

Abstract
Mutations in the human mitochondrial genome are known to cause an array of diverse disorders, most of which are maternally inherited, and all of which are associated with defects in oxidative energy metabolism. It is now emerging that somatic mutations in mitochondrial DNA (mtDNA) are also linked to other complex traits, including neurodegenerative diseases, ageing and cancer. Here we discuss insights into the roles of mtDNA mutations in a wide variety of diseases, highlighting the interesting genetic characteristics of the mitochondrial genome and challenges in studying its contribution to pathogenesis.
Mitochondria — from the Greek mitos (thread-like) and khondros (grain or granule) — are bacterium-sized organelles found in all nucleated cells. They have a myriad of functions, but it is fair to say that what sets them apart is their role in energy production. To reiterate an oft-used cliché, mitochondria are the ‘powerhouses of the cell’ because they generate more than 90% of a typical cell’s ATP, which is the ‘energy currency’ of the cell. Mitochondria contain their own DNA (mitochondrial DNA (mtDNA)), which is linked to their origin as eukaryotic endosymbionts1. Unlike nuclear DNA (nDNA), mtDNAs are maternally inherited and are present in multiple copies per cell; the number varies according to the bioenergetic needs of each particular tissue and can range over three orders of magnitude, depending on the cell type2. As such, mitochondrial diseases caused by mutations in mtDNA follow the laws of population genetics. This is a crucial distinction from diseases that follow Mendelian genetics and underlies many of the unique, and often perplexing, features of these disorders.
Over the past 25 years, the identification of pathogenic mtDNA mutations has evolved in parallel with advances in sequencing and cell biology technologies. The first decade was mainly devoted to identifying new pathogenic mutations associated with various syndromes (BOX 1) using standard sequencing techniques. This effort, which is now based on automated microarray-based sequencing and high-throughput sequencing strategies, was aided enormously by the use of cybrid technology (BOX 2). Beginning in 1995, mutations in nuclear genes affecting oxidative phosphorylation (OXPHOS) function were identified at an escalating pace initially on the basis of linkage analysis and homozygosity mapping, then on monochromosomal transfer techniques, gene complementation studies and sequencing of candidate genes, and more recently on scanning entire patient genomes through whole-exome sequencing3. These studies have revealed that mitochondrial disorders can result from mutations not only in genes encoding subunits of the OXPHOS complexes but also in genes required for their translation and assembly, for providing the proper lipid milieu and for determining the spatial and temporal relationships of the organelles to their subcellular environment (‘mitodynamics’). However, as mtDNA mutations are now being linked to several complex traits, we need to improve our understanding of mitochondrial genetics at the same time as learning more about the nuclear genome.
Box 1. Primary mitochondrial DNA-related diseases.
Leber’s hereditary optic neuropathy (LHON)
This is the most common mitochondrial DNA (mtDNA)-related disorder, causing subacute loss of central vision in young adults, predominantly men. LHON is usually due to homoplasmic mutations in one of three genes encoding complex I subunits (m.11778G→A in NADH dehydrogenase 4 (ND4), m.3460G→A in ND1 and m.14484T→C in ND6). Although mutations are predominantly homoplasmic, the retinal ganglion cells are affected selectively. The marked predominance of LHON in men, which is not accounted for by differences in mitochondrial genetics between the sexes, has been attributed to the relative abundance of oestrogen receptors120 and may be the first example of the role of epigenetics in the expression of mtDNA-related diseases.
Leigh’s syndrome
This disease is more commonly due to nuclear DNA (nDNA) mutations than to mtDNA mutations. The mutations are most frequently in subunits of complex I or in assembly factors of complex IV. Despite the striking variety of specific aetiologies, Leigh’s syndrome has common clinical features (developmental delay or regression, respiratory abnormalities, recurrent vomiting, nystagmus, ataxia, dystonia and early death) and characteristic radiological changes (bilateral symmetrical lesions affecting mostly basal ganglia and the brainstem), a reflection of the devastating effects of energy shortage on the developing nervous system.
Mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS)
This is a multisystem disorder in which brain, muscle and the endocrine system are predominantly involved and is often fatal in childhood or in young adulthood121. Unique to MELAS are the transient eponymous stroke-like episodes, which are most often due to infarcts in the temporal and occipital lobes and are associated with hemiplegia and cortical blindness. Although we still do not understand the cause, their occurrence highlights the fact that MELAS is also an angiopathy, making it almost unique among the mitochondrial diseases122. Although the most common causal mutation is m.3243A→G in tRNALeu(UUR), more than a dozen other mutations have been associated with MELAS, affecting both tRNA and protein-coding genes.
Myoclonus epilepsy and ragged red fibres (MERRF)
For unknown reasons, MERRF is due almost exclusively to mutations in tRNALys. It is associated with a rather different set of signs and symptoms from the other diseases, often including cervical lipomas as well as myoclonus and epilepsy123. As in MELAS, the pathogenic cause seems to be a post-transcriptional failure to modify the tRNAs with taurine, thereby affecting translational efficiency124. Moreover, in MERRF and MELAS, there is mitochondrial proliferation not only in muscle (that is, the ragged red fibres (RRFs)) but also in blood vessels (strongly SDH-positive vessels (SSVs))125. Although blood vessel involvement can be found in patients with both disorders, angiopathy is less prevalent in MERRF. Interestingly, both the RRFs and SSVs in MERRF are negative for cytochrome c oxidase (COX) histochemical activity, whereas those in MELAS are typically COX-positive125,126 (BOX 3). Herein lies a paradox: why is it that MERRF, which has COX-negative SSVs, is not primarily an angiopathy, whereas MELAS, which has ostensibly ‘more normal’ COX-positive SSVs, is an angiopathy? One possible explanation is that it is the COX positivity that triggers the stroke-like episodes in MELAS, because the absolute amount of COX in these mitochondria-laden SSVs is far greater than normal126. As COX is known to bind nitric oxide (NO)127,128, which is a key signalling molecule for vasoconstriction (via the conversion of L-arginine to NO by nitric oxide synthase and the subsequent binding of NO to soluble guanylate cyclase129), it could be that in MELAS (but not in MERRF), NO is titrated out by supernormal levels of COX after a NO-mediated signal to dilate blood vessels. This could thereby prevent vasodilation and precipitate the strokes126. If this possibility is true, agents that increase compensatory NO levels to allow more normal vasodilation might be of therapeutic value. Indeed, treatment of patients with MELAS with L-arginine has been reported to mitigate the frequency and severity of strokes in this disorder130.
Neuropathy, ataxia and retinitis pigmentosa, and maternally inherited Leigh’s syndrome (NARP and MILS, respectively)
These are two possible clinical outcomes of mutations at the same nucleotide (m.8993T→G or, less frequently, m.8993T→C). In the family in which these were originally identified, three adults had one or more of the acronymic features, whereas a maternally related child had severe developmental delay, pyramidal signs, retinitis pigmentosa, ataxia and cerebellar and brainstem atrophy, features that are characteristic of Leigh’s syndrome. We now know that onset and severity depend on the mutation load: patients with NARP harbour ~70% mutant mtDNAs, whereas those with MILS harbour >90%.
Reversible respiratory chain deficiency
Although it is rare, mutation in another tRNA — a homoplasmic m.14674T→C mutation in the discriminator base of tRNAGlu — deserves to be mentioned. This mutation causes a highly unusual disorder, namely a ‘reversible’ form of OXPHOS deficiency in which infants are severely ill but, if sustained vigorously during the perinatal period, recover full mitochondrial function spontaneously within 2 years131. Interestingly, the disease can also be caused by mutations in TRMU132, a nucleus-encoded mitochondrial protein that catalyses the 2-thiolation of uridine at the wobble position of three mt-tRNAs133, including tRNAGlu. These mutations may also modify the expression of a deafness-associated mutation at nucleotide 1,555 in 12S ribosomal RNA134.
Sporadic deletions of mtDNA
Kearns–Sayre syndrome (KSS)25 is defined by the onset before age 20 of ophthalmoplegia (that is, paralysis of the muscles that move the eyeballs), ptosis (that is, droopy eyelids), pigmentary retinopathy and at least one of the following: complete heart block, cerebrospinal fluid protein levels above 100 mg dL−1 and cerebellar ataxia. In this multisystemic disorder, partially deleted mtDNAs (Δ-mtDNAs) are present in all examined tissues (implying that the deletion event occurred in the germ line or soon after fertilization). In chronic progressive external ophthalmoplegia (CPEO)26 — a myopathy with the defining features of ophthalmoplegia and ptosis — Δ-mtDNAs are found only in muscle (implying that the deletion event occurred after fertilization in the muscle lineage of mesoderm). In Pearson’s syndrome27, which is characterized by sideroblastic anaemia and exocrine pancreas dysfunction, Δ-mtDNAs are initially abundant in haematopoietic cells. Fascinatingly, patients with Pearson’s syndrome who survive their anaemia often develop KSS as teenagers: as the Δ-mtDNA load declines in blood (a rapidly dividing tissue), the Δ-mtDNA load increases in more terminally differentiated tissues (for example, in the muscles or brain). This phenomenon demonstrates two other features of mitochondrial genetics: namely, the different thresholds for dysfunction in various tissues and the autonomy of mitochondrial division (and mtDNA replication) even within terminally differentiated cells.
Box 2. Cybrids.
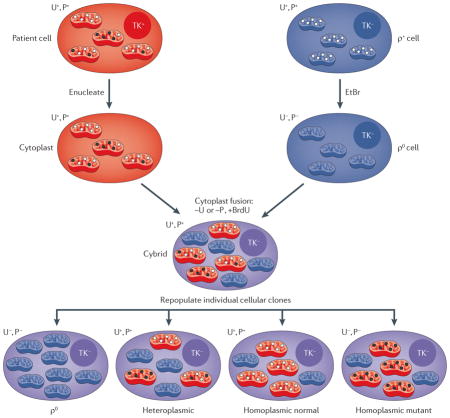
Because there is currently no technology available for introducing exogenous DNA into mammalian mitochondria stably and heritably, the pathogenicity of many primary mtDNA mutations discussed in this Review has been established using cytoplasmic hybrid, or cybrid, technology. In this approach, patient mitochondria and mitochondrial DNAs (mtDNAs) are transferred to a host ρ0 cell containing mitochondria that are devoid of endogenous mtDNAs135 (see the figure). Thus, cybrids can be assayed for donor mitochondrial function in a ‘standard’ nuclear background93.
In the most common approach for making ρ0 cells, a transformed cell line that is deficient in thymidine kinase activity (TK−) is exposed to ethidium bromide (EtBr), which is an inhibitor of mtDNA replication (mitochondria have a high membrane potential and take up EtBr, as it is ionizable). The ρ0 line is auxotrophic for uridine and pyruvate (U−, P−). The ρ0 cells are repopulated by forming hybrids with cytoplasts (cells that lack nuclei but still contain mitochondria) from a patient cell line. After fusion, cells are plated in media containing bromodeoxyuridine (+BrdU) and lacking either pyruvate or uridine (−U or −P), which permits only the growth of ρ0 cells that have fused with cytoplasts containing functional mitochondria (TK+ donor cells are not able to grow in the presence of BrdU). If heteroplasmic cells are used as donors, it is possible to isolate cybrid clones that harbour varying proportions of mutated mtDNAs, ranging from 0% mutant (that is, 100% wild type) to 100% mutant (that is, 0% wild type). Both the parental ρ0 line and homoplasmic cybrid lines harbouring nonsense- or frameshift-mutated mtDNAs must be grown in media supplemented with uridine (+U; ~200 μM) and pyruvate (+P; ~1 mM)136.
In addition to being of interest to scientists, mtDNA-related diseases may pose a public health concern. Epidemiological studies of mtDNA mutations in adults have shown a minimum prevalence of approximately 1 in 5,000, making mitochondrial diseases among the most common genetic disorders and a major burden for society4.
There have been several recent excellent reviews describing the role of mtDNA mutations in disease5–7, and so we discuss primary pathogenic mtDNA mutations only in broad outline to demonstrate some of the unique aspects of mitochondrial genetics and function. We focus on more speculative roles of mtDNA mutations — both germline and somatic — in complex phenotypes, such as ageing, neurodegenerative diseases and cancer.
Mitochondrial function and dysfunction
The human mitochondrial genome is a tiny 16.6 kb circle containing only 37 genes: 13 of these genes encode proteins and the remaining 24 — consisting of 2 ribosomal RNAs (rRNAs) and 22 tRNAs — are used for translation of those 13 polypeptides8 (FIG. 1a). Remarkably, all 13 polypeptides are components of a single metabolic entity: the OXPHOS system (FIG. 1b). Pathogenic mutations resulting in respiratory chain deficiency can arise from mutations in either genome9. In addition to these structural subunits, there are more than 50 nDNA-encoded ancillary gene products required for the proper assembly and functioning of the OXPHOS system (FIG. 1b). Although mitochondrial OXPHOS disorders are quite varied, they typically display a number of ‘canonical’ biochemical and morphological features (BOX 3).
Figure 1. Mitochondrial DNA and the human respiratory chain/oxidative phosphorylation system.

a | The human mitochondrial genome. The 37 mitochondrial DNA (mtDNA)-encoded genes include seven subunits of complex I (ND1, 2, 3, 4, 4L, 5 and 6), one subunit of complex III (cytochrome b (Cyt b)), three subunits of complex IV (Cyt c oxidase (COX) I, II and III), two subunits of complex V (A6 and A8), two rRNAs (12S and 16S) and 22 tRNAs (one-letter code). Also shown are the origins of replication of the heavy strand (OH) and the light strand (OL), and the promoters of transcription of the heavy strand (HSP) and light strand (LSP). b | Oxidative phosphorylation (OXPHOS) complexes. The system is comprised of complex I (NADH dehydrogenase-CoQ reductase), complex II (succinate dehydrogenase), complex III (ubiquinone-Cyt c oxidoreductase), complex IV (COX) and complex V (ATP synthase; F0 and F1 denote the proton-transporting and catalytic subcomplexes, respectively), plus two electron carriers, coenzyme Q (CoQ; also called ubiquinone) and Cyt c. Complexes I–IV pump NADH- and FADH2-derived protons (produced by the tricarboxylic acid (TCA) cycle and the β-oxidation ‘spiral’) from the matrix across the mitochondrial inner membrane to the intermembrane space to generate a proton gradient while at the same time transferring electrons to molecular oxygen to produce water. The proton gradient, which makes up most of the mitochondrial transmembrane potential (Δψm), is used to do work by being dissipated across the inner membrane in the opposite direction through the fifth complex (ATP synthase), thereby generating ATP from ADP and free phosphate. Complexes I, III, IV and V contain both mtDNA- and nuclear DNA (nDNA)-encoded subunits, whereas complex II, which is also part of the TCA cycle, has only nDNA-encoded subunits. Note that CoQ also receives electrons from dihydroorotate dehydrogenase (DHOD), an enzyme of pyrimidine synthesis. Polypeptides encoded by nDNA are in blue (except for DHOD, which is in pink); those encoded by mtDNA are in colours corresponding to the colours of the polypeptide-coding genes (shown in a). The ‘assembly’ proteins are all nDNA-encoded. IMS, intermembrane space; MIM, mitochondrial inner membrane. Part a is modified from REF. 149 © (2006) Wiley.
Box 3. Biochemical and morphological features of mitochondrial disease.
Although mitochondrial oxidative phosphorylation (OXPHOS) disorders are quite varied, they typically display a number of ‘canonical’ biochemical and morphological features. An obvious feature is respiratory chain deficiency, which typically manifests itself as reduced enzymatic function in one or more respiratory chain complexes, with a concomitant reduction in cellular oxygen consumption and ATP synthesis. In addition, patients often have increased resting lactic acid levels in the blood and cerebrospinal fluid9. Under normal conditions, pyruvate — the end product of anaerobic glycolysis — is transported into mitochondria for further oxidation in the tricarboxylic acid (TCA) cycle to produce the reducing equivalents (namely, NADH and FADH2) that are required for proton pumping by the respiratory chain. However, if the respiratory chain is compromised, NADH and pyruvate accumulate in the cytosol. Excess cytosolic pyruvate is converted to lactate by lactate dehydrogenase, thereby accounting for the increased lactic acid found in many mitochondrial diseases and especially in those affecting infants or young children9.
A prominent morphological feature of OXPHOS disease is the ragged red fibre (RRF), which reflects a massive proliferation of OXPHOS-defective mitochondria29 in muscle; this can be visualized with the modified Gomori trichrome stain137. The accumulation can also be observed using nitro-blue tetrazolium to detect the activity of succinate dehydrogenase (SDH; that is, complex II)138, which contains only nuclear DNA (nDNA)-encoded subunits. In this case, the RRFs appear dark blue (see the figure). The figure shows RRFs and strongly SDH-positive vessels (SSVs) in serial sections of skeletal muscle from patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) and myoclonus epilepsy and ragged red fibres (MERRF) visualized by histochemistry to detect the activities of SDH (left) and cytochrome c oxidase (COX; right). Note that RRFs and SSVs are COX-negative in MERRF, whereas they are COX-positive in MELAS. RRFs are a sufficient but not necessary feature for determining mitochondrial disease (as is increased lactate in the blood or cerebrospinal fluid).
The vast majority of OXPHOS mutations impair the cell’s ability to produce ATP in amounts that are sufficient for maintaining viability, but sometimes OXPHOS problems can leave ATP synthesis intact and yet can be deleterious by, for example, stimulating the overproduction of reactive oxygen species (ROS)139 or inducing an autoimmune response140.
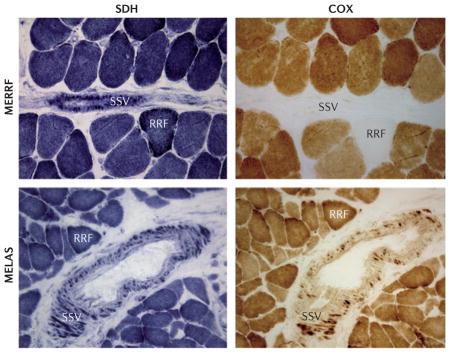
Photographs are provided courtesy of Eduardo Bonilla and Kurenai Tanji, Columbia University, New York, USA.
Pathogenic mtDNA mutations: distribution and threshold effects
Primary mtDNA-related diseases may be defined as those disorders that arise directly from a mutation in mtDNA — either a point mutation or a DNA rearrangement (that is, a deletion, duplication or inversion) — that directly compromises OXPHOS function. Since 1988, more than 270 point mutations have been described, affecting every mtDNA gene (examples are provided in TABLE 1). Remarkably, more than half of these mutations are located in tRNA genes, even though tRNAs comprise only about 10% of the total coding capacity of the genome. Conversely, the polypeptide-coding genes, comprising almost 70% of the genome, account for only about 40% of the mutations, and the two rRNA genes (15% of coding capacity) account for only about 2%.
Table 1.
mtDNA mutations in human primary respiratory chain disorders
| Gene product | Number of mutations | Main disorder |
|---|---|---|
| rRNAs | ||
| 12S rRNA | 5 | Deafness |
| 16S rRNA | 1 | Atypical MELAS |
| tRNAs | ||
| tRNAAla | 3 | Myopathy |
| tRNAArg | 2 | Various |
| tRNAAsn | 5 | Myopathy |
| tRNAAsp | 2 | Various |
| tRNACys | 3 | Various |
| tRNAGln | 3 | Various |
| tRNAGlu | 7 | Reversible respiratory chain deficiency |
| tRNAGly | 3 | Various |
| tRNAHis | 4 | Various |
| tRNAIle | 14 | PEO |
| tRNALeu(CUN) | 8 | Myopathy |
| tRNALeu(UUR) | 23 | MELAS |
| tRNALys | 14 | MERRF |
| tRNAMet | 2 | Various |
| tRNAPhe | 14 | Myopathy |
| tRNAPro | 5 | Multisystem |
| tRNASer(AGY) | 4 | Myopathy |
| tRNASer(UCN) | 12 | Myopathy; deafness |
| tRNAThr | 2 | Various |
| tRNATrp | 12 | Encephalomyopathy |
| tRNATyr | 4 | Myopathy |
| tRNAVal | 6 | Multisystem |
| Polypeptides | ||
| ATP synthase 6 | 13 | NARP or MILS |
| ATP synthase 8 | 2 | Various |
| COX I | 10 | Various |
| COX II | 8 | Various |
| COX III | 6 | Myopathy |
| Cytochrome b | 21 | Sporadic myopathy |
| ND1 | 16 | MELAS; LHON |
| ND2 | 3 | Various |
| ND3 | 5 | Leigh’s syndrome |
| ND4 | 5 | LHON |
| ND4L | 1 | LHON |
| ND5 | 12 | MELAS |
| ND6 | 11 | LHON |
This is a ‘minimal’ list that was compiled by the authors. A more comprehensive list (>300 mutations) is available at MITOMAP, which contains confirmed pathogenic mutations (including those selected for this table), those that are possibly associated with pathology and those that appear to confer a higher risk of developing disease or are modifiers of disease presentation. COX, cytochrome c oxidase; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; MERRF, myoclonus epilepsy and ragged red fibres; MILS, maternally inherited Leigh’s syndrome; mtDNA, mitochondrial DNA; NARP, neuropathy, ataxia and retinitis pigmentosa; ND, NADH-ubiquinone oxidoreductase; PEO, progressive external ophthalmoplegia; rRNA, ribosomal RNA.
This unexpected distribution is likely to be due to the fact that most patients with mtDNA mutations are heteroplasmic, which means that they harbour a mixture of normal and mutated mtDNAs. Moreover, with one exception10, all of these heteroplasmic mtDNA mutations are ‘recessive’: an extremely high amount of mutation is required before a clinical phenotype evinces itself. For unknown reasons, this ‘threshold effect’ varies among mutation types, and in skeletal muscle the mutation load for any particular tRNA (~90%)11 is typically higher than that for large-scale partial deletions of mtDNA (~70–80%)12. The threshold for mutation load in polypeptide-coding genes can be similarly broad, with low levels of mutation causing one type of clinical presentation and higher levels causing another. An excellent example of this behaviour is the m.8993T→G mutation in the ATP synthase 6 (ATP6) gene: at mutation loads above 90%, patients present with maternally inherited Leigh’s syndrome (MILS), which is a fatal encephalopathy13 (BOX 1), whereas at mutation loads in the range of 70–90% patients present with neuropathy, ataxia and retinitis pigmentosa (NARP)14, which is a late-onset, debilitating but non-fatal disorder. By contrast, a patient with 70% mutation in a tRNA will rarely display overt disease. Thus, tRNA mutations are ‘tolerated’ over a broader range of mutant load than are mutations in polypeptide-coding genes. Of course, after the mutation load has risen above 90–95%, no matter what the type of mutation is, OXPHOS deficiency occurs, usually with devastating results.
These high thresholds for pathogenicity also mask the actual frequency of potentially pathogenic mtDNA mutations in the population. Although the prevalence of mtDNA-related disease is about 1 in 5,000, the population frequency of the ten most common pathogenic mtDNA mutations is much higher — approximately 1 in 200 — suggesting that many normal individuals carry subthreshold levels of potentially harmful mtDNA mutations15.
The skewed distribution of mutations towards tRNA errors initially implies that maternally inherited mutations in structural subunits of the respiratory chain are selected against, presumably during embryogenesis, whereas mutations in tRNAs are somehow less ‘severe’ during embryogenesis. Indeed, maternally inherited mutations in tRNAs might be under less-stringent purifying selection through the germ line than are mutations in polypeptide-coding genes16. However, it is easy to imagine that the reverse might also hold true: namely, that mutations in rRNA and tRNA genes that disrupt global protein synthesis would have a greater deleterious effect than mutations in single respiratory chain subunits. The paucity of mutations in rRNA genes supports this alternative view: there is only one known mutation in 16S rRNA associated with a primary mitochondrial disease, and none of the five known mutations in the 12S rRNA gene is lethal, but they do cause non-syndromic deafness17 for reasons related to an effect on 12S rRNA methylation and subsequent nuclear E2F1-mediated transcription18 rather than to the role that 12S rRNA has in protein synthesis. Thus, in addition to differential degrees of purifying selection, the predilection of mutations for tRNA genes may be due to other, currently unknown reasons.
Examples of point mutations in mtDNA
Among the point mutations (TABLE 1), the most common are: an A→G transition at position 3,243 in the tRNALeu(UUR) gene, which causes mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS); an A→G transition at position 8,344 in tRNALys, which causes myoclonus epilepsy and ragged red fibres (MERRF) syndrome; and the aforementioned T→G transversion at position 8,993 in ATP6, which causes NARP and MILS (BOX 1). These three disorders highlight the diversity of biochemical, morphological and clinical presentations that are a hallmark of mitochondrial disorders and that not only challenge the diagnostic capabilities of clinicians but also pose fundamental questions regarding basic mitochondrial biology.
NARP and MILS highlight additional aspects of mitochondrial disease and behaviour. These disorders are due to mutations in the membrane-spanning (so-called F0) portion of ATP synthase (FIG. 1b) that compromise ATP synthesis. Whereas all of the mutations described above affect respiratory chain integrity, thereby reducing the mitochondrial transmembrane potential (Δψm)19,20, ATP synthase mutations typically leave the respiratory chain intact but decouple proton flow from ATP generation21. As such, Δψm in NARP or MILS is either unchanged or is actually higher than normal21. This crucial difference in transmembrane potential will render cells from patients with NARP or MILS refractory to treatment strategies that are predicated on selectively eliminating mitochondria with a low Δψm (REFS 19,20).
Examples of large-scale deletions of mtDNA
The most easily observable mtDNA mutations are the large-scale partial deletions: mtDNAs with these deletions are designated Δ-mtDNAs. Discovered in 1988 (REFS 22,23), these mutations remove ~2–10 kb of mtDNA, and the deletion breakpoints are often flanked by short (5–13 bp) direct repeats24. The deletions are typically sporadic (that is, only the proband has the mutation; mothers and siblings do not), and although they vary among patients, each patient harbours only a single species of deletion. These facts imply that the deletion event occurred either in the germ line of the mother or in early embryogenesis of the proband; the deletion event arose randomly in a single mtDNA molecule, and this single molecule of Δ-mtDNA was massively amplified during pre- and postnatal development through clonal expansion. The deletions can be located almost anywhere in the mitochondrial genome24, but they all cause one of three pathogenically similar disorders, as distinguished by their tissue proclivities9: Kearns– Sayre syndrome (KSS)25; chronic progressive external ophthalmoplegia (CPEO, or simply PEO)26; or Pearson’s syndrome27 (BOX 1).
In all three syndromes, the deleted region encompasses at least one tRNA gene. Indeed, this is the basis for the underlying pathogenesis of these syndromes and demonstrates two additional aspects of mitochondrial behaviour. First, the loss of a single tRNA gene is sufficient to compromise the translation of all 13 mtDNA-encoded polypeptides28. Second, this failure to translate mt-mRNAs occurs even in heteroplasmic cells but only if the fraction of Δ-mtDNA within each cell (or muscle segment) is above ~80% and the amount of normal mtDNAs is insufficient to complement the defect by providing the ‘missing’ tRNAs20,29,30. If the mutation load is below this threshold, complementation will occur20, as normal mRNAs and polypeptides can move fairly freely, not only within individual organelles but also as a result of the constant fusion and fission of mitochondria. By contrast, the mtDNAs themselves are more ‘confined’, as they are bound in mtDNA–protein complexes called nucleoids that are attached to the inner face of the inner membrane and operate as semi-autonomous genetic elements20.
