

Abstract
Prenatal diagnosis of disorders due to mitochondrial DNA (mtDNA) tRNA gene mutations is problematic. Experience in families harboring the protein-coding ATPase 6 m.8993T>G mutation suggests that the mutant load is homogeneous in different tissues, thus allowing prenatal diagnosis. We have encountered a novel protein-coding gene mutation, m.10198C>T in MT-ND3. A baby girl homoplasmic for this mutation died at 3 months after severe psychomotor regression and respiratory arrest. The mother had no detectable mutation in accessible tissues. The product of a second pregnancy showed only wild-type mt genomes in amniocytes, chorionic villi, and biopsied fetal muscle. This second girl is now 18 months old and healthy. Our observations support the concept that the pathogenic mutation in this patient appeared de novo and that fetal muscle biopsy is a useful aide in prenatal diagnosis.
Keywords: mtDNA, complex I deficiency, prenatal diagnosis, mitochondria
One of the most frustrating problems—for patients and clinicians alike—in dealing with mitochondrial DNA (mtDNA)-related disorders is that of prenatal diagnosis. For diseases due to mutations in tRNA genes, which include some of the most severe clinical syndromes, such as MELAS and MERRF, prenatal diagnosis is perilous because the mutation load varies in different fetal tissues and is not necessarily reflected by the degree of heteroplasmy in amniocytes or chorionic villi. This is why efforts are underway to prevent altogether the transmission of maternal mtDNA through pro-nuclear transfer from a patient’s zygote to an enucleated normal oocyte.1 However, the situation appeared to be different for at least 1 mtDNA-related disorder, due to mutations in the gene encoding ATPase 6, namely, neuropathy, ataxia, retinitis pigmentosa/maternally inherited Leigh syndrome (NARP/MILS), because there is good evidence that the mutations causing this disease do not show tissue- or age-related variations.2,3 This was confirmed by preimplantation genetic diagnosis (PGD). Interestingly, blastomeres from in vitro–fertilized embryos were homoplasmic either for the mutant or the wild-type gene,4,5 consistent with the skewed segregation of the NARP mutation found in human oocytes.6
Another difficulty in prenatal diagnosis regards our ability to document de novo mutations in protein-coding mtDNA genes. A serendipitous event allowed us to show that a mutation in MT-ND3, which encodes subunit 3 of complex I, was, in fact, new in an affected infant. Prenatal diagnosis in her sister was greatly aided by fetal muscle biopsy.
Materials and Methods
Subjects
Patient 1
This baby girl with intrauterine growth retardation was born at 38 weeks of gestation by spontaneous vaginal delivery to healthy nonconsanguineous parents (Figure 1). Birth weight was 2319 g and Apgar scores were 5, 7, and 8 at 1, 5, and 10 minutes, respectively. Pregnancy had been complicated by preterm labor at 24 weeks, treated with bed-rest, terbutalin, and corticosteroids. At birth, the baby was noted to have a scaphoid abdomen and mild respiratory distress. Diaphragmatic hernia was diagnosed, which was repaired uneventfully at the sixth day of life. At 4 weeks, the parents noted arching, screaming, and limb stiffening, which was attributed to gastroesophageal reflux and was treated with ranitidine. She was also noted to have torticollis. At 7 weeks, she developed episodes of grunting, head turning, and eye rolling that lasted from 10 to 30 seconds and occurred every 2 to 3 days. At 8 weeks, Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain showed bilateral lesions of the basal ganglia and thalamus. Physical examination was normal and developmental milestones included smiling and tracking. Laboratory examinations showed increased blood lactate and pyruvate. Despite a normal electroencephalography (EEG), she was placed on phenobarbital and discharged home. By 11 weeks, the neurologic examination had become abnormal: she was awake and responded to tactile stimulation but did not cry. She stopped fixing and following and developed roving eye movements and nystagmus. After a respiratory arrest, she was intubated and transferred to the intensive care unit, where she died after a few days.
Figure 1.
