholar]