

Abstract
Objective:
To assess the safety and tolerability of high-dose creatine, the feasibility of enrolling premanifest and 50% at-risk subjects in a prevention trial, and the potential of cognitive, imaging, and blood markers.
Methods:
Sixty-four eligible consenting participants were randomly allocated (1:1) to 15 g twice daily of creatine monohydrate or placebo for a 6-month double-blind phase followed by a 12-month open-label extension. Subjects included premanifest (tested) and at-risk (not tested) individuals without clinical symptoms or signs of Huntington disease (HD). Primary outcomes were safety and tolerability. Exploratory endpoints included fine motor, visuospatial, and memory performance; structural and diffusion MRI; and selected blood markers.
Results:
Forty-seven HD carriers and 17 non-HD controls were enrolled. Fifteen discontinued treatment (2 assigned to placebo); all were followed for the entire study period. Primary analysis was by intent to treat. The most common adverse events were gastrointestinal. Neuroimaging demonstrated treatment-related slowing of cortical and striatal atrophy at 6 and 18 months.
Conclusion:
We describe a design that preserves the autonomy of subjects not wanting genetic testing while including controls for assessing the specificity of treatment effects. Our results demonstrate the feasibility of prevention trials for HD and the safety of high-dose creatine, provide possible evidence of disease modification, support future studies of creatine, and illustrate the value of prodromal biomarkers.
Classification of evidence:
This study provides Class I evidence that high-dose creatine is safe and tolerable.
Huntington disease (HD) is a progressive autosomal dominant inherited disorder that typically manifests clinically in the third to fifth decades. Genetic testing enables the unambiguous identification of those destined to develop HD and the possibility of initiating preventive treatments during the disease prodrome. Emerging neuroprotective therapies have set the stage for secondary prevention trials with the goal of delaying the onset of symptoms. Demonstrating prevention is a major challenge because very large trials are necessary to demonstrate reduced rates of clinical onset. Furthermore, while subtle cognitive and motor dysfunction and changes on neuroimaging measures occur as early as 2 decades before symptoms,1,2 reliable markers of prodromal progression remain elusive.
PRECREST (Creatine Safety and Tolerability in Premanifest HD) is a secondary prevention trial using up to 30 g daily of creatine monohydrate, a high-energy phosphate buffer that restores adenosine triphosphate from adenosine diphosphate. We used a novel study design that included asymptomatic individuals who were either 50% at risk on the basis of an affected first-degree relative but not wishing to know their genetic status or who had undergone predictive testing and were known to have the HD mutation. The former greatly enlarged the pool of HD carriers for PRECREST and the matched non-HD control group negated coercion of genetic testing and permitted an assessment of the specificity of effects of creatine. PRECREST included a 6-month randomized, double-blind, placebo-controlled phase followed by 12 months of open-label creatine, which promoted recruitment and retention and provided further longitudinal data for assessing potential clinical, cognitive, and biological markers of the HD prodrome.3
METHODS
Participants.
Sixty-four asymptomatic participants, at least 26 years of age, who were either premanifest (PHD) individuals with a known genetic test result or who were at 50% risk of HD on the basis of an affected first-degree relative were recruited for this single-site study at the Massachusetts General Hospital. Nineteen participants were known genetic carriers and 45 participants were 50% at risk and intended to remain blinded to their genotype. Double-blind genotyping confirmed the diagnosis in the known HD carriers and established that the participant pool was composed of 47 premanifest HD carriers and 17 healthy age-matched controls (HCs) without a CAG repeat expansion. The study biostatistician had exclusive access to genotypes, which were not disclosed to the participants or investigators.
Standard protocol approvals, registrations, and patient consents.
The study protocol was approved by the Partners Human Research Committee and monitored by an independent data and safety monitoring board approved by the National Institute of Neurological Disorders and Stroke. ClinicalTrials.gov: NCT00592995.
Randomization and masking.
Treatment was unstratified by genotype and individuals were randomized in a 1:1 schema to receive either the active drug (creatine) or matching placebo for the placebo-controlled phase. Four-digit enrollment identification numbers, corresponding to specific study drug boxes, were developed by the study biostatistician. Treatment assignments were randomly and sequentially assigned at baseline. The treatment allocation for the placebo-controlled phase was not disclosed for the duration of the study (CONSORT [Consolidated Standards of Reporting Trials], figure 1). The creatine group was composed of 25 PHD and 7 HC subjects; the placebo group was composed of 22 PHD and 10 HC subjects. There were no significant baseline differences between the randomized groups (table e-1 on the Neurology® Web site at Neurology.org).
