

Abstract
A fundamentally new approach to asymmetric catalysis in organic chemistry is described based on the in vitro evolution of enantioselective enzymes. It comprises the appropriate combination of gene mutagenesis and expression coupled with an efficient high-throughput screening system for evaluating enantioselectivity (enantiomeric excess assay). Several such cycles lead to a “Darwinistic” process, which is independent of any knowledge concerning the structure or the mechanism of the enzyme being evolved. The challenge is to choose the optimal mutagenesis methods to navigate efficiently in protein sequence space. As a first example, the combination of error-prone mutagenesis, saturation mutagenesis, and DNA-shuffling led to a dramatic enhancement of enantioselectivity of a lipase acting as a catalyst in the kinetic resolution of a chiral ester. Mutations at positions remote from the catalytically active center were identified, a surprising finding, which was explained on the basis of a novel relay mechanism. The scope and limitations of the method are discussed, including the prospect of directed evolution of stereoselective hybrid catalysts composed of robust protein hosts in which transition metal centers have been implanted.
Asymmetric catalysis plays a profound role in modern organic chemistry (1–3). Numerous therapeutic drugs, plant-protecting agents, and fragrances and most natural products are chiral, many (or most) exerting a specific biological effect only in one enantiomeric form. The practicing organic chemist needing to perform a catalytic asymmetric transformation has several options, including synthetic chiral transition metal complexes (1–4), organocatalysts (5), catalytic antibodies (6), and enzymes (7), none of which are general.
Enzymes are products of evolution and might therefore be expected to function with high enantioselectivity only with natural substrates under physiological conditions. However, it is well known that this is not the case, because a surprisingly large number of unnatural compounds are converted with high enantioselectivity (7), even in organic solvents (8). Nevertheless, the problem of substrate specificity persists. In such cases several approaches to enhance enzyme stereoselectivity have been described, including site-specific mutagenesis based on theoretical considerations (7–9).
Several years ago we proposed a more general concept that does not require any knowledge of the structure or the mechanism of the enzyme, namely in vitro evolution of enantioselectivity (10–12). It entails the combination of known methods of random gene mutagenesis and expression (13–16) coupled with an appropriate high-throughput screening system (17) for evaluating the enantioselectivity of thousands of mutant enzymes (Fig. 1). In the first round of mutagenesis/screening the most enantioselective enzyme variant is identified, and the inferior variants are discarded. By subjecting the gene encoding the hit to another cycle of mutagenesis/screening, a “Darwinistic” process is triggered that can be repeated as often as necessary until the desired degree of enantioselectivity has evolved in a given reaction of interest. Typically, in each cycle ≈3,000 potentially enantioselective mutants are screened.
Fig. 1.

Directed evolution of an enantioselective enzyme (11, 12, 41).
After application of an appropriate mutagenesis method, a variety of which are available (13–16), the mutant genes are inserted into a bacterial host that is plated on agar plates. Individual bacterial colonies are transferred to 96or 384-well culture plates with a colony picker. In the final step the culture supernatants are transferred to the wells of microtiter plates where the reaction of interest occurs.
The challenge in enhancing the enantioselectivity of an enzyme in a given reaction of interest involves two problems: (i) choosing the optimal gene mutagenesis protocols from the list of currently known methods, and (ii) developing efficient high-throughput enantiomeric excess (ee) assays.
Mutagenesis Methods
Although the possibility of in vitro evolution was suggested in the 1980s, it was not until later that molecular biologists began to devise efficient methods for gene mutagenesis. A milestone in this ongoing area of research concerns the error-prone PCR (epPCR) (18). By varying the reaction conditions empirically, “errors” in DNA amplification can be induced so as to cause one, two, three, or more amino acid substitutions in the encoded protein (18–20). Most studies in the area of directed evolution of enzymes begin with epPCR (10–16), although it is not truly random (21).
Another important method for introducing point mutations is saturation mutagenesis, a generalized term pertaining to the substitution or insertion of codons encoding all possible 20 proteinogenic amino acids at any predetermined position of the enzyme (22). Usually an excess of 300–400 clones needs to be picked to be certain that the 20 enzyme variants are in fact in the library. The problem of choosing the appropriate position in the enzyme needs to be solved first.
A rather different way to generate mutants concerns recombinative methods such as DNA-shuffling, a novel process pioneered by Stemmer and coworkers (14). Accordingly, two or more genes are fragmented and then reassembled. Several other gene-assembly methods have since been developed, most of them requiring the availability of the respective wild-type genes (23–25). We have devised a method for efficient gene recombination that does not require the genes themselves but only information regarding their sequences (26). The process is based on the assembly of appropriately designed synthetic oligonucleotides, which is guided by information derived from gene alignment and computer analysis (Fig. 2).
Fig. 2.

General concept of assembly of designed nucleotides (26); two strategies for the linking of fragments are possible (cases I and II). In case I the two genes, A and B, that are to be virtually shuffled are aligned, the different colored stars refer to information that encodes different amino acids, whereas oligonucleotide fragments with both colored stars in the same position of the parent gene denote the synthetic oligonucleotide fragment with degenerate nucleotides. The gray blocks denote conserved regions of sequence that can be used as the linking part with homologous recombination. Case II shows no homology between flanking oligos, which can be assembled by ligation between single-stranded DNA with unknown terminal sequences.