Secondary mtDNA defects: multiple deletions and depletion of mtDNA
Secondary mtDNA-related disorders are those in which mtDNA integrity is compromised but not via a ‘direct hit’ on the mitochondrial genome. Rather, mtDNA mutations in these disorders (secondary mtDNA mutations) are caused ‘indirectly’ as a consequence of a Mendelian-inherited mutation in a nuclear gene that encodes a protein required for the proper replication and/or maintenance of the mitochondrial genome, ultimately resulting in mtDNA instability. This instability can manifest itself through the generation of multiple point mutations or large-scale Δ-mtDNAs that arise over the lifetime of the patient31; loss of the entire mitochondrial genome (that is, mtDNA depletion32) can also occur. Often, all three types of error coexist within tissues of the same patient33. Strictly speaking, these syndromes are not considered to be primary mtDNA diseases. However, because at a basic level these diseases are defects in the dialogue between the nucleus and the mitochondria, they share many of the features of the primary disorders, including their population-genetic behaviour (for example, heteroplasmy and threshold effects).
There are various mutations that can cause mtDNA instability, including those in nDNA-encoded proteins required for maintenance of mitochondrial nucleotide pools34, nucleotide transport35, DNA replication36 and RNA transcription37. For example, PEO can occur with Mendelian inheritance owing to mutations in a nucleus-encoded mitochondrial DNA helicase called Twinkle37; these mutations result in the generation and accumulation in muscle of multiple large-scale Δ-mtDNAs that are structurally similar to the ‘clonal’ Δ-mtDNAs observed in patients with sporadic PEO and KSS. Depending on the loads of depleted and mutated mtDNAs and their tissue distributions, secondary mtDNA-related disorders can have various presentations. Examples of syndromes include mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)34, Alpers’ sydrome38 and PEO in isolation or as a part of syndromes that include PEO plus other features.
Ageing and neurodegeneration
Accumulation of mtDNA mutations in normal ageing
The mutation load of the clonal Δ-mtDNAs found in sporadic KSS, PEO and Pearson’s syndrome is extremely high (>80% in affected issues)39. Interestingly, these mutations also accumulate during the lifetime of normal people, albeit at extremely low levels that are detectable only by PCR40 and with different amounts in different tissues41. It appears that these Δ-mtDNAs accumulate during our lifetimes, especially in postmitotic tissues such as the brain, heart and muscle42–44. Although they arise spontaneously as the result of somatic mutation, each individual species of Δ-mtDNA has the capacity to expand clonally45; this expansion is particularly noticeable in muscle from aged normal individuals46 (and in patients with Mendelian PEO)47. Although each individual species of Δ-mtDNA may represent less than 0.1% of total mtDNA43,44, sufficient numbers of different Δ-mtDNA species accumulate so that, depending on their tissue distribution and concentration48, they can have deleterious consequences49.
Although Δ-mtDNAs clearly accumulate in ageing, the evidence that the same holds for point mutations is weaker43. There are many ongoing studies aimed at detecting somatic mtDNA mutations, but it still remains uncertain whether these mutations cause overt disease or whether they have a major role in normal ageing. Perhaps studies of experimental models that age more rapidly than humans (for example, flies, worms and even yeast) will shed light on this question.
mtDNA mutations in late-onset neurodegenerative disease
There is a growing interest in the potential role of mitochondrial dysfunction in general, and of mtDNA mutations in particular, in late-onset neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s diseases, amyotrophic lateral sclerosis and hereditary spastic paraplegias50. Although there seems to be little doubt of a mitochondrial connection, it seems to relate more to problems of mitochondrial dynamics (for example, fission, fusion, distribution and anchorage) or quality control (for example, autophagy) than to mutations in mtDNA50. However, there is one neurodegenerative disorder in which mtDNA mutations may have a role (albeit probably a secondary one), and that is Parkinson’s disease. Studies of proteins mutated in familial Parkinson’s disease — for example, PINK1, a mitochondrially targeted kinase51, and parkin, a cytosolic ubiquitin ligase that translocates to low-Δψm dysfunctional mitochondria52 — have implicated altered mitochondrial quality control as an underlying element in the pathogenesis of both the familial and sporadic forms of the disease. However, it has also been found that the substantia nigra — the brain region that is most affected in Parkinson’s disease (and the main site of dopaminergic neuron degeneration in patients with Parkinson’s disease) — harbours substantial levels of the types of Δ-mtDNAs found in KSS and in normal ageing53,54. This finding has led to the hypothesis that these somatically produced Δ-mtDNAs may eventually cause bioenergetic deficit, leading to the disease. This hypothesis has received indirect support from studies of mice in which mtDNA deficiency was induced specifically in the substantia nigra and that displayed many of the pathological features of Parkinson’s disease55. We note, however, that patients with KSS who have high levels of Δ-mtDNAs do not, as a rule, have features of Parkinson’s disease and that only one mutation in mtDNA has been associated with such features56. Further analysis in model systems, including mice harbouring clonal57 or multiple58 Δ-mtDNAs, or analysis of PINK1- or PARK2 (also known as parkin)-mutated mice for the presence of Δ-mtDNAs, may help to resolve this issue.
Inherited mtDNA variants in complex traits
In addition to the potential roles of somatic mtDNA mutations in ageing and neurodegenerative diseases, are there mtDNA haplotypes or specific polymorphisms that are associated with predisposion to neurodegenerative diseases or that are associated with other complex traits? Different ethnic groups have distinct mitochondrial haplotypes that have resulted from the accumulation of variations from the mtDNA of our ancestral ‘mitochondrial Eve’59. Some of these haplotypes may confer vulnerability to, or protection from, various common diseases. In addition, the genetic variation might be responsible for subtle biochemical differences among haplogroups. For example, it has been postulated that mtDNA haplotypes that favour a looser coupling between oxidation and phosphorylation — thereby generating a surplus of heat at the expense of ATP — may have favoured the settling of human populations in colder climates60; however, this hypothesis is controversial61,62.
Mitochondrial haplotypes as modifiers
It is clear that bona fide pathogenic mtDNA mutations can cause common disorders such as diabetes mellitus63, hearing loss64,65 or exercise intolerance66, but such mutations account for less than 2% of patients with these complex traits. What is less clear is the role of mtDNA haplotype variants in such common disorders. Theoretically, an mtDNA variant with mild functional consequences could interact with other polymorphisms in mtDNA or nDNA to produce complex traits synergistically67. A well-documented example is the effect of mtDNA haplotype on the expressivity of Leber’s hereditary optic neuropathy (LHON) mutations. The variable penetrance of these mutations cannot be accounted for by heteroplasmy, because the mtDNA defect is homoplasmic in most patients. Haplotype analyses of LHON mutations have revealed an increased risk of vision loss in the setting of haplotypes J1, J2 and K68,69. Analyses in vitro of cybrids (BOX 2) and fibroblast cells that harbour either the m.11778G→A or m.14484T→C mutations confirmed that haplogroup J conferred sensitivity to the neurotoxin n-hexane70, an environmental toxin that, like smoke and pesticides, has been implicated as an environmental trigger of optic neuropathy in patients with LHON70.
Susceptibility to complex disorders
Evidence for mtDNA susceptibility polymorphisms and/or haplogroups in complex disorders is highly controversial. There is evidence for and against such links in a number of diseases, including age-related macular degeneration, Alzheimer’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, hereditary spastic paraplegia, Huntington’s disease, ischaemic stroke, metabolic syndrome, myocardial infarction, psychiatric disorders, Parkinson’s disease, sarcopaenia and type 2 diabetes, among others71–83. Although it has been argued that those studies in which associations were not observed typically had a low statistical power owing to small sample sizes and the genotypic complexity of mtDNA lineages and sublineages, the fact remains that very few ‘primary’ mtDNA mutations (which have been found on diverse mtDNA haplotypic backgrounds) are associated with these disorders, and they are far fewer in number than are nuclear mutations found in these conditions50. Moreover, most of these studies were either correlative, descriptive or both and were also susceptible to technical difficulties that were unique to the analysis of mtDNA polymorphisms84. These included problems in determining the functional consequences of one haplotype as compared to another85 and in the misattribution of mutations to mtDNA resulting from the co-amplification of authentic mtDNA with nucleus-embedded mtDNA pseudogenes (described below). Taken together, we feel that although there may be important contributions of mtDNA haplotypes to complex traits and disorders, the evidence to date is too weak to support such a conclusion.
mtDNA mutations in cancer
Perhaps nowhere is the role of mtDNA mutations in pathology more contentious than in cancer. Moreover, the discovery of mtDNA mutations in tumours has also contributed to the recent resurgence of interest in the Warburg hypothesis of cancer, which states that tumours rely on ‘aerobic glycolysis’ rather than on OXPHOS in normoxic conditions to survive and even to thrive86.
In 1998, Bert Vogelstein’s group reported that colorectal cancers harbour numerous mtDNA mutations (typically, point mutations, not rearrangements, with many in the homoplasmic state); adjacent non-cancerous tissues had only normal mtDNAs87. Since then, this result has been replicated numerous times88 but, given our understanding of mitochondrial behaviour, especially in primary mitochondrial disease, these findings raise many questions. Why are there so many mutations, and why are many of the mutations in cis (that is, on the same mtDNA circle)? How do they arise rapidly? Why do some tumours have nonsense mutations, whereas others have synonymous or missense mutations, many of which are apparently functionally neutral? Why is there no apparent difference in tumour biology between those tumours that harbour homoplasmic mutations and those that harbour heteroplasmic mutations (sometimes below the threshold level typical of primary mitochondrial disease)? Fundamentally, these questions are really asking the same thing: what is the relationship of mtDNA mutations and OXPHOS function to cancer biology (BOX 4)?