Figure 1. CONSORT flow diagram.

CONSORT = Consolidated Standards of Reporting Trials; HC = healthy control; PHD = premanifest Huntington disease.
Procedures.
Routine clinical safety assessments were conducted at 2-month intervals during the placebo-controlled phase and every 4 months during the open-label phase. Additional assessments were completed at baseline (visit 1 or V1), 6 months (visit 5 or V5), the end of the placebo-controlled phase, and at 18 months (visit 8 or V8), the end of the open-label phase. Assessments at these visits included the Unified Huntington’s Disease Rating Scale to assess severity of symptoms and signs, Choice Reaction Task to evaluate subtle motor dysfunction,4 Hopkins Verbal Learning Test to evaluate episodic memory,5,6 and a computerized Mental Rotation Task to assess visuospatial processing.7 To evaluate potential effects of treatment on progressive brain pathology, structural and diffusion imaging (1.5T Avanto System; Siemens, Erlangen, Germany) were collected. Anatomical scans were analyzed using the FreeSurfer longitudinal processing stream, with high test-retest reliability and sensitivity,8 and the FSL Tract-Based Spatial Statistics and FSL Randomise were used for processing and analyses of diffusion tensor imaging data using threshold-free cluster enhancement for image-based correction for multiple comparisons.9 Results were adjusted for multiple comparisons using the false discovery rate criterion. Finally, blood was collected for evaluation of 8OH2dG, a marker of oxidative injury.10–12
Statistical analysis.
Primary outcome variables as per protocol, which included frequency of adverse events and tolerability, were analyzed according to intent to treat. Prevalent adverse events were compared between groups using Fisher exact test.
Differences at baseline on secondary endpoints between groups were evaluated using analysis of covariance, adjusting for age. Longitudinal analyses of secondary endpoints collected over time were performed according to actual treatment, using repeated-measures linear models. Visit, treatment group, and their interaction along with age at baseline were entered into the model. A compound symmetry variance-covariance structure was assumed. Longitudinal analyses included all available data without recourse to imputations.
RESULTS
Primary outcome measures.
Tolerability.
Participants were randomized to creatine vs placebo 32:32 and allowed to take their respective maximum tolerated dose, up to 30 g daily, throughout the study. More than two-thirds of those randomized to creatine tolerated the maximum dose, and more than three-quarters tolerated doses of 15 g or more. Approximately 13% of those randomized to placebo were unable to tolerate the study drug. Ten subjects discontinued the study drug during the placebo-controlled phase: interestingly, this included 1 HC and 9 PHD individuals (7 knew their genetic status, 2 had been assigned to placebo). During the open-label phase, an additional 5 subjects were unable to tolerate creatine (3 HCs and 2 PHD subjects). Reasons for discontinuation included mild gastrointestinal discomfort (8), the palatability of the oral formulation and the inconvenience of twice-daily dosing (1), and the psychological stress caused by treatment being a daily reminder of the risk of HD (6). There was no significant difference in tolerability to doses more than 15 g between those assigned to placebo vs creatine (p = 0.56).
Safety.
There were no significant clinical, laboratory, or ECG abnormalities during the study. Data were reviewed by an independent data and safety monitoring board with membership approved by the National Institute of Neurological Disorders and Stroke. No participant met the criteria for a clinical diagnosis, based on unequivocal motor signs, during the course of the study.
Adverse events.
The only significant differences between treatment groups were an increase in diarrhea and nausea associated with creatine (p = 0.007). Five serious adverse events occurred, none of which were expected or related to the study drug (fracture requiring repair [2], hysterectomy [the only serious adverse event during active treatment], nephrolithiasis, and miscarriage). Table e-2 summarizes the cumulative adverse events, by subject, with a frequency of >1.
Secondary endpoints.
Neuroimaging.
Using a general linear model, we found significant regional atrophy at baseline in the PHD group compared with HC subjects, particularly in sensorimotor, superior temporal, and portions of parietal and occipital cortex, in some regions by more than 0.3 mm, corresponding to as much as 10% thinner as compared with controls (figure 2A). Caudate and putamen volumes were also significantly smaller in the PHD group (p < 0.001). Diffusion measures were significantly different in the PHD subjects in whom fractional anisotropy was reduced, and mean diffusivity, axial diffusivity (AD), and radial diffusivity were increased, compared with HCs (figure 2B), consistent with widespread alterations in white matter integrity.
Figure 2. Baseline differences in neuroimaging between the control and PHD groups.

(A, left) At baseline, the PHD group demonstrated early regionally selective cortical atrophy in several cortical regions including inferior parietal, lateral occipital, precentral, precuneus, superior parietal, and superior temporal regions (the color range indicates p values, red to yellow, p < 0.05 to p < 0.001, respectively). (A, right) Magnitude maps demonstrating cortical thinning in the PHD group as compared with HCs in the corresponding regions. Red reflects >0.1 mm thinning, corresponding to approximately 3% thinner cortex in the PHD group. (B) Voxel-based diffusion maps at baseline suggest extensive early white matter alterations. FA was reduced and AD, MD, and RD were increased in the PHD group as compared with HCs (p < 0.05, corrected), including the corpus callosum, corticospinal tracts, corona radiata, and superior longitudinal fasciculus. AD = axial diffusivity; CN = control; FA = fractional anisotropy; HC = healthy control; MD = mean diffusivity; PHD = premanifest Huntington disease; RD = radial diffusivity.
At the end of the placebo-controlled phase, using a surface-based, within-subject, longitudinal approach, we found that the rate of cortical thinning in several cortical regions, including portions of precentral, superior and middle temporal, superior and middle frontal, precuneus, posterior parietal, and occipital, was significantly slower in the PHD-creatine group compared with the PHD-placebo group, in whom thinning progressed as much as 5% per year (p < 0.0001 in select regions, figure 3).
Figure 3. Surface-based maps showing cortical thinning to be slowed by creatine.
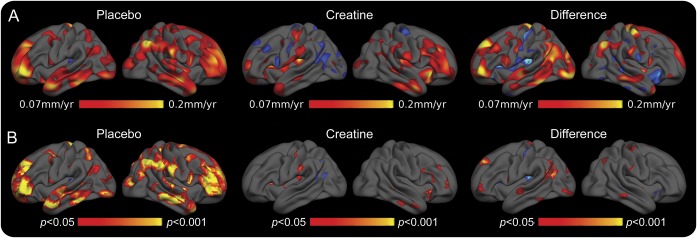
(A) Rate maps, according to randomization. (B) Corresponding significance maps. Premanifest Huntington disease (PHD) subjects randomized to placebo demonstrated progressive thinning at rates as high as 0.2 mm/year in several cortical regions, including portions of frontal, temporal, and parietal cortex; these were significant compared with baseline. In contrast, in the PHD subjects randomized to creatine, cortical thinning primarily involved portions of the frontal and temporal cortex and was not significant compared with baseline. The differences in the rates of thinning in the creatine-treated and the placebo-treated groups were significant in a number of regions. These data suggest a beneficial effect of creatine on cortical thinning.
The partial crossover design necessitated use of a repeated-measures linear model for evaluating select regions of interest. At the end of the placebo-controlled phase, there was no significant change in white matter (−0.3551 cm3) and gray matter (−4.55 cm3) volumes in the PHD group during creatine treatment. In contrast, there was progressive volume loss (white matter −6.29 cm3, gray matter −9.52 cm3) in the PHD group randomized to placebo, although differences between the groups were not significant. Similarly, there was no significant change in basal ganglia volumes during creatine treatment, whereas those randomized to placebo demonstrated progressive atrophy (caudate −0.07 cm3 creatine, −0.29 cm3 placebo; putamen −0.11 cm3 creatine, −0.49 cm3 placebo) (figure 4A). Creatine was also associated with significantly less cortical thinning in several regions, including the precentral (p = 0.052), occipital (p = 0.028), superior frontal (p = 0.003), and supramarginal (p = 0.028) (figure 4B).
Figure 4. Repeated-measures modeling of 18-month changes confirms the slowing of cortical atrophy in the original placebo group.

(A) Volume. Between visit 1 (V1) and V5, the changes in white matter, gray matter, caudate, and putamen volumes in premanifest Huntington disease (PHD) subjects randomized to creatine were similar to changes in healthy control (HC) subjects. The reduction in these volumes tended to be more rapid in PHD subjects treated with placebo. (B) Cortical thinning. Between V1 and V5, the rates of cortical thinning in PHD subjects treated with creatine were not significantly different than HCs. In contrast, the rates of cortical thinning were significantly faster in the PHD subjects randomized to placebo (p < 0.05). Between V5 and V8, the rate of thinning in the PHD group randomized to placebo slowed significantly after creatine was started (p < 0.01, illustrated in select regions of interest; red arrow indicates the end of the placebo-controlled phase), again suggesting slowed atrophy. Blue = HCs; solid black lines = PHD subjects randomized to creatine at V1; black dashed lines = PHD subjects randomized to placebo who subsequently crossed over to open label.