High-Throughput ee Assays
The need to develop high-throughput screening systems for the rapid determination of the enantiopurity of a large number of samples arose from our desire to apply the methods of directed evolution to enantioselective enzymes (10). The first high-throughput ee assay was designed to monitor the hydrolytic kinetic resolution of chiral p-nitrophenol esters catalyzed by lipases or esterases. Specifically, it was used to study the enantioselective hydrolysis of the chiral ester 1 catalyzed by mutant lipases from Pseudomonas aeruginosa. Hydrolysis in buffered medium generates the two enantiomeric acids [(S)-2 and (R)-2] in addition to p-nitrophenolate (3), which shows a strong UV-visible absorption at 405 nm (Eq. 1) This finding means that the reaction can be performed on microtiter plates with thousands of lipase mutants as potentially enantioselective catalysts, a simple UV-visible plate reader measuring absorption as a function of time. However, since the racemate delivers information regarding only the overall rate, the (S)- and (R)-substrates were prepared and studied separately pairwise on 96-well microtiter plates (10). Whenever the slopes of the absorption/time curves differ considerably, a hit is indicated. The method allows ≈500–800 samples to be screened per day, which by today's standard is medium throughput.
This ee assay, although successful in the first case of directed evolution of an enantioselective enzyme (10) (see below), suffers from several disadvantages. Therefore, a good portion of our research has been directed toward developing more general, precise, and faster ee assays. Other groups have also contributed to this new area of research (reviewed in refs. 17 and 27). Select protocols for the most efficient ee assays based on MS (28, 29), NMR spectroscopy (30), and IR spectroscopy (31) have been published which allow 1,000–10,000 samples to be evaluated per day (32). Selection (33) rather than screening is a goal for the future, phage display constituting one possibility (34).
Directed Evolution of Enantioselective Lipases
In the mid-1990s we became intrigued by the possibility of applying the methods of directed evolution as a means to control enantioselectivity (Fig. 1 and refs. 10–12). As already mentioned, we chose the hydrolytic kinetic resolution of the ester 1 catalyzed by the lipase from P. aeruginosa as the model reaction (Eq. 1) (10). In addition to developing a screening system (see above), the problem of protein sequence space had to be considered. Since we wanted to use epPCR as the mutagenesis method in the early phase of the project, a decision regarding the mutation rate had to be made. The lipase from P. aeruginosa consists of 285 aa. On the basis of the algorithm n = 19M × 285!/[(285–M)! × M!], the size of the library, N, can be calculated as a function of M, the number of amino acid substitutions per enzyme molecule. In the case of M = 1, corresponding to the least amount of “ligand tuning,” the library would theoretically contain 5,415 unique members (10). However, because of the degeneracy of the genetic code, among other things, it is impossible to generate an epPCR library that contains all 5,415 variants (21). If the mutation rate is increased to yield an average of two amino acid exchanges per enzyme molecule (M = 2), then the number of mutant P. aeruginosa lipases predicted by this algorithm increases dramatically to ≈15 million. When M = 3, ≈52 billion enzyme variants are possible.
 |
Because of the efforts associated with screening for enantioselectivity, navigation in protein sequence space should be as efficient as possible, but even after 6 years of research it is still difficult to predict the optimal strategy when searching for enantioselectivity. We originally chose epPCR at a low mutation rate averaging only one amino acid substitution per enzyme molecule (10). On going through four cycles of mutagenesis/screening (2,000–3,000 mutants in each round), the selectivity factor in the hydrolytic kinetic resolution of the chiral ester 1 increased from E = 1.1 to E = 11.3 (variant A) in favor of (S)-2 (10). This result was accomplished by identifying the most enantioselective variant in each cycle, by isolating the corresponding mutant gene, and by using it as a template for the next round of epPCR. Fig. 3 shows the course of this enantioselective “evolution” together with the amino acid exchanges determined by DNA sequencing.
Fig. 3.

Increasing the E values of the lipase-catalyzed hydrolysis of the chiral ester 1 by cumulative mutations caused by epPCR (10, 12).
It is possible to continue to improve enantioselectivity by performing additional rounds of epPCR, but such a strategy is not optimal. Rather, considerable efforts were invested to test other methods. One of these methods concerns the use of saturation mutagenesis (22), which requires an intelligent decision as to the choice of the position at which the epPCR is to be performed (35). We assumed that the identified sites of amino acid exchange are “hot spots” that are important for increasing enantioselectivity, but that the specific amino acids identified by sequencing are not necessarily optimal (12, 36). This assumption is reasonable because the size of the screened libraries was smaller than the theoretical number of enzyme variants and because the amino acid exchanges achieved by epPCR are biased (21). Therefore, several “mutation hot spots” were chosen for saturation experiments. One of the sensitive positions turned out to be 155. This strategy led to variant B displaying an E value of 20 (36). The gene encoding enzyme variant B was then subjected to another cycle of epPCR, resulting in an even better enzyme variant C having five mutations and a selectivity factor of E = 25.
We then considered DNA-shuffling (14), but it was not clear which mutant genes should be shuffled (37). DNA-shuffling of the mutant genes produced by low-mutation-rate epPCR failed to achieve significant improvements, leading us to conclude that higher gene diversity is necessary. Therefore, epPCR of the wild-type gene was repeated, this time at a higher mutation rate corresponding to three amino acid exchange events per enzyme molecule. We were delighted to discover that this process leads to the identification of several improved enzyme variants, D and E (E = 3 and E = 6.5) being the best ones (37). In the original project with a low mutation rate such selectivity factors were not obtained until the second cycle of epPCR (10). The genes encoding the variants C–E were then subjected to conventional DNA-shuffling. This process provided variant F, which had the highest enantioselectivity up to that point (E = 32) (37).
As part of yet another strategy we extended the region of accessible protein sequence space by developing a modified version of Stemmer's combinatorial multiple-cassette mutagenesis (37). The original form of combinatorial multiple-cassette mutagenesis is a special type of DNA-shuffling using the wild-type gene and cassettes composed of defined sequences to be randomized (38). In our version, two mutant genes encoding the enzyme variants D and E and a mutagenic oligocassette allowing simultaneous randomization at “hot spots” 155 and 162 were subjected to DNA-shuffling (Fig. 4). This resulted in the most enantioselective variant J to date, displaying a selectivity factor of E > 51 (37).
Fig. 4.

Extended combinatorial multiple-cassette mutagenesis in the evolution of an (S)-selective lipase variant (green star, position 20; purple star, position 161; yellow star, position 234; red circle, position 53; orange circle, position 180; blue circle, position 272) (37).
We also applied cassette mutagenesis, focusing on a region near the active site (positions 160–163), resulting in variant G with E = 30 (37). This strategy is a combination of rational design and directed evolution. The results of these and other experiments are summarized in Fig. 5. A total of only 40,000 mutants were screened (37). It is likely that on exploring larger portions of protein space efficiently, more and even better lipase variants can be created. The assumption that millions of potential variants are highly enantioselective is not unfounded. However, efficient directed evolution is not a matter of generating huge libraries that then require considerable efforts in screening for the desired property. The goal is to create a maximum in structural diversity while minimizing the size of the libraries.
Fig. 5.

Schematic summary of the directed evolution of enantioselective enzymes (lipase variants) catalyzing the hydrolytic kinetic resolution of ester 1 (37). CMCM, combinatorial multiple-cassette mutagenesis.
One of the traditional limitations of enantioselective enzyme catalysis has to do with the fact that only one enantiomer is usually accessible. It was therefore an intriguing goal to try to invert the sense of enantioselectivity in the kinetic resolution of ester 1 (11). By screening for the (R)-product and applying similar strategies based on epPCR, saturation mutagenesis, and DNA-shuffling, a mutant showing an (R)-selectivity (E = 30) was in fact evolved (39). It has 11 amino acid substitutions that are quite different from those of the (S)-selective enzyme variants.
Learning from Directed Evolution
Most of these results were obtained without prior knowledge of the 3D structure of the enzyme or of its mechanism (10–12, 36, 39). Rather, we relied on the Darwinistic nature of the concept. Despite the fundamental differences between rational design and our approach, we are convinced that directed evolution is an ideal tool in learning lessons concerning functional enzymes, especially when dealing with enantioselectivity, because this parameter is a particularly sensitive probe.
The lipase from P. aeruginosa is a hydrolase having the usual catalytic triad composed of aspartate, histidine, and serine (Fig. 6) (40). In a proton shuttle serine is activated and attacks the ester with formation of an oxyanion that is stabilized by H-bonding. Collapse of the tetrahedral intermediate leads to an acyl enzyme intermediate, which in turn reacts with water by means of a similar oxyanion, liberating the acid and regenerating the lipase. In the chiral ester 1, stereoselectivity is determined in the first step.
Fig. 6.

Although x-ray structural characterization of the (S) and (R)-selective mutants of the lipase from P. aeruginosa would provide ideal information in attempts to unveil the source of enhanced enantioselectivity, such data are not yet available. However, consideration of the crystal structure of the wild-type lipase (40), is in itself illuminating. The structure shows a conserved α/β hydrolase fold typical of lipases and the presence of a lid and the usual catalytic triad composed of aspartate (D229), histidine (H251), and serine (S82) (37).
We made a surprising discovery when analyzing the mutation sites of one of the lipase mutants (variant C) showing an intermediate degree of enantioselectivity in the kinetic resolution of ester 1 (E = 26) (36). It has five mutations (S149G, S155F, V47G, S164G, and V55G), most of them occurring at sites remote from the active center (S82) (12, 36, 41). This observation leads to a change in paradigm, because all previous attempts to influence enantioselectivity of an enzyme by using site-specific mutagenesis had focused on amino acid substitutions near the active center (42, 43). Such protein engineering was designed to “carve” an appropriate chiral pocket at the active center, in line with Fischer's “lock-and-key” hypothesis (44) or modified versions such as Koshland's induced fit (45). We hoped to obtain even better lipase variants and thus postponed the analysis.
Having indeed created by directed evolution a mutant (variant J) displaying even higher enantioselectivity in the kinetic resolution of ester 1 (E > 51) (37), we initiated a detailed molecular modeling/quantum mechanical study (46). Variant J has six mutations(D20N, S53P, S155M, L162G, T180I, and T234S), most of which are again remote (37, 46) (Fig. 7).
Fig. 7.
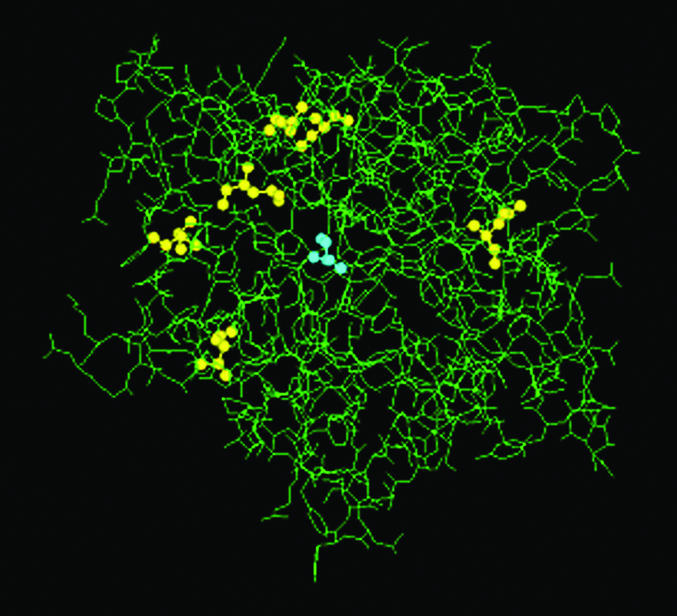
Crystal structure of the wild-type lipase from P. aeruginosa (40) with the active center (blue) and six mutations (yellow) of variant J (46).
The first step was the quantum mechanical calculation of the force fields involved in the oxyanion, followed by extensive molecular dynamics calculations by using the moloc and charmm programs (46). The initial geometries for the molecular dynamics simulations were derived from the x-ray structure of the wild-type lipase from P. aeruginosa having a phosphonate inhibitor bound covalently at the active site (S82). The inhibitor was “replaced” by the (S)- and (R)-ester 1 with formation of the respective Michaelis–Menten complexes and the tetrahedral oxyanions, respectively. Thereafter, the amino acid exchanges of the mutants were considered. After assigning the appropriate protonation state, the systems were relaxed by a series of minimizations and subsequently subjected to 1-ns molecular dynamics simulations (46). Solvation by water was considered.
To uncover the source of (S)-enantioselectivity, not only the wild-type lipase and the best mutant (variant J) were considered, but also eight other (real and hypothetical) mutants for the purpose of studying the influence of single mutations and the possible role of cooperative effects. This theoretical analysis resulted in some remarkable insights (46). The most important conclusion focuses on two of the six mutations, namely, S53P and L162G, which were demonstrated by the calculations to act cooperatively in enhancing (S)-enantioselectivity.
In the wild-type (Fig. 8 Left) S161, S53, and H83 form a strong H-bond network. The mutation S53P disrupts this network, allowing the side chain of histidine at position 83 to adapt different conformations. This adaptation is readily possible only if L162 mutates to G162, which in fact is the experimental result of directed evolution. The molecular dynamics calculations clearly show that the side chain of histidine 83 can then undergo motion toward the active site to provide additional stabilization of the oxyanion by means of a new H bond (46). This relay mechanism is only possible with the (S)-ester 1 (Fig. 8 Right). In the analogous tetrahedral intermediate originating from (R)-ester 1, the presence of the methyl group at the stereogenic center results in severe steric repulsion, which prevents the analogous stabilizing H bond (46). Thus, a chiral pocket has been created by directed evolution which not only accommodates the (S)-ester geometrically, but also provides additional stabilization of the oxyanion and therefore most likely of the transition state. The mutation at position 155 was also shown to contribute to the observed (S)-selectivity (46).
Fig. 8.

Comparison between the wild-type (Left) and the double mutant S53P+L162G (Right) (46).
The theoretical analysis reveals yet another notable feature. The mutation L162G leads to the creation of a new binding pocket, specifically to house the “carboxylic acid part” of long-chain esters (46). The “alcohol part” of the ester is located in a different binding pocket that was not affected by the evolutionary process. The role of the other three mutational changes still needs to be studied in detail. The next major step should be the quantum mechanical calculation of the energies of the transition states.
In summary, the combination of directed evolution and theoretical analysis has led to the identification of a remarkable effect that would never have been discovered by traditional protein design. The presence of a previously nonexisting binding pocket and the operation of a relay mechanism led to the prediction that mutant J should show higher activity in the hydrolysis of (S)-ester 1. Indeed, this mutant is two orders of magnitude more active than the wild type (41). The results of the theoretical study also predict that relative to the wild type the mutant lipase J should show higher activity in the hydrolysis of other branched esters as well. This has yet to be studied experimentally. Finally, our theoretical study suggests that further mutagenesis experiments are in order, e.g., the generation of a variant in which mutations S53P and L162G are the only structural changes that can be introduced by site-specific mutagenesis. We believe that the fusion of the two very different ways to perform protein engineering, namely, directed evolution and rational design, constitutes a powerful strategy in the quest to create enantioselective enzymes. This conclusion is related to studies of Hilvert and coworkers concerning binary pattering (47).
Intruiging lessons are emerging from studies concerning directed evolution of enantioselective enzymes, and we can expect many more in the future. Remote amino acid substitutions have also been shown to influence other enzyme properties such as stability and activity (48–53), although the number of reported cases is still limited and explanations at a molecular level are rare. Zhao and Arnold (48) have enhanced the thermal stability of subtilisin E by directed evolution, sequence analysis of the thermally most stable mutant showing amino acid substitutions almost exclusively at positions remote from the active site (48). To explain the results, improved hydrogen bonding near a β-bulge and reduced cavity volume were postulated. Moreover, Benkovic, Hammes-Schiffer, and coworkers (51) have shown that amino acid exchange events at positions remote from the active site can shut down enzyme activity completely. Detailed explanations at a molecular level still need to be presented, but the exciting suggestion that coupled motions of the enzyme may be involved constitutes a challenge for advanced theory.
Remote mutations are, of course, statistically favored because numerically more amino acids are far away from the active site than close to it. Nevertheless, our studies show that the long-standing dogma regarding the necessity of amino acid substitutions exclusively at the active site to influence enantioselectivity no longer holds (10, 36, 39, 41). Directed evolution is capable of creating structural effects not easily foreseen by traditional approaches. However, we do not suggest that spatially close mutations are unimportant, nor do we propose that protein engineering should focus solely on remote effects when attempting to enhance enantioselectivity.
Directed Evolution of Other Enzymes
Further projects concerning the directed evolution of enantioselective enzymes are necessary. The development of alternative and perhaps even more efficient strategies for exploring protein sequence space are also needed. For example, we have proposed that systematic saturation mutagenesis at every position of a given enzyme constitutes a viable starting point for searching for enantioselectivity, providing a set of n libraries of single-site variants (where n is the number of amino acids in the wild type; ref. 54). Each library, consisting of an excess of 300–400 clones, contains 20 different enzyme variants corresponding to the introduction of the 20 proteinogenic amino acids at a given position. The set of n libraries, once on the shelf, can then be used to test any substrate that is converted to a chiral product by the particular enzyme. Although this set of libraries limits protein sequence space considerably, in favorable cases one or more hits may already display the desired degree of enantioselectivity. In other cases they form the starting point for the actual evolutionary optimization. An example concerns the hydrolytic desymmetrization of meso-cyclopentene-diacetate catalyzed by the lipase A from Bacillus subtilis, an enzyme composed of 181 aa (54). The wild-type enzyme Lip A leads to an ee value of only 38–45% (depending on the mode of expression). Although the systematic screening of the 181 small libraries has not yet been completed, a striking observation has already been made: namely, the identification of a mutant showing reversed enantioselectivity (ee = 85%) (H. Krumm, unpublished work). Since evolution is not yet involved at this stage, any other substrate can be screened in a similar manner by using the same set of lipase libraries without recourse to the generation of new mutants.