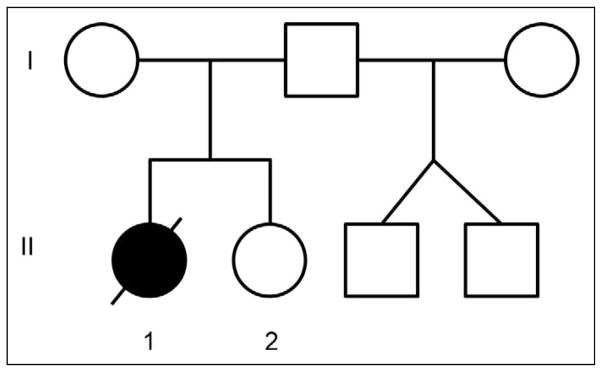
The pedigree shows the 2 biological sisters and the twin brothers born through heterologous artificial insemination.
Patient 2
The younger sister of patient 1 was born at term by cesarean section after an uncomplicated pregnancy. Her birth weight was 2633 g and her Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. She passed the newborn hearing screening and the results of the California Newborn Screening Panel were normal. She had normal growth and development. At 4 months, she weighed 6463 g, was 61 cm in length, and had a head circumference of 102 cm. She was cooing, reaching, and tracking. At her present age of 18 months, she walks alone, speaks in simple phrases, and is very social and responsive.
Tissues
From patient 1, we obtained a muscle biopsy at 11 weeks of age and postmortem specimens of skeletal muscle, cardiac muscle, liver, and brain. From patient 2, we obtained chorionic villus sampling at 11 weeks’ gestation, amniocytes (grown in culture by standard procedure and screened with 15 microsatellite markers to exclude maternal contamination) at 19 weeks’ gestation, cord blood, a portion of the placenta, and 20 hair follicles at 7 months of age. At 19 weeks’ gestation, a fetal muscle biopsy was performed without complication under direct ultrasound guidance.7 A 17-gauge introducer needle was inserted into the amniotic cavity, an 18-gauge biopsy needle was inserted, and 3 biopsies, each 2 to 3 mm long, were obtained from the fetal upper outer gluteal area. From the mother, we obtained venous blood, urinary sediment from first-morning-void urine, and 24 hair follicles. From the father, we obtained venous blood.
Muscle Histochemistry and Biochemistry
Histochemical studies of muscle using 8-μm-thick sections were carried out as described.8 Biochemical analysis was performed in 10% muscle extracts as described previously.9
Molecular Analysis
Total DNA was extracted from tissues using Puregene DNA Isolation Kit reagents (Qiagen Sciences, Valencia, California) according to the manufacturer’s recommended protocols. Direct sequencing of the ND genes was performed with a BigDye terminator cycle sequencing methodology on an ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, California). The m.10198C>T mutation was further investigated by PCR-RFLP analysis. The regions flanking the mutation site were PCR-amplified with forward primer 5′-ACCACAACTCAACGGCTACA-3′ and reverse primer 5′-TTGTAGGGCTCATGGTAGGG-3′. PCR conditions were 1 cycle at 94°C for 5 minutes, 30 cycles at 94°C for 1 minute, 68°C for 1 minute, 72°C for 1 minute, and 1 extension cycle at 72°C for 5 minutes. The resulting 169-bp fragment contains a unique restriction site for the endonuclease Cac81, which is absent in the mutant genomes. After digestion with Cac81, the normal pattern is 2 fragments of 101 and 68 bp, whereas in the presence of the mutation, the 169-bp fragment remains uncut. To assess the percentage of the mutation, we performed last cycle hot labeling10 and the digested product was electrophoresed in a 12% non-denaturing acrylamide gel and analyzed in a phospho-imager (Molecular Analyst, BioRad, Hercules, California) using Image-Quant software (Molecular Dynamics, Sunnyvale, California).
Microsatellite analysis on 15 STR loci (ABI, Foster City, California) was performed on DNA extracted from amniocytes and fetal muscle biopsy to verify the absence of contamination with maternal DNA.
Results
In patient 1, the muscle biopsy did not show any morphologic abnormalities: in particular, there were no ragged-red fibers (RRF), or cytochrome c oxidase (COX)–deficient fibers. However, biochemical analysis of the muscle extract showed decreased activity of complex I (27% of normal, referred to citrate synthase activity, data not shown). Direct sequencing of the mtDNA complex I genes showed a novel m.10198C>T mutation in the MT-ND3 gene. This mutation changes an alanine to a valine at amino acid position 47 (A47 V). PCR/RFLP analysis confirmed the presence of the mutation in the patient (Figure 1) and showed that it was homoplasmic in all tissues available (muscle biopsy [Figure 2], postmortem muscle, liver, heart, kidney, and brain). Analysis of accessible tissues from the mother failed to show the mutation in blood, urinary sediment, or in 24 individual hair samples (Figure 2). Similarly, no mutation was found in DNA extracted from the urinary sediment of the maternal grandmother. To reassure ourselves that we were not missing low levels of heteroplasmy in the tissues from the mother or from patient 2, we used 1 μL of digested PCR fragment as a template to amplify mutated mtDNA. Sequencing and digestion of the second PCR product failed to reveal any mutated fragment in these tissues.
Figure 2.
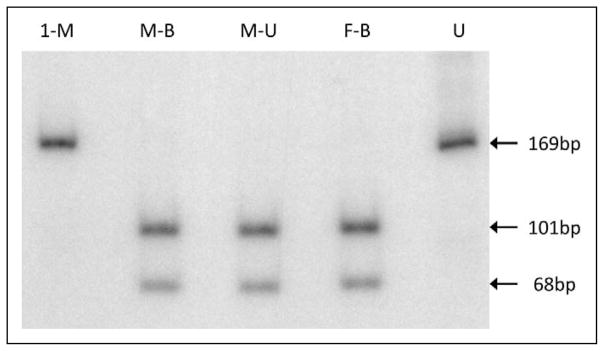
PCR/FFLP analysis of DNA from skeletal muscle of patient 1 (1-M), blood from the mother (M-B) and the father (F-B), and urine from the mother (M-U). U, uncut fragment.
In contrast to the molecular data in patient 1 and in agreement with those of the asymptomatic mother, the mutation was undetectable in patient 2, including amniocytes without maternal contamination, chorionic villus, cord blood, and—most importantly—a fetal muscle biopsy taken at 19 weeks of pregnancy (Figure 3). None of the 20 hair follicles examined had any detectable mutation. In keeping with these findings, patient 2 is a presently an 18-month-old healthy, developmentally normal, and thriving child. Although careful follow-up is justified, she has long surpassed the death age of her sister, and there is no clinical or genetic reason to suggest that she might deteriorate.
Figure 3.
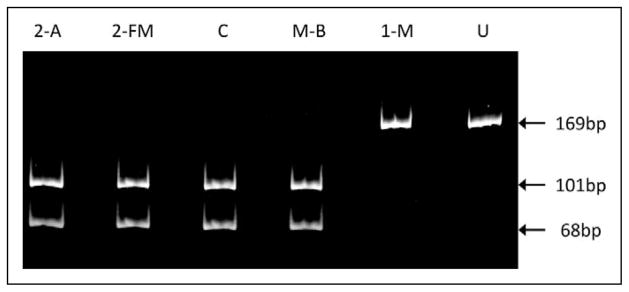
PCR/RFLP analysis of DNA extracted from patient 2’s amniocytes (2-A), fetal muscle (2-FM), control blood (C), mother’s blood (M-B), and patient 1’s skeletal muscle (1-M). U, uncut fragment.
Discussion
Despite the lack of maternal transmission and the noninformative muscle biopsy, the rapidly progressive infantile course of patient 1, her symmetrical basal ganglia lesions reminiscent of Leigh syndrome, and the biochemical data suggesting complex I deficiency, prompted us to sequence the 7 mtDNA genes encoding complex I (ND) proteins. We found a novel m.10198C>T mutation in the MT-ND3 gene, converting a highly conserved alanine to a valine at codon 47 (A47 V).
This mutation fits canonical criteria for mtDNA mutation pathogenicity. First, it segregated with disease severity, as it was homoplasmic in all tissues of the patient but undetectable in accessible tissues from the asymptomatic mother. Second, it was accompanied by an isolated defect of complex I activity in muscle (30% of normal with the NADH-CoQ assay). Third, it was not reported as a common polymorphic variant in the Mitomap database.11 Fourth, it affected an evolutionarily highly conserved site (Figure 4).
Figure 4.

Evolutionary comparison of the mutant site, showing remarkable preservation in multiple species.
Interestingly, the alanine that is converted to valine by this mutation (A47 V) is adjacent to the alanine converted to threonine (A47 T) by another pathogenic mutation (m10197G>A).12 Also interestingly, 4 of 5 pathogenic mutations reported thus far in MT-ND3 affect the same functionally important extramembrane loop between 2 transmembrane helices12 and only 1 did not.13
All 5 mutations caused remarkably similar clinical phenotypes in 14 of the 15 patients reported12–20: severe, early-onset rapidly progressive psychomotor deterioration, signs of brainstem or basal ganglia involvement, and symmetrical, bilateral neuroimaging features compatible with Leigh syndrome. Only 1 patient was a 42-year-old man with a progressive history of epilepsy, stroke-like episodes, optic atrophy, and dementia. Most patients had increased lactic acid levels in blood, CSF, or both. Characteristically, the muscle biopsy showed only mild, if any, mitochondrial changes without typical ragged-red fibers (RRF), contrasting with the severe complex I deficiency documented biochemically. Most children with Leigh-like syndrome died in infancy, often before 1 year of age. The mutation load in skeletal muscle varied between 83% and 100%. Although these mutations were often defined as “de novo,”13,15,19 small amounts of mutations were found in blood12,18 or fibroblasts15 of asymptomatic mothers, 1 of whom had a mutation load of 50% in blood (Sarzi and others 2007). Only 2 mothers were symptomatic and had substantial mutation loads in blood or muscle.12
The clinical features of our patient 1 coincided with those of most reported infants with severe Leigh-like syndrome. The parents were counseled extensively regarding the complex issues involved in prenatal diagnosis of an mtDNA mutation, and they opted to use donor oocytes: that second pregnancy resulted in healthy twin boys. However, when faced with a new and unplanned pregnancy, the parents decided to have the fetus evaluated for mutation load in amniocytes, chorionic villus, and fetal muscle biopsy, although they were well aware that no prenatal diagnosis could be offered. In agreement with the undetectable levels of the mutation in fetal tissues and in cord blood, the new infant at 18 months of age appears healthy and thriving.
The question of prenatal diagnosis in families harboring mtDNA mutations is thorny. It is generally accepted that tRNA mutations replicate stochastically in different tissues, such that a low mutation load in amniocytes or chorionic villus is no guarantee that other fetal tissues will have equally low levels of the mutation or that the mutation load may not increase during embryogenesis. However, experience with the m.8993T>G mutation associated with NARP/MILS has repeatedly shown that prenatal diagnosis is possible because the mutation load found in amniocytes or chorionic villi is similar in all fetal tissues and does not seem to drift substantially with time.2,3,21–23
We asked ourselves whether mutations in other mtDNA protein-coding genes might also be amenable to prenatal diagnosis. When the mother of patient 1 faced an unplanned spontaneous second pregnancy, we had a chance to prove that this was, in fact, the case. There are 2 precedents of prenatal diagnosis for the m10158T>C mutation (S34P) in ND3. In 2004, McFarland et al found 80% residual activity of complex I in chorionic villi and the baby was normal.15 In 2006, Lebon et al found normal complex I activity and no trace of the mutation in amniocytes and chorionic villi of a second pregnancy, and that infant was also normal.19
These findings suggest that mutations in MT-ND3 either occur de novo or are present in similar amounts in different accessible and nonaccessible tissues. One explanation could be that the germline bottleneck—irrespective of whether it happens during oogenesis or folliculogenesis24—in mothers carrying a very light mutation load favors the development of homoplasmic wild-type embryos and only occasionally does a mutation-carrying oocyte slip through the tight bottleneck. In practical terms, this experience suggests that prenatal diagnosis of the Leigh-like syndrome resulting from MT-ND3 mutation is feasible when mothers are clinically unaffected and harbor few or no mutations in accessible tissues. Sadly, this is possible only after 1 affected child is diagnosed with the mutation, as in the present family.