Box 4. Mitochondrial function in cancer.
Mitochondrial function is altered in numerous ways in tumours, allowing them to survive and to circumvent cell-death pathways141. Some of these changes are summarized below.
Loss of p53 function
Among its numerous functions, the tumour suppressor p53 regulates respiratory chain function and glycolysis via positive transcriptional regulation of synthesis of cytochrome oxidase 2 (SCO2), which encodes a complex IV assembly protein, and negative regulation of TP53-induced glycolysis regulator (TIGAR; also known as C12orf5). When TP53 (the gene that encodes p53) is mutated or lost in tumours, glycolysis (and the mitochondrial tricarboxylic acid (TCA) cycle) is stimulated142 and the respiratory chain is depressed. This phenomenon is known as the Crabtree effect.
Evasion of hypoxia-mediated cell death
Poorly vascularized, rapidly dividing tumour cells become hypoxic, stimulating cell-death pathways mediated by the transcription factor hypoxia-inducible factor 1 (HIF1). Among other effects, this change promotes vascularization, cell survival and invasion. HIF1 subunit-α is normally degraded by the proteasome after hydroxylation by prolyl hydroxylase and subsequent ubiquitylation by the von Hippel–Lindau E3 ubiquitin ligase, but increased levels of TCA-derived succinate in tumours inhibit the hydroxylase, thus stabilizing HIF1. Notably, mutations in nuclear genes encoding two TCA cycle enzymes — namely, succinate dehydrogenase (SDH) and fumarase, both of which result in increased cytosolic succinate levels — can give rise to cancer. These cancers are pheochromocytomas and paragangliomas in the case of SDH143 and leiomyomas in the case of fumarase144. These observations lend support to the idea that HIF1α has a role in tumorigenesis and to the conceptual underpinnings of the Warburg hypothesis145.
Evasion of apoptosis
Cell death by apoptosis is regulated by a balance between interacting pro-apoptotic factors (for example, BAX and the truncated form of BH3-interacting domain death agonist (tBID)) and anti-apoptotic factors (for example, BCL-2). In cancer, this balance is shifted towards anti-apoptotic factors. For example, the glycolytic enzyme hexokinase, which binds to voltage-dependent anion-selective channel (VDAC1) on the mitochondrial outer membrane, is upregulated in tumours; this prevents the interaction of VDAC1 with tBID146,147.
Role in DNA replication
In addition to the requirement of a respiratory chain to support pyrimidine synthesis (see text), it was recently shown that purine biosynthesis in tumours is also mediated by mitochondria via pathways requiring glycine. In mitochondria, glycine is converted to tetrahydrofolate (by the glycine cleavage system), which provides one-carbon units for incorporation into the purine ring. Although glycine is made both in the cytosol (de novo from carbon dioxide and ammonia and salvaged by serine and tetrahydrofolate) and in mitochondria (also from serine and tetrahydrofolate), tumours (but not rapidly dividing normal cells) consume glycine that is metabolized specifically by the mitochondrial pathway148. Thus, DNA replication using both purine and pyrimidine precursors — the sine qua non of tumour growth — rely on mitochondrial function.
This brief overview vastly understates the complicated interplay between mitochondrial and tumour biology141 but gives some sense of the potential effects of a possible role that mitochondria and mitochondrial DNA mutations might have in cancer.
Although there are dozens of studies documenting mtDNA mutations in cancer, almost none of them has asked whether a specific mtDNA genotype affects mitochondrial function and whether such dysfunction has a role in tumorigenesis. There have been a few reports of tumours that are, for example, cytochrome c oxidase (COX)-deficient5,89, but it is not clear whether the deficiency is due to a mutation in an mtDNA-encoded COX gene, or whether this is the result of a general downregulation of respiratory chain function or even the result of reduced mtDNA copy number. Moreover, it is noteworthy that of the known pathogenic point mutations causing primary mitochondrial disease (TABLE 1), none is unequivocally associated with cancer. Thus, mtDNA mutations seem more likely to be a marker of cancer and/or to arise as a secondary effect of altered tumour biology than to be a cause of cancer.
An alternative idea is that mtDNA mutations result in downregulation of OXPHOS function, and this process somehow promotes tumour survival. One idea in support of this view is that defects in OXPHOS lead to activation of the AKT cell survival pathway, including downregulation of apoptosis via a mechanism mediated by cellular NADH, which is increased when mitochondrial respiration is blocked90. However, defective bioenergetics in tumours, either through mtDNA mutation or reduction in mtDNA copy number91 (but the issue of copy number is contentious92), raises a conceptual problem: mitochondrial respiratory chain function is indispensable for the synthesis of the pyrimidines that cells — especially highly proliferating tumour cells — need to divide, and in the absence of a functioning respiratory chain, cells eventually die93. This is because a key reaction in de novo pyrimidine synthesis — the conversion of dihydroorotate to orotate (a precursor of uridine monophosphate (UMP)) — is catalysed by the mitochondrial enzyme dihydroorotate dehydrogenase, which depends on an intact respiratory chain for its function94; more specifically, the electron acceptor for this redox reaction is reduced ubiquinone (that is, CoQ) and, through complexes III and IV, molecular oxygen (FIG. 1b). If a similar effect operates in tumours that contain homoplasmic loss-of-function mtDNA mutations, it would mitigate against, not for, the generation of mtDNA mutations. We note in this regard that cells that are completely devoid of mtDNA (called ρ0 cells (BOX 2)) do not form tumours when they are injected into mice subcutaneously95,96, but they are tumorigenic when injected intramuscularly96. This is perhaps because the subcutaneous site is less vascularized than the muscle and cannot take up uridine from the circulation (uridine is ~5 μM in plasma97).
Are tumour mtDNA mutations artefacts?
If some baseline level of normal OXPHOS function is required for tumours to be viable, why have so many mtDNA mutations been detected? One possibility is that at least some of these could be technical artefacts. If a low-stringency BLAST of mtDNA is run against the human genome, it is possible to detect ~1,000 nucleus-embedded mitochondrial sequences (NUMTs), ranging in size from ~50 bp to >15 kb (that is, almost full-length)98,99. These NUMTs have been relocalized from the mitochondria to the nucleus in evolutionary time98 and have subsequently been subjected to genetic drift, with individual NUMTs picking up polymorphic mutations in different human lineages100,101 (that is, they are essentially mtDNA-derived pseudogenes). What is less appreciated is that NUMTs can also enter the nucleus during the lifetime of an individual, especially under conditions of stress. This rapid relocalization of mtDNA sequences has been documented in plants102, yeast103, rodents104 and humans105–107. Of course, in these cases, the sequences of authentic mtDNA and of recently relocalized NUMTs will be extremely similar or possibly identical.
Chromosomal instability in tumours has the potential to increase the NUMT copy number in several ways: the NUMTs could be located in unstable regions that lead to aneuploidy; they could be located in homogeneously staining regions (HSRs), which are tandemly repeated regions of the genome with increased copy number; and they can be selectively amplified within small regions of the chromosome as double-minute DNAs (dmDNAs)108. In the cases of HSRs and dmDNAs, the copy number in the repeated regions can vary, ranging from ~3 to ~30 repeats, with a median value of ~10 repeats108,109. Because the typical dmDNA is about 1–3 Mb in size, it will contain on average one NUMT (on the basis of there being ~1,000 NUMTs in a human genome) and will be amplified selectively by approximately tenfold over its pre-cancerous level110,111. We therefore speculate that NUMT targets can increase in tumours in ways that vary from individual to individual and from tumour to tumour.
When using PCR of total cellular DNA to analyse mtDNA in normal tissues, the number of authentic mtDNA molecules far exceeds the number of NUMTs detectable by any particular pair of PCR primers, and the PCR preferentially amplifies the normal mtDNA. However, in tumours, these PCR reactions might amplify one or more NUMTs along with (or even in preference to) authentic mtDNA owing to a reduction in the mtDNA/NUMT target DNA ratio. There are at least three possible reasons for this: a reduction in mtDNA copy number91; the presence of large numbers of potentially highly polymorphic and diverged NUMTs in tumours; and the potential to amplify specific NUMTs selectively in dmDNAs. Therefore, depending on the choice of primers (FIG. 2), mutations in tumour NUMT DNA may be misattributed to those in authentic tumour mtDNA112. We note that when mitochondria were purified before PCR amplification, no pathogenic mtDNA mutations were found in mouse brain tumours113, and the number of mtDNA mutations in human colorectal tumours was actually lower than that in the surrounding tissue114.
Figure 2. Example of the potential to amplify inadvertently a nucleus-embedded mitochondrial sequence.

Comparison of a region of the 16S rRNA gene within authentic mitochondrial DNA (mtDNA) (top lines; numbering according to the standard ‘Cambridge’ mtDNA sequence8) to that of a nucleus-embedded mitochondrial sequence (NUMT) located on chromosome 17p11.2 (bottom lines; genome numbering according to the February 2009 release of the human genome (GRCh37/hg19)). The primer pairs used (Forward and Reverse 1, and Forward and Reverse 2)112 have the potential to amplify both mtDNA and the NUMT. Sequence differences between mtDNA (black) and NUMT DNA (red) are shown in bold.