The idea of applying systematic saturation was recently also described by scientists at Diversa (San Diego) in their efforts to improve the enantioselectivity of a nitrilase in the desymmetrization of the prochiral dinitrile 4 (Eq. 2 and ref. 55). This catalytic reaction is of special industrial interest because the ethyl ester of (R)-5 is an intermediate in the synthesis of the cholesterol-lowering drug Lipitor. The wild-type nitrilase has 330 aa and leads to an enantioselectivity of ee = 87.8% in favor of (R)-5. On performing systematic saturation mutagenesis, several improved mutants were found displaying ee values between 95.4% and 98.1% (55). A total of 31,584 clones were evaluated for enantioselectivity by using the Mülheim MS-based screening system (28, 56) as the ee assay, in this case, 15N-labeled 4 being used as the substrate. In a laboratory-scale reaction one of the best mutants performed impressively well, even at high substrate concentration (3 M). The mixture of 4 (1 g, 9 mmol), the mutant nitrilase (30 mg), and a phosphate buffer (2.03 ml) was stirred at 20°C for 15 h to furnish the desired product (R)-5 (1.1 g, 96%, 98.5% ee) with a 619 g·liter–1·d–1 volumetric productivity (54).
 |
Screening an initial mutant library (or set of libraries) can lead to improvements in the ee as in the examples above and in some other cases (57, 58), although true directed evolution along the lines of Fig. 1 is not involved. An impressive example of the use of directed evolution to control the enantioselectivity of enzymes concerns the use of a hydantoinase in the production of unnatural methionine as reported by May et al. (59). By applying several rounds of mutagenesis, reversal of enantioselectivity was achieved. In yet another study the substrate acceptance of an aldolase was altered to include the reaction of a given aldehyde that is not converted at all by the wild-type enzyme, enantioselectivity being maintained at 100% (60).
Preliminary results from our laboratories concerning epoxide hydrolases are also encouraging. In the hydrolytic kinetic resolution of glycidyl phenyl ether (6) with the epoxide hydrolase from Aspergillus niger as the catalyst, enantioselectivity was more than doubled in going from E = 4.6 to E = 10.8 in favor of (S)-7 (Eq. 3) (61).
 |
This enantioselectivity was achieved on the basis of a single cycle of epPCR at high mutation rate, which does not yet constitute directed evolution. Three mutations were identified, two of which (K332E and A390E) occur at remote positions and one (A217V) closer to the active site (A192). A preliminary model suggests that the larger valine side chain at residue 217 disfavors the (R)-substrate because of steric congestion. Before embarking on actual directed evolution, an improved expression system was developed (62).
The kinetic resolution of substrate 6 can be carried out more efficiently with synthetic transition metal catalysts described by Jacobsen (63). We are therefore turning our attention to substrates that are not converted by the Jacobsen catalysts or epoxide hydrolases (64).
Finally, we believe that enzymecatalyzed enantioselective partial oxidations will become more important in the future, especially when synthetic catalysts fail completely (65). Preliminary studies in our laboratories concerning the directed evolution of a cyclohexanone monooxygenase as a biocatalyst in the Baeyer–Villiger reaction of 4-hydroxycyclohexanone show that the enantioselectivity of this synthetically interesting transformation can be increased substantially from ee = 9% to ee = 90%. It was also possible to invert the sense of enantioselectivity. Moreover, a mutant was evolved that converts 4-methoxycyclohexanone with ee = 98.5%. No presently known synthetic transition metal catalyst can even come close to this performance. Another perspective that we are interested in concerns cytochrome P450-catalyzed hydroxylation of steroids and other substrates, the aim being to exploit the possibility of directed evolution in the quest to control simultaneously regio- and diastereoselectivity of this important type of carbon-hydrogen activation. Goals of this type are ideally suited for directed evolution of selective enzymes.
Directed Evolution of Hybrid Catalysts
Directed evolution of enantioselective enzymes constitutes a previously uninvestigated approach to asymmetric catalysis in organic chemistry, but several limitations exist. No enzyme can catalyze transition metal-mediated hydroformylation, allylic substitution, hydrovinylation, or Heck reactions, to mention only a few reaction types that form the heart of organic chemistry. If an enzyme were capable of such catalysis, then the methods of directed evolution as described herein could be applied to tune regio- and stereoselectivity and catalyst activity. In addressing this question, we have proposed the concept of directed evolution of hybrid catalysts in which a protein, acting as a host for a synthetic catalyst, is chemically modified to introduce a metal/ligand center at a predetermined position (41, 66–68). The covalent or noncovalent attachment of certain catalytically active species to proteins has already been described by using wild-type proteins (69–72). However, this affords only a single catalyst in a defined protein environment, which may not actually exert a positive effect in catalysis. For example, why should a diphosphine/rhodium complex be more active as a hydrogenation catalyst in a protein than in its normal environment? The answer is “no,” unless of course the appropriate amino acids are positioned spatially correctly around the active (synthetic) site to stabilize the transition state of the reaction in a way that was originally proposed by Pauling for real enzymatic reactions (73). Additional binding of the metal by an amino acid of the protein may also affect the coordination chemistry and therefore the catalytic profile. We have suggested that it may be possible to achieve such activating effects (and stereoselectivity) by using directed evolution to produce thousands of mutants of an appropriately chosen protein as a host, to modify them chemically en masse, and then to perform a given transition metal-catalyzed reaction (41, 66–68). The gene encoding the best hit with respect to activity (and enantioselectivity) can then be subjected again to the process, creating an “evolutionary pressure” on the system (Fig. 9). Although in preliminary studies we were successful in performing chemical modification with introduction of ligands of the type salen, dipyridine and diphosphine, and several transition metals in large-scale reactions using a special wild-type enzyme (papain) (67), parallelization, and miniaturization of expression, protein purification, and chemical modification have yet to be accomplished. As an alternative to covalent chemical modification, we are also pursuing Whitesides' concept (69) of using noncovalent introduction of a rhodium–diphosphine complex by the avidin (or strepavidin)–biotin interaction (67). Attachment of metals directly to cysteine is yet another possibility.