For the group crossing over from placebo to creatine at V5, atrophy rates slowed, replicating the initial treatment benefit. Evaluating the cumulative effect of the average annual rate of change associated with creatine exposure compared with change due to placebo, during the double-blind portion of the study, demonstrated significant differences in caudate (p = 0.04), precentral (p = 0.008), superior frontal (p = 0.003), supramarginal (p = 0.01), and occipital (p = 0.022), and a trend in putamen (p = 0.06) and white matter (p = 0.08).
Modeling of AD in white matter regions, including the corpus callosum, corticospinal tracts, corona radiata, and superior longitudinal fasciculus, demonstrated no treatment effects on diffusion measures during the study. Representative AD maps are shown in figure 5.
Figure 5. Axial diffusivity not affected by creatine treatment.

Repeated-measures modeling of 18-month change. There was no effect of creatine on axial diffusivity for any region of interest (red arrow indicates the end of the placebo-controlled phase, p < 0.01 for all regions of interest). Blue = healthy controls; solid black lines = premanifest Huntington disease (PHD) subjects randomized to creatine at visit 1 (V1); black dashed lines = PHD subjects randomized to placebo who subsequently crossed over to open label.
Cognitive measures.
At baseline, PHD participants performed less well than HC subjects on the Unified Huntington’s Disease Rating Scale cognitive battery, consistent with earlier reports, less well on the Mental Rotation Task, suggesting subtle deficits in visuospatial function, and less well on the Hopkins Verbal Learning Test, suggesting early deficits in episodic recall and recognition memory. There was no significant effect of creatine on cognitive measures, compared with baseline, at either V5 or V8 (table e-3).
Blood analyses.
At baseline, a nonsignificant trend of higher 8OH2dG was observed in PHD as compared with HC subjects. At V5, there was a trend toward lower 8OH2dG concentrations in the PHD-creatine group compared with PHD-placebo, primarily because of a reduction of levels in those who had baseline elevations above the mean control value (p = 0.06).
DISCUSSION
PRECREST is the first secondary prevention trial in prodromal HD. It examined the feasibility of a novel design that included 50% at-risk individuals; the safety and tolerability of creatine in doses higher than previously reported; and candidate cognitive, blood, and imaging markers of the HD prodrome that could be useful for future studies.
A major hurdle for prevention trials is that only a small fraction13,14 of individuals with a genetic risk of HD have undergone genetic testing because of its attendant psychological stress and social risks (genetic discrimination).15,16 Thus, relatively few premanifest individuals are available for clinical trials, and requiring genetic testing as a prerequisite for participation could be considered coercive. In designing PRECREST, we included 50% at-risk individuals, rather than limit enrollment to confirmed carriers, so that individuals who had not desired genetic testing would have an opportunity to participate and to incorporate a matched non-HD control group for examining the specificity of treatment effects. This strategy prevented coercion of genetic testing as a condition for participating and greatly expanded the pool of potential participants, because the vast majority (>90% in the United States) of individuals at risk of HD have abstained from genetic testing.16 The HCs were crucial for examining age effects and for assessing whether treatment affects neuroimaging and other markers in controls. We and our institutional review board considered exposing healthy participants without the HD mutation to the risks of a treatment to be acceptable because the expected toxicities from creatine are modest and reversible, and also because these subjects are HD family members with a significant stake and potential for benefit for their families from the knowledge gained compared with naive controls.
PRECREST also allowed us to better understand some of the factors affecting the participation of at-risk and PHD subjects. We postulated that side effects and toxicity might be less acceptable to healthy premanifest individuals who might have full time jobs or young families, or that the lack of symptoms in premanifest individuals might make the potential benefits difficult to conceptualize, especially for experimental treatments of uncertain efficacy and toxicity. We were able to enroll a sizable group of otherwise healthy individuals at a single site who were able to participate despite work, childcare, and other demands. Interestingly, there was a higher dropout rate among known gene-expanded carriers compared with those at 50% risk, even though the former group might be expected to be more motivated by the increased certainty of future illness. Known carriers were also less willing to tolerate mild gastrointestinal difficulties and were more concerned about the risk of inadvertent disclosure of genetic information. Possible explanations were that participation was more difficult for those certain to develop clinical HD because of a higher level of anxiety or because the HD prodrome depleted their psychological, cognitive, or social resources. The unexpectedly high rate of intolerability and the reasons for discontinuing study drug in the known gene-expanded group may be important for future trial design considerations.
