

Abstract
An autocatalytic reaction in which the reaction product serves as a catalyst to produce more of itself and to suppress production of its enantiomer serves as a mechanistic model for the evolution of homochirality. The Soai reaction provided experimental confirmation of this concept, nearly 50 years after it was first proposed. This Perspective offers a rationalization of the Soai autocatalytic reaction; accounting for enantiomeric excess and rate observations, that is both simple as well as gratifying in its implications for the chemical origin of life.
Counted among the most noteworthy findings of the last decade in asymmetric catalysis research must certainly be Soai and coworkers' (1–3) discovery of asymmetric amplification in the autocatalytic alkylation of pyrimidyl aldehydes with dialkylzincs (Scheme 1), in which the reaction rate is accelerated by addition of catalytic amounts of its alcohol product. Autocatalysis is a well known phenomenon, and asymmetric versions of autocatalytic reactions have been reported (4). What made Soai's (1) discovery truly remarkable was that it provided the first example such a reaction employing very low enantiomeric excess (ee) catalyst and yielding very high ee catalyst as product. Some 40 years earlier (5), asymmetric amplification in autocatalysis had been implicated in a theoretical rationalization of the evolution of homochirality from a racemic environment. Thus, the ramifications of Soai's work (1) have the potential to speak to the heart of some of our most intriguing questions about the chemical origin of life on Earth.
Scheme 1.

The Soai autocatalytic reaction.
It is important to view the significance of Soai's discovery (1) in the context of the intensive search that has taken place to find an experimental system that would confirm the theoretical conclusions concerning asymmetric amplification in autocatalysis outlined in Frank's 1953 paper (5). This work provided a “simple and sufficient life model” for a chemical rationalization of the evolution of high ee in organic molecules from a small initial imbalance, and Frank (5) concluded with these provocative words: “A laboratory demonstration may not be impossible.” In the intervening years, this “challenge to all red-blooded chemists” (6) has been taken up by several groups. Discussion of possible mechanisms through which high optical activity was achieved from a racemic or prochiral prebiotic environment have also implicated physical processes such as crystallization. For example, Kondepudi (7, 8) first reported spontaneous chiral symmetry breaking in crystallization little more than 10 years ago. However, the Soai system outlined in Scheme 1 was the first, and to date remains the only, successful chemical answer to this challenge.
Although it can hardly be argued that the particular reaction shown in Scheme 1 could itself have played an important role in an aqueous prebiotic environment, it serves as a model to help us understand the implications of asymmetric amplification in autocatalysis for rationalizing the chemical origin of life. This question has two separate but equally important parts: (i) can we rationalize the creation of an initial small imbalance of ee; and (ii) can we explain how an initial small imbalance may be propagated ultimately to produce a homochiral system?
Discussion of these two questions in the context of the Soai system (1) leads naturally to reflections on how what we have learned from this particular system may help us to frame the general challenges for the future.
Shattering the Mirror: Absolute Asymmetric Synthesis
The subject of absolute asymmetric synthesis has captivated scientists ever since the biological importance of l-amino acids and d-sugars was first recognized (9–12). We understand that when we go into the laboratory to prepare such molecules, we will arrive at a racemic mixture unless we employ a template; an asymmetric enzyme, reagent or catalyst, a chiral synthon, or some other physical or chemical force, to help direct the reaction toward the desired hand of the product. However, as Mislow (13) points out in a particularly lucid recent review on absolute asymmetric synthesis, it has long been known, if not generally recognized, that at least some degree of ee may be achieved without chiral intervention in such syntheses. The fact is that enantiomerically enriched products are bound to be formed from achiral precursors, merely as a result of statistical fluctuations (14–18). The historical development of these ideas is detailed by Mislow (13). He quotes reports as long as 100 years ago, where these ideas were discussed (19, 20), including the conclusion by Gilman in 1929 (21) that “the chemist actually does effect unwittingly direct asymmetric syntheses.” It is interesting, then, that the definition of absolute asymmetric synthesis originally proposed by Bredig (22) in 1923 placed an important emphasis on the need for the intervention of asymmetric physical forces to create an imbalance in ee, a point that has been echoed in numerous authoritative texts since. Mislow (13) proposes a new definition of absolute asymmetric synthesis as “the formation of enantiomerically enriched products from achiral precursors without the intervention of chiral chemical reagents or catalysts” (23–25).
Thus, our first question is answered: it becomes clear that the creation of a small imbalance in enantiomers by means of absolute asymmetric synthesis, or what Siegel (26) refers to as the “shattered mirror,” is occurring all of the time in our laboratories. Why then, does observation of spontaneous achiral symmetry breaking in chemical reactions elude us? The reason is that most of the time the shattered mirrors readily and repeatedly put themselves back together again. It is a rare case where the symmetry breaking is observable and persistent, as in the Soai reaction. The question is, what makes it observable in this case? In trying to find an answer, perhaps we have been focusing our attention on the wrong part of the problem: instead of pondering the origin of the initial imbalance, we need to understand how this imbalance can be propagated; that is, how can we prevent the mirror from repairing itself?
Here is where autocatalysis, such as in the example of the Soai reaction, has a role to play. To understand this idea, we take a closer look at Frank's (5) model for amplification in asymmetric autocatalysis. However, before we do this, we need to highlight a key point that has not received adequate attention in a number of the recent discussions of amplification of ee in asymmetric autocatalysis. This point is: autocatalysis alone is not enough.
Autocatalysis Alone
The failure of autocatalysis by itself to rationalize asymmetric amplification in autocatalytic reactions was discussed qualitatively by Girard and Kagan (27), and Blackmond (28) has provided a theoretical derivation. The argument is simple: let's consider an asymmetric catalytic reaction that yields a product of ee0, when carried out by using enantiopure catalyst (eecat = 1). For a simple catalytic reaction; that is, one exhibiting no nonlinear effects, the relationship between the product ee observed, eeprod, and the ee of the catalyst used, eecat, is linear:
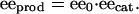 |
[1] |
Eq. 1 makes it easy to see that the product ee will be lower than the catalyst ee, even if the catalyst is only a tiny fraction less than perfect (ee0 < 1). Now, let's consider what happens in the case of an autocatalytic reaction: the newly formed product molecules; at an ee lower than their antecedents, serve as catalysts in further turnovers of substrate to product. Thus, in each cycle, the ee of the newly formed product decreases further. In terms of the total number of turnovers undertaken in an autocatalytic reaction (TON, defined as the moles of substrate converted per mole of catalyst initially present), the final ee product (eeprod,f) will be given by Eq. 2 (16):
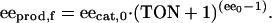 |
[2] |
Fig. 1 demonstrates the results of any simple asymmetric autocatalytic reaction. Even an autocatalyst of very high enantiopurity that is capable of producing itself in 99% ee will experience an erosion of ee; after only 200 turnovers, this catalyst ee will be <94%. An 88% ee catalyst that produces itself in 88% ee will show an erosion of product ee to <50% for the same number of turnovers. Thus, we may predict that in the eons of time because the first chiral molecules were formed, any process of pure autocatalytic self-replication would lead inexorably to a racemic world! Autocatalysis such as that demonstrated by the Soai reaction clearly needs something more if it is going to be part of our rationale for explaining the opposite trend; that is, an ee that increases over time.
Fig. 1.

Simple asymmetric autocatalytic reactions (unlike the Soai reaction) necessarily show an erosion of ee over time. Plotted is the final product ee as a function of turnover number in asymmetric autocatalytic reactions for two hypothetical of autocatalysts at different ee and different product enantioselectivity: a catalyst with an initial eecat,0 = 0.99 exhibiting an enantioselectivity of 0.99 (solid blue line), and a catalyst with an initial eecat,0 = 0.88 exhibiting an enantioselectivity of 0.88 (shaded magenta line).
Autocatalysis Plus Inhibition
The key to the Frank model (5) for spontaneous asymmetric synthesis by means of autocatalysis is the concept of an “anticatalyst” in self-replicating systems. Frank (5) termed this idea “mutual antagonism,” and it means that the autocatalyst must be capable not only of producing itself but also of suppressing, or stopping, production of its enantiomer. Simple catalytic or autocatalytic reactions offer no means of accomplishing this result; however, when we put autocatalysis together what we've learned from mechanistic studies of nonlinear effects in asymmetric catalysis, we can begin to see how product ee can increase over time in an autocatalytic system.
The Frank model (5) imagines a mixture of R and S species of unspecified initial proportions, each of which acts as a catalyst in its own self-production and acts either to suppress or stop production of its enantiomer. One mechanism corresponding to the mathematics of this model is the case where dimers may form from the enantiomers. This mechanism is outlined in Scheme 2. Specific mutual antagonism corresponds to the formation of an inactive heterochiral dimer SR (Scheme 2 A). This model provides us with an inhibition mechanism, because the dimer acts as a reservoir to “siphon off” a disproportionate fraction of the minor enantiomer, thus allowing an ee in the active monomer catalyst pool to build up over repeated autocatalytic cycles. If dimer formation is irreversible, the antagonistic interaction is lethal; a less severe suppression of self-replication rate ensues when a dimer/monomer equilibrium is allowed.
Scheme 2.

Models for including mutual antagonism in autocatalytic systems. (A) Specific mutual antagonism: enantiomeric R and S catalysts form a reservoir of inactive heterochiral dimers. If the initial ratio of S:R enantiomers is not 1:1, a greater fraction of the minor enantiomer is extracted into the dimer reservoir, which has total S:total R ratio equal to 1:1. (B) Unspecific mutual antagonism: enantiomeric R and S catalysts form a reservoir of inactive homochiral and heterochiral dimers in statistical proportions.
The Problem with Unspecific Mutual Antagonism
Mathematically, the concept of specific mutual antagonism works to explain asymmetric amplification in autocatalysis, but chemical considerations cause us to wonder whether it would be more realistic to allow the formation of both homochiral and heterochiral dimers, as shown in Scheme 2B. Homochiral and heterochiral dimers figure in the Noyori model (29) for nonlinear effects in the catalytic alkylation of aldehydes with dialkylzincs, suggesting that this might be a good starting point for mechanistic conjecture in the Soai (1) system. Asymmetric amplification is achieved when the equilibria governing dimer formation are biased toward formation of SR. If there is no preference; that is, if the equilibrium concentrations of RR, SS, and SR are statistically determined, then neither enantiomer is “pulled” preferentially out of the active catalyst pool. This result means the system has no way of altering the initial imbalance between R and S. In Noyori's catalytic system (29), this situation would lead to a linear relationship between catalyst and product ee; in an autocatalytic system, this situation is called unspecific mutual antagonism, and under these conditions, Frank (5) noted, any initial disproportion in enantiomers is preserved, but not amplified.