These caveats are speculative, and for that reason they should not be allowed to obscure the very real possibility that authentic mtDNAs are indeed mutated in tumours (especially as the rapid cellular division in cancers may allow for the accumulation and fixation of spontaneous mtDNA mutations115). Moreover, even if the mutated DNAs are artefacts derived from amplified NUMTs116 and are less biologically informative, they are nevertheless still extremely useful as early markers of cancer117,118, and thus they may have clinical use.
Concluding remarks
The notion that mutations in mtDNA, and the subsequent alterations in mitochondrial oxidative energy metabolism, are confined to a small group of pathologies no longer holds true. Together, primary mtDNA-related diseases, which were previously deemed to be extremely rare, have now been shown to be as frequent as some Mendelian disorders, such as Duchenne’s muscular dystrophy or cystic fibrosis. Moreover, mtDNA mutations — either partial deletions or point mutations — that are present at high levels in primary mitochondrial disorders have now been identified at low levels in infant cord blood15, at somewhat higher levels in the ageing population49,54 and at fairly high levels within target tissues in some of the most common pathologies that afflict humankind53. Although we have made some progress in using the basic concepts of mitochondrial biology and genetics to deduce the pathogenesis of primary mitochondrial OXPHOS disease, we are only now beginning to identify and to understand the role of secondary mtDNA mutations in ageing, in Parkinson’s disease and perhaps in other disorders as well.
Two technological issues will determine the pace of further progress in the field. With respect to understanding the relationship of underlying mtDNA haplotypes to complex traits, the application of recent technological advances — such as whole-exome sequencing, whole-genome sequencing, high-throughput whole-transcriptome shotgun sequencing and various ‘omics’ approaches (especially proteomics, lipidomics and metabolomics) — to this problem may enable us to understand more clearly the relationship of genotype to phenotype. However, there is currently no proven technology that will allow us to introduce exogenous mtDNA into mammalian mitochondria in a stable and heritable way. Thus, we cannot make targeted mtDNA mutations to study basic issues in mitochondrial biogenesis (for example, dissecting mtDNA elements controlling replication, transcription or translation) nor can we introduce targeted mutations into mitochondria to generate cellular or animal models of mtDNA-based disorders, be they primary, secondary or complex. However, after this major technical hurdle has been overcome, the field will be poised to enter a revolutionary new phase in our understanding of mitochondrial biology and disease. To the question posed in 2000 regarding the phenotypic consequences of mtDNA mutations — ‘are we scraping the bottom of the barrel?’119 — the answer is a definite ‘no’.
Acknowledgments
The authors were supported by grants from the US National Institutes of Health (HD32062), the US Department of Defense (W911NF-12-1-0159), the Muscular Dystrophy Association, the Ellison Medical Foundation, the Alzheimer Drug Discovery Foundation and the Marriott Mitochondrial Disorder Clinical Research Fund.
Glossary
- Microarray-based sequencing
DNA sequencing based on the ability of the target DNA to hybridize to an ordered set of defined oligonucleotides immobilized on a chip
- Monochromosomal transfer
Typically, the transfer of individual human chromosomes (or parts of chromosomes) to rodent cells in order to identify and map human genes responsible for disease
- Mitodynamics
The morphological properties of mitochondria (for example, fusion, fission, distribution, anchorage, positioning). These properties can change both in space and in time within cells
- Primary pathogenic mtDNA mutations
Mutations that can compromise OXPHOS function and cause disease. They arise in mtDNA through processes such as random errors during normal mtDNA replication
- Nystagmus
An involuntary, quick, rhythmic movement of the eyeball, which can be horizontal, vertical or rotary
- Hemiplegia
Paralysis of the left or right side of the body owing to a brain lesion affecting motor pathways
- Strongly SDH-positive vessels (SSVs)
Abnormal, massive accumulation of mitochondria in blood vessels, as visualized by enzyme histochemistry to detect the activity of succinate dehydrogenase (SDH; complex II of the respiratory chain)
- Heteroplasmic
A term that describes the presence of two or more mtDNA genotypes within a cell, tissue or organism
- Lactic acidosis
The abnormal accumulation of lactic acid causing lower pH in blood in a resting individual (that is, not during or immediately after exercise), which is seen in many patients with mitochondrial disease
- Myoclonus
Involuntary jerky movement of an area of the body (usually a limb)
- Mitochondrial transmembrane potential (Δψm)
This is the electrical potential generated across the mitochondrial inner membrane due to differences in the distribution of ions (for example, H+, Ca2+, Na+, K+ and ionized ATP species) between the matrix and the intermembrane space
- Secondary mtDNA mutations
Mutations that can compromise OXPHOS function and cause disease and that arise in mtDNA owing to a mutation in the nuclear genome that compromises mtDNA integrity (for example, a mutation in mitochondrial DNA polymerase-γ causing systemic errors in mtDNA replication)
- Homoplasmic
A term that describes the presence of a single mtDNA genotype within a cell, tissue, or organism
- Aerobic glycolysis
The conversion of glucose to lactate (and the production of ATP by substrate level phosphorylation) even in the presence of oxygen, especially in tumours
- Double minute DNAs (dmDNAs)
Tiny fragments of extrachromosomal nuclear DNA found in many tumours, derived from the amplification of small regions of chromosomal DNA
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
References
- 1.Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14:255–274. doi: 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Rodriguez LJ. Appendix 1. Basic properties of mitochondria. Methods Cell Biol. 2007;80:809–812. doi: 10.1016/S0091-679X(06)80040-3. [DOI] [PubMed] [Google Scholar]
- 3.Calvo SE, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. This was one of the first applications of sequencing the ‘mitochondrial exome’ (that is, ~1,500 nuclear genes encoding mitochondrial-targeted proteins) to identify pathogenic mutations causing mitochondrial disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaefer AM, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 5.Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 6.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 7.Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44:41–59. doi: 10.3109/07853890.2011.598547. This is a comprehensive overview of mitochondrial respiratory chain disorders. [DOI] [PubMed] [Google Scholar]
- 8.Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 9.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 10.Sacconi S, et al. A functionally dominant mitochondrial DNA mutation. Hum Mol Genet. 2008;17:1814–1820. doi: 10.1093/hmg/ddn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoneda M, Miyatake T, Attardi G. Heteroplasmic mitochondrial tRNALys mutation and its complementation in MERRF patient-derived mitochondrial transformants. Muscle Nerve. 1995;3:S95–S101. doi: 10.1002/mus.880181420. [DOI] [PubMed] [Google Scholar]
- 12.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 13.Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol. 1993;34:827–834. doi: 10.1002/ana.410340612. [DOI] [PubMed] [Google Scholar]
- 14.Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. This was the first large-scale epidemiological survey of the prevalence of common pathological mtDNA mutations in the normal population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart JB, et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prezant TR, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nature Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 18.Raimundo N, et al. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012;148:716–726. doi: 10.1016/j.cell.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilkerson RW, et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilkerson RW, Schon EA, Hernandez E, Davidson MM. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol. 2008;181:1117–1128. doi: 10.1083/jcb.200712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sgarbi G, et al. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem J. 2006;395:493–500. doi: 10.1042/BJ20051748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 23.Lestienne P, Ponsot G. Kearns-Sayre syndrome with muscle mitochondrial DNA deletion. Lancet. 1988;331:885–886. doi: 10.1016/s0140-6736(88)91632-7. [DOI] [PubMed] [Google Scholar]
- 24.Damas J, et al. Mitochondrial DNA deletions are associated with non-B DNA conformations. Nucleic Acids Res. 2012;40:7606–7621. doi: 10.1093/nar/gks500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol. 1958;60:280–289. [PubMed] [Google Scholar]
- 26.Moraes CT, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns–Sayre syndrome. N Engl J Med. 1989;320:1293–1299. doi: 10.1056/NEJM198905183202001. [DOI] [PubMed] [Google Scholar]
- 27.Pearson HA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95:976–984. doi: 10.1016/s0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- 28.Nakase H, et al. Transcription and translation of deleted mitochondrial genomes in Kearns–Sayre syndrome: implications for pathogenesis. Am J Hum Genet. 1990;46:418–427. This was one of the first studies providing insight into the pathomechanism of mtDNA deletion disorders. [PMC free article] [PubMed] [Google Scholar]
- 29.Mita S, Schmidt B, Schon EA, DiMauro S, Bonilla E. Detection of “deleted” mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc Natl Acad Sci USA. 1989;86:9509–9513. doi: 10.1073/pnas.86.23.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moraes CT, et al. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nature Genet. 1992;1:359–367. doi: 10.1038/ng0892-359. [DOI] [PubMed] [Google Scholar]
- 31.Ashley N, et al. Defects in maintenance of mitochondrial DNA are associated with intramitochondrial nucleotide imbalances. Hum Mol Genet. 2007;16:1400–1411. doi: 10.1093/hmg/ddm090. [DOI] [PubMed] [Google Scholar]