Individuals at risk of HD who either know or do not know their genetic status typically have high levels of concern about the confidentiality of their genetic risk.17,18 For those not wanting genetic testing, it was crucial to provide assurance that their genotyping would not be disclosed. For premanifest participants that had not yet been diagnosed clinically with HD, it was crucial that their identity as HD family members be protected from the medical record to help preserve their autonomy over their genetic information. To separate subject identities from activities generating medical data, we developed an alternative set of identifiers for the purposes of this study. Our experience suggests that preserving autonomy over genetic testing and genetic risk information is a critical consideration for designing future clinical trials in this vulnerable population.
A major goal in PRECREST was to evaluate potential markers of progression for prodromal HD. While we found significant differences between HCs and gene-expanded carriers at baseline in many cognitive measures, most failed to provide the necessary sensitivity to usefully measure change over the duration of PRECREST. This has been a consistent finding in other studies evaluating the feasibility of using cognitive measures in trials in this population,2,4,19 and speaks to the limitations of applying cross-sectional models to predict longitudinal change. It can also be understood when considering that the HD prodrome is slowly progressive over more than 2 decades before discernable clinical symptoms, signs, or manifestations occur. The 8OH2dG level, a plasma marker of oxidative stress, trended toward normal with creatine treatment in subjects with baseline elevations. However, because 8OH2dG is elevated in only a subset of PHD individuals,20 this may not be a useful pharmacodynamic marker for PHD subjects.21
There were significant changes in brain anatomy and white matter integrity in premanifest individuals at baseline compared with controls, consistent with previous studies.4,22,−,24 Using longitudinal models, we demonstrated that at the end of the placebo-controlled phase (6 months), HD carriers receiving creatine had significantly slower rates of cortical thinning, suggesting potentially beneficial effects of creatine on prodromal progression. This was replicated by the reduced slopes in the PHD group originally randomized to placebo after crossing over to creatine at V5. Brain volumes in HC subjects were not affected by creatine treatment, indicating that reductions in atrophy in PHD subjects could not be explained by the nonspecific effects of creatine treatment. Longitudinal diffusion measures were not significantly changed in response to treatment; they may not be as sensitive for measuring change as anatomical measures. This study illustrates the potential utility of longitudinal MRI measures of subclinical progression for studying PHD in clinical trials. While slowed atrophy suggests that creatine could slow preclinical progression, the potential clinical impact of these findings on delaying the onset of HD is unknown and must be defined by an efficacy study designed to measure it.
This study illustrates the feasibility of designing and implementing a phase II study in PHD, the value of including high-value 50% at-risk subjects whose autonomy and genetic privacy can be protected, and the utility of using neuroimaging to seek preliminary evidence of disease modification. Our study also underscores the importance of clinically relevant, sensitive, and reliable biomarkers for PHD and suggests that anatomical neuroimaging measures may provide a critical biomarker in future clinical trials of this population.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to the individuals who participated in this study, who so generously contributed their time and energy to this work, and without whom it would not have been possible. The work is dedicated to J.H.R., who lived a life helping those in need. The authors thank the members of the data and safety monitoring board and the individuals who helped recruit and assess subjects for this study, including Alex Bender, Susan Maya, Puja Turakhia, Rachel Goldstein, Angela Hu, Jamie Hill, and Matt Germat. The Avicena Group Inc. (Palo Alto, CA) provided the study drug.
GLOSSARY
- AD
axial diffusivity
- HC
healthy control
- HD
Huntington disease
- PHD
premanifest Huntington disease
- PRECREST
Creatine Safety and Tolerability in Premanifest HD
Editorial, page 824
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
H.D.R. is the co-principal investigator for the PRECREST study. Her responsibilities have included obtaining funding, study design and conception, protocol development, clinical assessments, data analyses and interpretation, and authoring the manuscript. G.D. was the study statistician who was responsible for the data analysis and interpretation and contributed to authoring and reviewing the manuscript and generation of figures. S.G. was involved in the study design, protocol development, data management and quality control, data analysis and interpretation, and reviewing the manuscript. K.M. was involved in the protocol development, assistance with cognitive ratings, data analysis and interpretation, and reviewing the manuscript. M.R. was responsible for the development of the magnetic resonance processing analytical stream, data quality control, and reviewing the manuscript. J.-P.C. was responsible for the diffusion analyses, quality control, and contributed to the editing of the manuscript. T.D.T. and P.J.W. were responsible for the implementation of the scanner protocols, magnetic resonance data analyses, data quality control, and contributed to the editing of the manuscript. W.M. was responsible for the 8OH2dG analysis, data interpretation, and contributed to the editing of the manuscript. D.H.S. was responsible for the analysis and interpretation of data and contributed to the editing of the manuscript. S.M.H. was co-principal investigator and was responsible for obtaining funding, study design and conception, data interpretation, and had a key role in authoring the manuscript.