Studies of the Soai Reaction: A Paradox Emerges
In the first detailed mechanistic studies of the Soai reaction, Blackmond et al. (30) made an interesting observation. They noticed that for reactions carried out under identical conditions, the initial rate for a racemic catalyst was observed to be approximately one-half that of an enantiopure catalyst; more importantly, that ratio was maintained throughout the reaction, as shown in Fig. 2. Why is that significant? Let's consider the implications of this result in the context of the models described above. The Soai reaction proceeds in high ee, so for all practical purposes, we can consider an enantiopure system consisting of almost entirely R species throughout, whereas a racemic system maintains equal amounts of R and S throughout the reaction. As an autocatalytic reaction proceeds, the catalyst doubles in concentration, then doubles again, and again. This reaction is necessarily reflected in the rate, which is proportional to catalyst concentration. If the racemic catalyst exhibits a bias toward the heterochiral dimer; as it must if we are to observe amplification of ee, then part of the R and S enantiomer pool is shunted off, not only into the inactive RR and SS reservoirs as it is for the enantiopure catalyst but also into the inactive SR reservoir that is significant for nonenantiopure catalysts only. Thus, the rate of production in the racemic system should start to lag behind that of the enantiopure. Instead, as Figs. 2 and 3 show, the Blackmond et al. (30) study revealed that the racemic system keeps pace excellently with its enantiopure counterpart! This finding reveals unequivocally that the reaction rate data agree with what we have described here as the Frank model (5) for unspecific mutual antagonism. Now for the paradox: the rate data fit a model that says there should be no asymmetric amplification in ee; but amplification in ee is the very phenomenon that is the hallmark of the Soai reaction system! Indeed, asymmetric amplification was observed experimentally in the Blackmond et al. (30) studies when nonenantiopure catalyst was used.
Fig. 2.

Experimental reaction rate as a function of fraction conversion of the aldehyde 1b in the Soai autocatalytic reaction shown in Scheme 1 employing enantiopure and racemic catalyst. The experimental rate for the racemic catalyst has been multiplied by a factor of ≈2 (1.93).
Fig. 3.

Schematic depicting how the catalyst concentration increases for enantiopure and racemic catalysts as the autocatalytic reaction of Scheme 1 proceeds, as predicted by the experimentally measured reaction rates shown in Fig. 2.
The Solution: Dimers Are the Catalysts
The paradox is resolved if we allow the Soai reaction to follow a mechanism related to Scheme 2B, but with a twist: what whether it is not the monomers, but instead the dimers, that serve as catalysts? This mechanism is shown in Scheme 3. In fact, the dimers dominate: the kinetic studies suggest that the Soai system (1) is driven toward stochastic formation of homochiral and heterochiral dimers (unspecific mutual antagonism), and the R and S monomers themselves are present only in kinetically insignificant concentrations. This twist takes us full circle back to the earliest theoretical work on nonlinear effects in asymmetric catalysis by Kagan and coworkers (31, 32). The ML2 model, modified in this case for autocatalysis (33), allows us to account for amplification of ee by adding one simple constraint: the homochiral and heterochiral species must have different activities as catalysts. All we need to assume to explain the amplification of ee is that the homochiral species is better at catalyzing the reaction than is the heterochiral species. As we discuss in the next section, this hybrid model offers a rationalization of the Soai autocatalytic reaction; accounting for ee and rate observations, that is both simple as well as gratifying in its implications for the chemical origin of life.
Scheme 3.

ml2 model for autocatalysis. The monomeric R and S enantiomers form homochiral (RR and SS) and heterochiral (SR) dimers that themselves serve as the active catalysts in the autocatalytic reaction.
Statistics and a Stroke of Luck
With this hybrid dimer model, we have an elegant and simple solution to the mystery of the evolution of homochirality: First, we can now accept the experimentally observed statistical formation of the dimers (Frank's unspecific antagonistic interactions; ref. 5). This solution makes sense when we consider that what we are trying to describe is a world before a chiral pool existed. Design of specific stereochemical bias by modern methods generally requires access to an asymmetric template, and today we can draw on the chiral pool for our building blocks. Our proficiency in tuning desired properties is reflected in the large number of efficient chiral ligands that have been discovered for a wide variety of chemical transformations. In a prebiotic soup of simple molecules, however, the resources for pulling off such a feat would have been meager.
Second, we don't have to give up on asymmetric amplification, just because the dimers are capable only of statistical formation. Indeed, accounting for amplification of ee by assuming different activities for the homochiral and heterochiral dimers makes sense because they are diastereomeric species. It may be fortunate that the relative reactivities turned out that SR is less active, and not the reverse, in the Soai reaction; indeed, if things had gone the other way, we would ultimately have found ourselves with a racemic product. Perhaps other attempts at such stochastically determined dimer catalysts have gone unmarked because of reversed heterochiral/homochiral relative reactivities. However, in this sense, luck; and perhaps perseverance, may have played the same role in the prebiotic chemistry that ultimately established homochirality. Statistics, and one stroke of luck; a difference in reactivity that ended up working for us instead of against us, are all that is required for us to find ourselves in our homochiral world today.