Fig. 9.

Concept of directed evolution of hybrid catalysts (41, 66–68).
A different approach is to use metalloenzymes in synthetic reactions not intended by nature. For example, copper-containing enzymes could serve as catalysts in conjugate addition reactions or cyclopropanations, whereas zinc enzymes are candidates for Diels–Alder reactions. The challenge is to manipulate the wild-type enzymes by directed evolution so that efficient catalysts will be formed for application in the laboratories of organic chemists.
Conclusions
Since our original proposal that the methods of directed evolution may provide a new way to control the enantioselectivity of enzymes for use in organic chemistry (10), a great deal of progress has been made. The most intensively studied case involves the directed evolution of a lipase acting as a catalyst in the hydrolytic kinetic resolution of a chiral ester. The selectivity factor E was increased from 1.1 to >51 in favor of one enantiomer (12, 37). Reversal of enantioselectivity was also achieved (E = 30 in favor of the opposite enantiomer; ref. 39). Several efficient high-throughput ee assays were developed (17, 27–32), so that today the analytical problems of almost any project on directed evolution of enantioselective enzymes can be solved. One challenge for the future is development of selection (rather than screening) systems for enantioselectivity.
The practical results of our investigations are independent of any knowledge concerning the structure or the mechanism of the enzyme. Nevertheless, directed evolution is an excellent tool in enriching our knowledge of how enzymes function, specifically when analyzing the evolved mutants by molecular modeling/quantum mechanical methods. Indeed, effects that would never have been uncovered by conventional rational design were discovered, e.g., remote effects in controlling asymmetric induction (41, 46). In the future the combination of directed evolution and rational design may turn out to be the most effective way to improve the catalytic profile of enantioselective enzymes as catalysts in organic chemistry.
Several other enzymes have also been subjected to random mutagenesis to enhance their enantioselectivity, although directed evolution was not always involved (55, 57, 59–62). A single round of epPCR is not an evolutionary process.
Directed evolution is not only a method with which to enhance the activity, stability, and enantioselectivity of enzymes, but it can also potentially be used to solve such classical problems as product inhibition, which is known to cause difficulties in some (but certainly not all) large-scale industrial processes. Indeed, directed evolution of enantioselectivity is on its way to become an established science, and industrial applications are emerging. Finally, the prospect of applying directed evolution in controlling activity and selectivity of hybrid catalysts in reactions usually considered to be the domain of classical transition metal catalysis may also offer new perspectives (66–68).
Acknowledgments
We thank K.-E. Jaeger (Universität Düsseldorf, Düsseldorf, Germany), B. W. Dijkstra (University of Groningen, Groningen, The Netherlands), W. J. Quax (University of Groningen), R. M. Furstoss (Université de la Mediterranee, Marseille, France), J. C. Baratti (Université de la Mediterranee), M. Arand (Universität Würzburg, Würzburg, Germany), M. M. Kayser (University of New Brunswick, Saint John, Canada), and W. Thiel (Mülheim) for collaborations. This work was supported by European Union Projects QLK3-2000-00426 and QLRT-2001-00519, the Fonds der Chemischen Industrie, and the Max Planck Society.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: epPCR, error-prone PCR; ee, enantiomeric excess.
References
- 1.Knowles, W. S. (2002) Angew. Chem. Int. Ed. Engl. 41, 1998–2007. [Google Scholar]
- 2.Noyori, R. (2002) Angew. Chem. Int. Ed. Engl. 41, 2008–2022. [PubMed] [Google Scholar]
- 3.Sharpless, K. B. (2002) Angew. Chem. Int. Ed. Engl. 41, 2024–2032. [PubMed] [Google Scholar]
- 4.Jacobsen, E. N., Pfaltz, A. & Yamamoto, H., eds. (1999) Comprehensive Asymmetric Catalysis (Springer, Berlin), Vols. 1–3.
- 5.List, B. (2002) Tetrahedron 58, 5573–5590. [Google Scholar]
- 6.Schultz, P. G., Yin, J. & Lerner, R. A. (2002) Angew. Chem. Int. Ed. Engl. 41, 4427–4437. [DOI] [PubMed] [Google Scholar]
- 7.Drauz, K. & Waldmann, H., eds. (2002) Enzyme Catalysis in Organic Synthesis: A Comprehensive Handbook (VCH, Weinheim, Germany), 2nd Ed., Vols. 1–3.
- 8.Klibanov, A. M. (2001) Nature 409, 241–246. [DOI] [PubMed] [Google Scholar]
- 9.Reetz, M. T. (2002) Curr. Opin. Chem. Biol. 6, 145–150. [DOI] [PubMed] [Google Scholar]
- 10.Reetz, M. T., Zonta, A., Schimossek, K., Liebeton, K. & Jaeger, K.-E. (1997) Angew. Chem. Int. Ed. Engl. 36, 2830–2832. [Google Scholar]
- 11.Reetz, M. T. & Jaeger, K.-E. (2000) Chem. Eur. J. 6, 407–412. [DOI] [PubMed] [Google Scholar]
- 12.Reetz, M. T. (2000) Pure Appl. Chem. 72, 1615–1622. [Google Scholar]
- 13.Arnold, F. H. (2001) Nature 409, 253–257. [DOI] [PubMed] [Google Scholar]
- 14.Powell, K. A., Ramer, S. W., del Cardayré, S. B., Stemmer, W. P. C., Tobin, M. B., Longchamp, P. F. & Huisman, G. W. (2001) Angew. Chem. Int. Ed. Engl. 40, 3948–3959. [DOI] [PubMed] [Google Scholar]
- 15.Brakmann, S. & Johnsson, K. (2002) Directed Molecular Evolution of Proteins (or How to Improve Enzymes for Biocatalysis) (Wiley-VCH, Weinheim, Germany).