- 32.Moraes CT, et al. Mitochondrial DNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991;48:492–501. This was the first description of a quantitative defect in mtDNA copy number causing disease. [PMC free article] [PubMed] [Google Scholar]
- 33.Nishigaki Y, Marti R, Copeland WC, Hirano M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J Clin Invest. 2003;111:1913–1921. doi: 10.1172/JCI17828. This was the first description of a secondary mitochondrial disorder causing mtDNA point mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 35.Kaukonen J, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 36.Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 37.Spelbrink JN, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nature Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 38.Naviaux RK, Nguyen KV. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 39.Moraes CT, et al. Phenotype-genotype correlations in skeletal muscle of patients with mtDNA deletions. Muscle Nerve. 1995;3:S150–S153. doi: 10.1002/mus.880181429. [DOI] [PubMed] [Google Scholar]
- 40.Cortopassi GA, Shibata D, Soong NW, Arnheim N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc Natl Acad Sci USA. 1992;89:7370–7374. doi: 10.1073/pnas.89.16.7370. This was among the first reports that called attention to the accumulation of somatic mtDNA deletions in normal ageing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corral-Debrinski M, et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 42.Meissner C, et al. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp Gerontol. 2008;43:645–652. doi: 10.1016/j.exger.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Pallotti F, Chen X, Bonilla E, Schon EA. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am J Hum Genet. 1996;59:591–602. [PMC free article] [PubMed] [Google Scholar]
- 44.Simonetti S, Chen X, DiMauro S, Schon EA. Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim Biophys Acta. 1992;1180:113–122. doi: 10.1016/0925-4439(92)90059-v. [DOI] [PubMed] [Google Scholar]
- 45.Oldfors A, et al. Mitochondrial DNA deletions and cytochrome c oxidase deficiency in muscle fibres. J Neurol Sci. 1992;110:169–177. doi: 10.1016/0022-510x(92)90025-g. [DOI] [PubMed] [Google Scholar]
- 46.Bodyak ND, Nekhaeva E, Wei JY, Khrapko K. Quantification and sequencing of somatic deleted mtDNA in single cells: evidence for partially duplicated mtDNA in aged human tissues. Hum Mol Genet. 2001;10:17–24. doi: 10.1093/hmg/10.1.17. [DOI] [PubMed] [Google Scholar]
- 47.Vu TH, et al. Analysis of mtDNA deletions in muscle by in situ hybridization. Muscle Nerve. 2000;23:80–85. doi: 10.1002/(sici)1097-4598(200001)23:1<80::aid-mus10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 48.Petruzzella V, et al. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt-3243. Hum Mol Genet. 1994;3:449–454. doi: 10.1093/hmg/3.3.449. [DOI] [PubMed] [Google Scholar]
- 49.Bua E, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schon EA, Przedborski S. Mitochondria: the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vives-Bauza C, et al. Control of mitochondrial integrity in Parkinson’s disease. Prog Brain Res. 2010;183:99–113. doi: 10.1016/S0079-6123(10)83006-7. [DOI] [PubMed] [Google Scholar]
- 52.Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genet. 2006;38:515–517. doi: 10.1038/ng1769. This was the first report of correlating a neurodegenerative disease with the accumulation of mtDNA mutations in the clinically relevant target tissue. [DOI] [PubMed] [Google Scholar]
- 54.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 55.Ekstrand MI, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Coo IF, et al. A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann Neurol. 1999;45:130–133. doi: 10.1002/1531-8249(199901)45:1<130::aid-art21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 57.Inoue K, et al. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nature Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- 58.Tyynismaa H, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci USA. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 60.Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- 61.Amo T, Brand MD. Were inefficient mitochondrial haplogroups selected during migrations of modern humans? A test using modular kinetic analysis of coupling in mitochondria from cybrid cell lines. Biochem J. 2007;404:345–351. doi: 10.1042/BJ20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saxena R, et al. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet. 2006;79:54–61. doi: 10.1086/504926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kadowaki T, et al. A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl J Med. 1994;330:962–968. doi: 10.1056/NEJM199404073301403. This was an important survey that demonstrated the importance of a specific mtDNA mutation as a cause of diabetes mellitus. [DOI] [PubMed] [Google Scholar]
- 64.Bitner-Glindzicz M, et al. Prevalence of mitochondrial 1555A→G mutation in European children. N Engl J Med. 2009;360:640–642. doi: 10.1056/NEJMc0806396. [DOI] [PubMed] [Google Scholar]
- 65.Vandebona H, et al. Prevalence of mitochondrial 1555A→G mutation in adults of European descent. N Engl J Med. 2009;360:642–644. doi: 10.1056/NEJMc0806397. [DOI] [PubMed] [Google Scholar]
- 66.Andreu AL, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341:1037–1044. doi: 10.1056/NEJM199909303411404. [DOI] [PubMed] [Google Scholar]
- 67.Elson JL, Majamaa K, Howell N, Chinnery PF. Associating mitochondrial DNA variation with complex traits. Am J Hum Genet. 2007;80:378–382. doi: 10.1086/511652. author reply 382–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carelli V, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet. 2006;78:564–574. doi: 10.1086/501236. This paper provieds a good example of the ‘synergistic’ relationship between specific mtDNA haplotypes and the severity of pathogenic mtDNA point mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hudson G, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007;81:228–233. doi: 10.1086/519394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghelli A, et al. The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS ONE. 2009;4:e7922. doi: 10.1371/journal.pone.0007922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anderson CD, et al. Common mitochondrial sequence variants in ischemic stroke. Ann Neurol. 2011;69:471–480. doi: 10.1002/ana.22108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arning L, et al. Mitochondrial haplogroup H correlates with ATP levels and age at onset in Huntington disease. J Mol Med. 2010;88:431–436. doi: 10.1007/s00109-010-0589-2. [DOI] [PubMed] [Google Scholar]
- 73.Ingram CJ, et al. Analysis of European case-control studies suggests that common inherited variation in mitochondrial DNA is not involved in susceptibility to amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:341–346. doi: 10.3109/17482968.2012.654394. [DOI] [PubMed] [Google Scholar]
- 74.Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol. 2008;43:24–33. doi: 10.1016/j.exger.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khusnutdinova E, et al. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson’s disease. Ann NY Acad Sci. 2008;1147:1–20. doi: 10.1196/annals.1427.001. [DOI] [PubMed] [Google Scholar]
- 76.Mancuso M, Filosto M, Orsucci D, Siciliano G. Mitochondrial DNA sequence variation and neurodegeneration. Hum Genom. 2008;3:71–78. doi: 10.1186/1479-7364-3-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishigaki Y, Fuku N, Tanaka M. Mitochondrial haplogroups associated with lifestyle-related diseases and longevity in the Japanese population. Geriatr Gerontol Int. 2010;10:S221–S235. doi: 10.1111/j.1447-0594.2010.00599.x. [DOI] [PubMed] [Google Scholar]
- 78.Rose G, et al. No evidence of association between frontotemporal dementia and major European mtDNA haplogroups. Eur J Neurol. 2008;15:1006–1008. doi: 10.1111/j.1468-1331.2008.02222.x. [DOI] [PubMed] [Google Scholar]
- 79.SanGiovanni JP, et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS ONE. 2009;4:e5508. doi: 10.1371/journal.pone.0005508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanchez-Ferrero E, et al. Mitochondrial DNA polymorphisms/haplogroups in hereditary spastic paraplegia. J Neurol. 259:246–250. doi: 10.1007/s00415-011-6155-1. [DOI] [PubMed] [Google Scholar]
- 81.Santoro A, et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE. 2012;5:e12037. doi: 10.1371/journal.pone.0012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sequeira A, et al. Mitochondrial mutations and polymorphisms in psychiatric disorders. Front Genet. 2012;3:103. doi: 10.3389/fgene.2012.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tranah GJ. Mitochondrial-nuclear epistasis: implications for human aging and longevity. Ageing Res Rev. 2012;10:238–252. doi: 10.1016/j.arr.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McRae AF, Byrne EM, Zhao ZZ, Montgomery GW, Visscher PM. Power and SNP tagging in whole mitochondrial genome association studies. Genome Res. 2008;18:911–917. doi: 10.1101/gr.074872.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Battersby BJ, Shoubridge EA. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet. 2001;10:2469–2479. doi: 10.1093/hmg/10.22.2469. [DOI] [PubMed] [Google Scholar]
- 86.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Polyak K, et al. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nature Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 88.Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89:65–71. doi: 10.1016/j.lfs.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 89.Melnick PJ. Enzyme patterns of tumors demonstrated histochemically in cryostat sections. Ann NY Acad Sci. 1965;125:689–715. doi: 10.1111/j.1749-6632.1965.tb45422.x. [DOI] [PubMed] [Google Scholar]
- 90.Pelicano H, et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yu M, et al. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life. 2007;59:450–457. doi: 10.1080/15216540701509955. [DOI] [PubMed] [Google Scholar]
- 92.Bai RK, et al. Mitochondrial DNA content varies with pathological characteristics of breast cancer. J Oncol. 2011;2011:496189. doi: 10.1155/2011/496189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.King MP, Koga Y, Davidson M, Schon EA. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNALeu(UUR) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12:480–490. doi: 10.1128/mcb.12.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller RW. Dihydroorotate-quinone reductase of Neurospora crassa mitochondria. Arch Biochem Biophys. 1971;146:256–270. doi: 10.1016/s0003-9861(71)80063-2. [DOI] [PubMed] [Google Scholar]