STUDY FUNDING
Support by the NIH (grants P01NS058793, NS042861, NS058793, AT000613, and FD003359 to H.D.R. and S.M.H., and NS058793 to G.D.).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Tabrizi SJ, Reilmann R, Roos RA, et al. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol 2012;11:42–53 [DOI] [PubMed] [Google Scholar]
- 2.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79:874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huntington Study Group Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord 1996;11:136–142 [DOI] [PubMed] [Google Scholar]
- 4.Tabrizi SJ, Langbehn DR, Leavitt BR, et al. Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solomon AC, Stout JC, Johnson SA, et al. Verbal episodic memory declines prior to diagnosis in Huntington's disease. Neuropsychologia 2007;45:1767–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aretouli E, Brandt J. Episodic memory in dementia: characteristics of new learning that differentiate Alzheimer's, Huntington's, and Parkinson's diseases. Arch Clin Neuropsychol 2010;25:396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohr E, Brouwers P, Claus JJ, Mann UM, Fedio P, Chase TN. Visuospatial cognition in Huntington's disease. Mov Disord 1991;6:127–132 [DOI] [PubMed] [Google Scholar]
- 8.Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 2012;61:1402–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith SM, Jenkinson M, Johansen-Berg H, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage 2006;31:1487–1505 [DOI] [PubMed] [Google Scholar]
- 10.Hersch SM, Gevorkian S, Marder K, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology 2006;66:250–252 [DOI] [PubMed] [Google Scholar]
- 11.Stack EC, Ferrante RJ. Huntington's disease: progress and potential in the field. Expert Opin Investig Drugs 2007;16:1933–1953 [DOI] [PubMed] [Google Scholar]
- 12.Long JD, Matson WR, Juhl AR, et al. 8OHdG as a marker for Huntington disease progression. Neurobiol Dis 2012;46:625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myers RH. Huntington's disease genetics. NeuroRx 2004;1:255–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hersch S, Jones R, Koroshetz W, Quaid K. The neurogenetics genie: testing for the Huntington's disease mutation. Neurology 1994;44:1369–1373 [DOI] [PubMed] [Google Scholar]
- 15.Bombard Y, Palin J, Friedman JM, et al. Beyond the patient: the broader impact of genetic discrimination among individuals at risk of Huntington disease. Am J Med Genet B Neuropsychiatr Genet 2012;159B:217–226 [DOI] [PubMed] [Google Scholar]
- 16.Quaid KA, Sims SL, Swenson MM, et al. Living at risk: concealing risk and preserving hope in Huntington disease. J Genet Couns 2008;17:117–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayden MR, Bombard Y. Psychosocial effects of predictive testing for Huntington's disease. Adv Neurol 2005;96:226–239 [PubMed] [Google Scholar]
- 18.Harper PS, Gevers S, de Wert G, Creighton S, Bombard Y, Hayden MR. Genetic testing and Huntington's disease: issues of employment. Lancet Neurol 2004;3:249–252 [DOI] [PubMed] [Google Scholar]
- 19.Stout JC, Jones R, Labuschagne I, et al. Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington's disease. J Neurol Neurosurg Psychiatry 2012;83:687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hersch SM, Rosas HD. Biomarkers to enable the development of neuroprotective therapies for Huntington's disease. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton, FL: CRC Press; 2011 [Google Scholar]
- 21.Borowsky B, Warner J, Leavitt BR, et al. 8OHdG is not a biomarker for Huntington disease state or progression. Neurology 2013;80:1934–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 2005;65:745–747 [DOI] [PubMed] [Google Scholar]
- 23.Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004;63:66–72 [DOI] [PubMed] [Google Scholar]
- 24.Rosas HD, Lee SY, Bender AC, et al. Altered white matter microstructure in the corpus callosum in Huntington's disease: implications for cortical “disconnection.” Neuroimage 2010;49:2995–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.