- 16.Arnold, F. H. & Georgiou, G. (2003) Directed Enzyme Evolution: Screening and Selection Methods (Humana Press, Totowa, NJ).
- 17.Reetz, M. T. (2001) Angew. Chem. Int. Ed. Engl. 40, 284–310. [PubMed] [Google Scholar]
- 18.Leung, D. W., Chen, E. & Goeddel, D. V. (1989) Technique (Philadelphia) 1, 11–15. [Google Scholar]
- 19.Cadwell, R. C. & Joyce, G. F. (1992) PCR Methods Appl. 2, 28–33. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki, M., Christians, F. C., Kim, B., Skandalis, A., Black, M. E. & Loeb, L. A. (1996) Mol. Diversity 2, 111–118. [DOI] [PubMed] [Google Scholar]
- 21.Eggert, T., Jaeger, K.-E. & Reetz, M. T. (2004) in Enzyme Functionality: Design, Engineering, and Screening, ed. Svendsen A. (Marcel Dekker, New York), pp. 375–390.
- 22.Barettino, D., Feigenbutz, M., Valcarcel, R. & Stunnenberg, H. G. (1994) Nucleic Acids Res. 22, 541–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostermeier, M., Shim, J. H. & Benkovic, S. J. (1999) Nat. Biotechnol. 17, 1205–1209. [DOI] [PubMed] [Google Scholar]
- 24.Sieber, V., Martinez, C. A. & Arnold, F. H. (2001) Nat. Biotechnol. 19, 456–460. [DOI] [PubMed] [Google Scholar]
- 25.Richardson, T. H., Tan, X., Frey, G., Callen, W., Cabell, M., Lam, D., Macomber, J., Short, J. M., Robertson, D. E. & Miller, C. (2002) J. Biol. Chem. 277, 26501–26507. [DOI] [PubMed] [Google Scholar]
- 26.Zha, D., Eipper, A. & Reetz, M. T. (2003) ChemBioChem 4, 34–39. [DOI] [PubMed] [Google Scholar]
- 27.Reetz, M. T. (2003) Methods Mol. Biol. 230, 259–282. [DOI] [PubMed] [Google Scholar]
- 28.Reetz, M. T., Becker, M. H., Klein, H.-W. & Stöckigt, D. (1999) Angew. Chem. Int. Ed. Engl. 38, 1758–1761. [DOI] [PubMed] [Google Scholar]
- 29.Schrader, W., Eipper, A., Pugh, D. J. & Reetz, M. T. (2002) Can. J. Chem. 80, 626–632. [Google Scholar]
- 30.Reetz, M. T., Eipper, A., Tielmann, P. & Mynott, R. (2002) Adv. Synth. Catal. 344, 1008–1016. [Google Scholar]
- 31.Tielmann, P., Boese, M., Luft, M. & Reetz, M. T. (2003) Chem. Eur. J. 9, 3882–3887. [DOI] [PubMed] [Google Scholar]
- 32.Reetz, M. T. (2003) Methods Mol. Biol. 230, 283–290. [DOI] [PubMed] [Google Scholar]
- 33.Reetz, M. T. & Rüggeberg, C. J. (2002) Chem. Commun. 1428–1429. [DOI] [PubMed]
- 34.Dröge, M. J., Rüggeberg, C. J., van der Sloot, A. M., Schimmel, J., Dijkstra, R. S., Verhaert, R. M. D., Reetz, M. T. & Quax, W. J. (2003) J. Biotechnol. 4, 19–28. [DOI] [PubMed] [Google Scholar]
- 35.Reetz, M. T. & Jaeger, K.-E. (1999) Top. Curr. Chem. 200, 31–57. [Google Scholar]
- 36.Liebeton, K., Zonta, A., Schimossek, K., Nardini, M., Lang, D., Dijkstra, B. W., Reetz, M. T. & Jaeger, K.-E. (2000) Chem. Biol. 7, 709–718. [DOI] [PubMed] [Google Scholar]
- 37.Reetz, M. T., Wilensek, S., Zha, D. & Jaeger, K.-E. (2001) Angew. Chem. Int. Ed. Engl. 40, 3589–3591. [DOI] [PubMed] [Google Scholar]
- 38.Crameri, A. & Stemmer, W. P. C. (1995) BioTechniques 18, 194–196. [PubMed] [Google Scholar]
- 39.Zha, D., Wilensek, S., Hermes, M., Jaeger, K.-E. & Reetz, M. T. (2001) Chem. Commun. 2664–2665.
- 40.Nardini, M., Lang, D. A., Liebeton, K., Jaeger, K.-E. & Dijkstra, B. W. (2000) J. Biol. Chem. 275, 31219–31225. [DOI] [PubMed] [Google Scholar]
- 41.Reetz, M. T. (2002) Tetrahedron 58, 6595–6602. [Google Scholar]
- 42.Rotticci, D., Rotticci-Mulder, J. C., Denman, S., Norin, T. & Hult, K. (2001) ChemBioChem 2, 766–770. [DOI] [PubMed] [Google Scholar]
- 43.Hirose, Y., Kariya, K., Nakanishi, Y., Kurono, Y. & Achiwa, K. (1995) Tetrahedron Lett. 36, 1063–1066. [Google Scholar]
- 44.Fischer, E. (1894) Ber. Dtsch. Chem. Ges. 27, 2985–2036. [Google Scholar]
- 45.Fersht, A. (1997) Structure and Mechanism in Protein Science (Freeman, New York).