Outlook
The first mechanistic studies on the Soai reaction thus implicated a trimeric species as the transition state for the reaction; that is, two molecules of alcohol/alkoxide in the dimeric catalysts and one molecule of prochiral aldehyde substrate, as shown in Scheme 4A. However, more recent work by Buono and Blackmond (34) shows that this conclusion is a coincidence of the fact that the reactions were carried out at equimolar ratios of aldehyde to dialkylzinc. When the conditions were expanded to include other substrate/reagent ratios, the observed kinetics implicated a tetrameric template, as shown in Scheme 4B. The structure of these proposed species are not known, and ongoing work is aimed at elucidating these answers, but the stoichiometries involved in the catalytic event may be deduced from the kinetic data. Higher-order species might also be imagined, in which the catalyst is in a state of further agglomeration. Recent work (35) suggests that physical processes such as selective precipitation might also play a role in the asymmetric amplification that led to the evolution of homochirality. In addition, the role of transport phenomena in the “open system” (5, 36) represented by the probiotic world is a consideration that we often neglect when we carry out reactions in simple laboratory vials. The flux of reactants and products in and out of reactor “pools” may have contributed to the evolution of homochirality. The possibility of precipitation-dissolution processes contributing to asymmetric amplification in such open systems has been discussed by Welch (36).
Scheme 4.

Proposed models for the Soai reaction based on trimeric (A) and tetrameric (B) transition states, where the order refers to the number of prochiral aldehyde plus ex-aldehyde (alcohol/alkoxide or nascent alkoxide species) that are brought together (although not simultaneously) in the reaction event.
The collection of chiral subunits into higher-order species is an important consideration in understanding the development of homochirality in biological function. The “minimal systems” for self-replication developed by von Kiedrowski (37, 38) and others for nucleic acids (39), peptides (40, 41), small organic molecules (42), and ribozymes (43) all represent autocatalytic systems based on core templates larger than those in the Soai reactions. However, none of those systems involve prochiral molecules, meaning that the origin of the first imbalance in asymmetric centers is not at issue in these cases; when we examine von Kiedrowski's (37, 38) autocatalytic system based on DNA hexamers, for example, we know we are already firmly committed and residing in the camp of the l-amino acids and the d-sugars. We might agree with Cintas and coworkers' (44) view that the Soai system represents a “triumph of reductionism” in helping us to understand the chemical origin of life in molecular terms, but we also accept his statement that there remains a “large gap between molecular chirality and molecular evolution.” Thus, further studies ought to be aimed at trying to provide information that might help close this gap.
Another point comes to mind when the Soai reaction is compared to complex systems exhibiting self-replication, such as those involving nucleic acids and peptides. We can see from the discussion above that the Soai reaction has provided considerable food for thought concerning possible mechanisms for the evolution of biological homochirality from a small imbalance of enantiomeric precursors. However, as mentioned before, the particular chemistry of this reaction; that involving dialkylzinc compounds, is unlikely in itself to have been of importance in an aqueous prebiotic soup. Therefore, the search continues for other organic transformations that could provide a closer model for how asymmetric amplification in the prebiotic world could have occurred. Here, we might turn to enzymes for inspiration to help us take lessons from the ways in which simple organic molecules are constructed and combine these with concepts we have developed about how asymmetric amplification might be effected in such systems.