- 95.Hayashi J, Takemitsu M, Nonaka I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat Cell Mol Genet. 1992;18:123–129. doi: 10.1007/BF01233159. [DOI] [PubMed] [Google Scholar]
- 96.Morais R, et al. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 1994;54:3889–3896. [PubMed] [Google Scholar]
- 97.Peters GJ, et al. In vivo inhibition of the pyrimidine de novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (DUP-785; NSC 368390) in mice and patients. Cancer Res. 1990;50:4644–4649. [PubMed] [Google Scholar]
- 98.Mourier T, Hansen AJ, Willerslev E, Arctander P. The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Mol Biol Evol. 2001;18:1833–1837. doi: 10.1093/oxfordjournals.molbev.a003971. [DOI] [PubMed] [Google Scholar]
- 99.Ramos A, et al. Nuclear insertions of mitochondrial origin: database updating and usefulness in cancer studies. Mitochondrion. 2011;11:946–953. doi: 10.1016/j.mito.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 100.Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lang M, et al. Polymorphic Numts trace human population relationships. Hum Genet. 2012;131:757–771. doi: 10.1007/s00439-011-1125-3. [DOI] [PubMed] [Google Scholar]
- 102.Wang D, Lloyd AH, Timmis JN. Environmental stress increases the entry of cytoplasmic organellar DNA into the nucleus in plants. Proc Natl Acad Sci USA. 2012;109:2444–2448. doi: 10.1073/pnas.1117890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng X, Ivessa AS. The migration of mitochondrial DNA fragments to the nucleus affects the chronological aging process of Saccharomyces cerevisiae. Aging Cell. 2010;9:919–923. doi: 10.1111/j.1474-9726.2010.00607.x. This is a fascinating example of how mtDNA fragments can be transferred to the nucleus in ‘real time’ (that is, during the lifetime of an individual) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caro P, et al. Mitochondrial DNA sequences are present inside nuclear DNA in rat tissues and increase with age. Mitochondrion. 2010;10:479–486. doi: 10.1016/j.mito.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 105.Goldin E, et al. Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Hum Mutat. 2004;24:460–465. doi: 10.1002/humu.20094. [DOI] [PubMed] [Google Scholar]
- 106.Turner C, et al. Human genetic disease caused by de novo mitochondrial-nuclear DNA transfer. Hum Genet. 2003;112:303–309. doi: 10.1007/s00439-002-0892-2. [DOI] [PubMed] [Google Scholar]
- 107.Willett-Brozick JE, Savul SA, Richey LE, Baysal BE. Germ line insertion of mtDNA at the breakpoint junction of a reciprocal constitutional translocation. Hum Genet. 2001;109:216–223. doi: 10.1007/s004390100564. [DOI] [PubMed] [Google Scholar]
- 108.Storlazzi CT, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: origin and structure. Genome Res. 2010;20:1198–1206. doi: 10.1101/gr.106252.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Valsesia A, et al. Network-guided analysis of genes with altered somatic copy number and gene expression reveals pathways commonly perturbed in metastatic melanoma. PLoS ONE. 2011;6:e18369. doi: 10.1371/journal.pone.0018369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hirano T, et al. Co-localization of mitochondrial and double minute DNA in the nuclei of HL-60 cells but not normal cells. Mutat Res. 1999;425:195–204. doi: 10.1016/s0027-5107(99)00037-8. [DOI] [PubMed] [Google Scholar]
- 111.Lee JH, Ryu TY, Cho CH, Kim DK. Different characteristics of mitochondrial microsatellite instability between uterine leiomyomas and leiomyosarcomas. Pathol Oncol Res. 2011;17:201–205. doi: 10.1007/s12253-010-9297-z. [DOI] [PubMed] [Google Scholar]
- 112.Ellinger J, et al. Circulating mitochondrial DNA in serum: A universal diagnostic biomarker for patients with urological malignancies. Urol Oncol. 2012;30:509–515. doi: 10.1016/j.urolonc.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 113.Kiebish MA, Seyfried TN. Absence of pathogenic mitochondrial DNA mutations in mouse brain tumors. BMC Cancer. 2005;5:102. doi: 10.1186/1471-2407-5-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ericson NG, et al. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012;8:e1002689. doi: 10.1371/journal.pgen.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. 2002;360:1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- 116.Parr RL, et al. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics. 2006;7:185. doi: 10.1186/1471-2164-7-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maki J, et al. Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am J Clin Pathol. 2008;129:57–66. doi: 10.1309/UJJTH4HFEPWAQ78Q. [DOI] [PubMed] [Google Scholar]
- 118.Parr RL, et al. Somatic mitochondrial DNA mutations in prostate cancer and normal appearing adjacent glands in comparison to age-matched prostate samples without malignant histology. J Mol Diagn. 2006;8:312–319. doi: 10.2353/jmoldx.2006.050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.DiMauro S, Andreu AL. Mutations in mtDNA: are we scraping the bottom of the barrel? Brain Pathol. 2000;10:431–441. doi: 10.1111/j.1750-3639.2000.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Giordano C, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain. 2011;134:220–234. doi: 10.1093/brain/awq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kaufmann P, et al. Natural history of MELAS associated with mitochondrial DNA m. 3243A>G genotype. Neurology. 2011;77:1965–1971. doi: 10.1212/WNL.0b013e31823a0c7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tay SH, et al. Aortic rupture in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Arch Neurol. 2006;63:281–283. doi: 10.1001/archneur.63.2.281. [DOI] [PubMed] [Google Scholar]
- 123.Chong PS, Vucic S, Hedley-Whyte ET, Dreyer M, Cros D. Multiple symmetric lipomatosis (Madelung’s disease) caused by the MERRF (A8344G) mutation: a report of two cases and review of the literature. J Clin Neuromuscul Dis. 2003;5:1–7. doi: 10.1097/00131402-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 124.Suzuki T, Nagao A, Suzuki T. Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. Wiley Interdiscip Rev RNA. 2011;2:376–386. doi: 10.1002/wrna.65. [DOI] [PubMed] [Google Scholar]
- 125.Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Cytochrome c oxidase activity is deficient in blood vessels of patients with myoclonus epilepsy with ragged-red fibers. Acta Neuropathol. 1993;85:280–284. doi: 10.1007/BF00227723. [DOI] [PubMed] [Google Scholar]
- 126.Naini A, et al. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox”. J Neurol Sci. 2005;229–230:187–193. doi: 10.1016/j.jns.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 127.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Torres J, Darley-Usmar V, Wilson MT. Inhibition of cytochrome c oxidase in turnover by nitric oxide: mechanism and implications for control of respiration. Biochem J. 1995;312:169–173. doi: 10.1042/bj3120169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rodriguez-Juarez F, Aguirre E, Cadenas S. Relative sensitivity of soluble guanylate cyclase and mitochondrial respiration to endogenous nitric oxide at physiological oxygen concentration. Biochem J. 2007;405:223–231. doi: 10.1042/BJ20070033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koga Y, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- 131.Horvath R, et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain. 2009;132:3165–3174. doi: 10.1093/brain/awp221. An unexpected connection is presented in this paper between an mtDNA mutation and a mitochondrial disorder with an unusual course. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Uusimaa J, et al. Reversible infantile respiratory chain deficiency is a unique, genetically heterogenous mitochondrial disease. J Med Genet. 2011;48:660–668. doi: 10.1136/jmg.2011.089995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Umeda N, et al. Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J Biol Chem. 2005;280:1613–1624. doi: 10.1074/jbc.M409306200. [DOI] [PubMed] [Google Scholar]
- 134.Yan Q, et al. Human TRMU encoding the mitochondrial 5-methylaminomethyl-2-thiouridylate-methyltransferase is a putative nuclear modifier gene for the phenotypic expression of the deafness-associated 12S rRNA mutations. Biochem Biophys Res Commun. 2006;342:1130–1136. doi: 10.1016/j.bbrc.2006.02.078. [DOI] [PubMed] [Google Scholar]
- 135.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. This was the first description of the use of cybrid technology in mammalian cells. [DOI] [PubMed] [Google Scholar]
- 136.King MP, Attardi G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996;264:304–313. doi: 10.1016/s0076-6879(96)64029-4. [DOI] [PubMed] [Google Scholar]
- 137.Engel WK, Cunningham GG. Rapid examination of muscle tissue. An improved trichrome method for fresh-frozen biopsy sections. Neurology. 1963;13:919–923. doi: 10.1212/wnl.13.11.919. [DOI] [PubMed] [Google Scholar]
- 138.Bonilla E, et al. New morphological approaches to the study of mitochondrial encephalomyopathies. Brain Pathol. 1992;2:113–119. doi: 10.1111/j.1750-3639.1992.tb00679.x. In this paper, a comprehensive survey is presented of morphological approaches used to diagnose and study mitochondrial diseases. [DOI] [PubMed] [Google Scholar]
- 139.Quinzii CM, et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010;24:3733–3743. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Loveland B, Wang CR, Yonekawa H, Hermel E, Lindahl KF. Maternally transmitted histocompatibility antigen of mice: a hydrophobic peptide of a mitochondrially encoded protein. Cell. 1990;60:971–980. doi: 10.1016/0092-8674(90)90345-f. [DOI] [PubMed] [Google Scholar]
- 141.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 142.Madan E, et al. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–957. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Nederveen FH, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tomlinson IP, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 145.Selak MA, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 146.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator — thinking outside the box. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 147.Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jain M, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases. Muscle Nerve. 2006;34:265–283. doi: 10.1002/mus.20598. [DOI] [PubMed] [Google Scholar]