- 46.Bocola, M., Otte, N., Jaeger, K.-E., Reetz, M. T. & Thiel, W. (2004) ChemBioChem. 5, 100–109. [DOI] [PubMed] [Google Scholar]
- 47.Taylor, S. V., Kast, P. & Hilvert, D. (2001) Angew. Chem. Int. Ed. Engl. 40, 3310–3335. [DOI] [PubMed] [Google Scholar]
- 48.Zhao, H. & Arnold, F. H. (1999) Protein Eng. 12, 47–53. [DOI] [PubMed] [Google Scholar]
- 49.Iffland, A., Tafelmeyer, P., Saudan, C. & Johnsson, K. (2000) Biochemistry 39, 10790–10798. [DOI] [PubMed] [Google Scholar]
- 50.Kumar, M., Kannan, K. K., Hosur, M. V., Bhavesh, N. S., Chatterjee, A., Mittal, R. & Hosur, R. V. (2002) Biochem. Biophys. Res. Commun. 294, 395–401. [DOI] [PubMed] [Google Scholar]
- 51.Agarwal, P. K., Billeter, S. R., Rajagopalan, P. T. R., Benkovic, S. J. & Hammes-Schiffer, S. (2002) Proc. Natl. Acad. Sci. USA 99, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold, F. H., Wintrode, P. L., Miyazaki, K. & Gershenson, A. (2001) Trends Biochem. Sci. 26, 100–106. [DOI] [PubMed] [Google Scholar]
- 53.Horsman, G. P., Liu, A. M. F., Henke, E., Bornscheuer, U. T. & Kazlauskas, R. J. (2003) Chem. Eur. J. 9, 1933–1939. [DOI] [PubMed] [Google Scholar]
- 54.Funke, S. A., Eipper, A., Reetz, M. T., Otte, N., Thiel, W. & van Pouderoyen, G. (2003) Biocatal. Biotransform. 21, 67–73. [Google Scholar]
- 55.DeSantis, G., Wong, K., Farwell, B., Chatman, K., Zhu, Z., Tomlinson, G., Huang, H., Tan, X., Bibbs, L., Chen, P., et al. (2003) J. Am. Chem. Soc. 125, 11476–11477. [DOI] [PubMed] [Google Scholar]
- 56.Reetz, M. T., Becker, M. H., Stöckigt, D. & Klein, H. W. (2000) Patent Appl. WO 00/58504.
- 57.Matcham, G. W. & Bowen, A. R. S. (1996) Chim. Oggi 14, 20–24. [Google Scholar]
- 58.Henke, E. & Bornscheuer, U. T. (1999) Biol. Chem. 380, 1029–1033. [DOI] [PubMed] [Google Scholar]
- 59.May, O., Nguyen, P. T. & Arnold, F. H. (2000) Nat. Biotechnol. 18, 317–320. [DOI] [PubMed] [Google Scholar]
- 60.Fong, S., Machajewski, T. D., Mak, C. C. & Wong, C.-H. (2000) Chem. Biol. 7, 873–883. [DOI] [PubMed] [Google Scholar]
- 61.Reetz, M. T., Torre, C., Eipper, A., Lohmer, R., Hermes, M., Brunner, B., Maichele, A., Bocola, M., Arand, M., Cronin, A., et al. (2004) Org. Lett. 6, 177–180. [DOI] [PubMed] [Google Scholar]
- 62.Cedrone, F., Niel, S., Roca, S., Bhatnagar, T., Ait-Abdelkader, N., Torre, C., Krumm, H., Maichele, A., Reetz, M. T. & Baratti, J. C. (2003) Biocatal. Biotransform. 21, 357–364. [Google Scholar]
- 63.Jacobsen, E. N. (2000) Acc. Chem. Res. 33, 421–431. [DOI] [PubMed] [Google Scholar]
- 64.Orru, R. V. A. & Faber, K. (1999) Curr. Opin. Chem. Biol. 3, 16–21. [DOI] [PubMed] [Google Scholar]
- 65.Carr, R., Alexeeva, M., Enright, A., Eve, T. S., Dawson, M. J. & Turner, N. J. (2003) Agnew. Chem. Int. Ed. Engl. 42, 4807–4810. [DOI] [PubMed] [Google Scholar]
- 66.Reetz, M. T. (2002) in Pharmacochemistry Library, Trends in Drug Research III, ed. van der Goot H. (Elsevier, Amsterdam), Vol. 32, pp. 27–37. [Google Scholar]
- 67.Reetz, M. T., Rentzsch, M., Pletsch, A. & Maywald, M. (2002) Chimia 56, 721–723. [Google Scholar]
- 68.Reetz, M. T. (2001) Patent Appl. DE-A 101 29187.6.
- 69.Wilson, M. E. & Whitesides, G. M. (1978) J. Am. Chem. Soc. 100, 306–307. [Google Scholar]
- 70.Hilvert, D. & Kaiser, E. T. (1987) Biotechnol. Genet. Eng. Rev. 5, 297–318. [DOI] [PubMed] [Google Scholar]
- 71.Qi, D., Tann, C.-M., Haring, D. & Distefano, M. D. (2001) Chem. Rev. 101, 3081–3111. [DOI] [PubMed] [Google Scholar]
- 72.Collot, J., Gradinaru, J., Humbert, N., Skander, M., Zocchi, A. & Ward, T. R. (2003) J. Am. Chem. Soc. 125, 9030–9031. [DOI] [PubMed] [Google Scholar]
- 73.Pauling, L. (1948) Nature 161, 707–709. [DOI] [PubMed] [Google Scholar]