Summary
The evolution of biological homochirality can be explained by a model in which an initial tiny imbalance of enantiomers is amplified. By using the remarkable Soai reaction as a model, this essay has described the key concepts necessary for this model to be viable. Although absolute asymmetric syntheses occur around us all of the time simply by virtue of statistics, the maintenance of such an imbalance, and its amplification by chemical means, requires a special kind of autocatalytic reaction. Amplification of ee can only result if a means exists to suppress the catalytic action of the “wrong” hand of the catalyst. Experimental studies of the Soai reaction reveal that statistical formation of dimer catalyst species coupled with lower activity of the heterochiral dimer is sufficient to rationalize the evolution of high ee from a tiny initial imbalance. This general mechanism could be effective in a world of simple organic molecules such as those likely to have been present in the prebiotic world.
Acknowledgments
I thank Prof. Kurt Mislow and Prof. George M. Whitesides for discussions. This work was supported by the Engineering and Physical Sciences Research Council and by Merck Research Laboratories.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: ee, enantiomeric excess.
References
- 1.Soai, K., Shibata, T., Morioka, H. & Choji, K. (1995) Nature 378, 767–768. [Google Scholar]
- 2.Shibata, T., Choji, K., Hayase, T., Aizu, Y. & Soai, K. (1996) Chem. Commun. 1235–1236.
- 3.Shibata, T., Morioka, H., Hayase, T., Choji, K. & Soai, K. (1996) J. Am. Chem. Soc. 118, 471–472. [Google Scholar]
- 4.Soai, K., Niwa, S. & Hori, H. (1990) J. Chem. Soc. Chem. Commun. 1990, 982–983. [Google Scholar]
- 5.Frank, F. C. (1953) Biochim. Biophys. Acta 11, 459–463. [DOI] [PubMed] [Google Scholar]
- 6.Wynberg, H., (1989) J. Macromol. Sci., Chem. 26, 1033–1041. [Google Scholar]
- 7.Kondepudi, D. K. (1990) Science 250, 975–976. [DOI] [PubMed] [Google Scholar]
- 8.Kondepudi, D. K. (2001) Acc. Chem. Res. 34, 946–954. [DOI] [PubMed] [Google Scholar]
- 9.Mason, S. (1988) Chem. Soc. Rev. 17, 347–359. [Google Scholar]
- 10.Avalos, M., Babiano, R., Cintas, P., Jimenez, J. L., Palacios, J. C. & Barron, L. D. (1998) Chem. Rev. (Washington, D.C.) 98, 2391–2402. [DOI] [PubMed] [Google Scholar]
- 11.Feringa, B. L. & van Delden, R. A. (1999) Angew. Chem. Int. Ed. Engl. 38, 3418–3438. [DOI] [PubMed] [Google Scholar]
- 12.Buschmann, H., Thede, R. & Heller, D. (2000) Angew. Chem. Int. Ed. Engl. 39, 4033–4036. [DOI] [PubMed] [Google Scholar]
- 13.Mislow., K. (2003) Collect. Czech. Chem. Commun. 68, 849–864. [Google Scholar]
- 14.Bonner, W. A. (1972) in Exobiology, ed. Ponnamperuma, C. (North–Holland, Amsterdam), p. 170.
- 15.Bonner, W. A. (1988) Top. Stereochem. 18, 1–96. [Google Scholar]
- 16.Frank, P., Bonner, W. A. & Zare, R. N. (2001) in Chemistry for the 21st Century, eds. Keinan, E. & Schechter, I. (Wiley, New York), p. 175.
- 17.MacDermott, A. J. (2002) in Chirality in Natural and Applied Science, eds. Lough, W. J. & Wainer, I. W. (Blackwell Scientific, Oxford), p. 23.
- 18.Quack, M. (2002) Angew. Chem. Int. Ed. Engl. 41, 4619–4630. [Google Scholar]
- 19.Pearson, K. (1898) Nature 58, 495. [Google Scholar]
- 20.Pearson, K. (1898) Nature 59, 30. [Google Scholar]
- 21.Gilman, H. (1929) Iowa State Coll. J. Sci. 3, 227. [PubMed] [Google Scholar]
- 22.Bredig, G. (1923) Ztschr. f. Angew. Chem. 36, 456. [Google Scholar]
- 23.Soai, K., Sato, I., Shibata, T., Komiya, S., Hayashi, M., Matsueda, Y., Imamura, H., Hayase, T., Morioka, H., Tabira, H., et al. (2003) Tetrahedron 14, 185–188. [Google Scholar]
- 24.Singleton, D. A. & Vo, L. K., (2003) Org. Lett. 5, 4337–4339. [DOI] [PubMed] [Google Scholar]
- 25.Gridnev, I. D., Serafimov, J. M., Quiney, H. & Brown, J. M. (2003) Org. Biomol. Chem. 1, 3811–3819. [DOI] [PubMed] [Google Scholar]
- 26.Siegel, J. S. (2002) Nature 419, 346. [DOI] [PubMed] [Google Scholar]
- 27.Girard, C. & Kagan, H. B. (1998) Angew. Chem. Int. Ed. Engl. 37, 2922–2959. [DOI] [PubMed] [Google Scholar]
- 28.Blackmond, D. G. (2002) Adv. Synth. Catal. 344, 156–158. [Google Scholar]
- 29.Kitamura, M., Suga, S., Oka, H. & Noyori, R. (1998) J. Am. Chem. Soc. 120, 9800–9809. [Google Scholar]
- 30.Blackmond, D. G., McMillan, C. R., Ramdeehul, S., Schorm, A. & Brown, J. M. (2001) J. Am. Chem. Soc. 123, 10103–10104. [DOI] [PubMed] [Google Scholar]
- 31.Puchot, C., Samuel, O., Duñach, E., Zhao, S., Agami, C. & Kagan, H. B. (1986) J. Am. Chem. Soc. 108, 2353–2357. [DOI] [PubMed] [Google Scholar]
- 32.Guillaneux, D., Zhao, S. H., Samuel, O., Rainford, D. & Kagan, H. B. (1994) J. Am. Chem. Soc. 116, 9430–9439. [Google Scholar]
- 33.Bailey, P. D. (1995) J. Chem. Soc. Chem. Commun. 1797–1799.
- 34.Buono. F. G. & Blackmond, D. G. (2003) J. Am. Chem. Soc. 125, 8978–8979. [DOI] [PubMed] [Google Scholar]
- 35.Buono, F. G., Iwamura, H. & Blackmond, D. G. Angew. Chem. Int. Ed. Engl., in press. [DOI] [PubMed]
- 36.Welch, C. J. (2001) Chirality 214, 425–427. [DOI] [PubMed] [Google Scholar]
- 37.von Kiedrowski, G. (1986) Angew. Chem. Int. Ed. Engl. 25, 932–933. [Google Scholar]
- 38.von Kiedrowski, G. (1993) in Bioorganic Frontiers, ed. Dugas, H. (Springer, New York), Vol. 3, p. 113–146. [Google Scholar]
- 39.Zielinski, W. S. & Orgel, L. E. (1987) Nature 327, 346–347. [DOI] [PubMed] [Google Scholar]
- 40.Yao, S., Ghosh, I., Zutshi, R. & Chmielewski, J. (1997) J. Am. Chem. Soc. 119, 10559–10560. [Google Scholar]
- 41.Saghatelian, A., Yokobayashi, Y., Soltani, K. & Ghadiri, M. R. (2001) Nature 409, 797. [DOI] [PubMed] [Google Scholar]
- 42.Wintner, E. A., Conn, M. M. & Rebek, J., Jr. (1994) J. Am. Chem. Soc. 116, 8877–8884. [Google Scholar]
- 43.Paul, N. & Joyce, G. P. (2002) Proc. Natl. Acad. Sci. USA 99, 12733–12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Avalos, M., Babiano, R., Cintas, P., Jimenez, J. L. & Palacios, J. C. (2000) Chem. Commun. 887–892.