We also note that, in our case no accessible tissue from the mother contained even trace amounts of the mutation, thus strongly suggesting that this MT-ND3 mutation had in fact arisen de novo in maternal germ cells or during early embryogenesis. Even though the lack of mutation in amniocytes and chorionic villi from the second pregnancy suggested that this fetus was not affected, this conclusion was greatly reinforced by the lack of mutation in skeletal muscle obtained through a fetal biopsy at 19 weeks of gestation. This procedure proved safe and resulted only in a barely visible scar in the gluteal area.
We conclude that Leigh syndrome due to de novo MT-ND3 mutations is amenable to prenatal diagnosis and is aided by prenatal muscle biopsy.
Acknowledgments
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the National Institutes of Health (HD32062) and by the Marriott Mitochondrial Disorder Clinical Research Fund (MMDCRF).
Footnotes
Reprints and permission: sagepub.com/journalsPermissions.nav
Author Contributions
SS was originally contacted by this family and acted as a genetic counselor; AN directed our diagnostic laboratory when this work was conducted and he supervised the biochemical analysis and the molecular genetic studies of JL; RHC performed the fetal muscle biopsy; HOA perfected the molecular work documenting conclusively the de novo nature of the mutation; MM verified that the DNA extracted from amniocytes was not contaminated with maternal DNA; MH and SD led the research and wrote the paper.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The research described here was approved by the IRB Office of Columbia University Medical Center (CUMC).
References
- 1.Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White SL, Shanske S, McGill JJ, et al. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue- or age-related variation. J Inherit Metab Dis. 1999;22:899–914. doi: 10.1023/a:1005639407166. [DOI] [PubMed] [Google Scholar]
- 3.White SL, Shanske S, Biros I, et al. Two cases of prenatal analysis for the pathogenic T to G substitution at nucleotide 8993 in mitochondrial DNA. Prenat Diagn. 1999;19:1165–1168. [PubMed] [Google Scholar]
- 4.Steffann J, Frydman N, Gigarel N, et al. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244–247. doi: 10.1136/jmg.2005.032326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feyereisen E, Steffann J, Romana S, et al. Five years’ experience of preimplantation genetic diagnosis in a Parisian Center: outcome of the first 441 started cycles. Fertil Steril. 2007;87:60–73. doi: 10.1016/j.fertnstert.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 6.Blok RB, Gook DA, Thorburn DR, Dahl HH. Skewed segregation of the mtDNA nt 8993 (T>G) mutation in human oocytes. Am J Hum Genet. 1997;60:1495–1501. doi: 10.1086/515453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans MI, Hoffman EP, Cadrin C, Johnson MP, Quintero RA, Golbus MS. Fetal muscle biopsy: collaborative experience with varied indications. Obstet Gynecol. 1994;84:913–917. [PubMed] [Google Scholar]
- 8.Tanji K, Bonilla E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods Cell Biol. 2001;65:311–332. doi: 10.1016/s0091-679x(01)65019-2. [DOI] [PubMed] [Google Scholar]
- 9.DiMauro S, Servidei S, Zeviani M, et al. Cytochrome c oxidase deficiency in Leigh syndrome. Ann Neurol. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- 10.Moraes CT, Ciacci F, Bonilla E, Ionasescu V, Schon EA, DiMauro S. A mitochondrial tRNA anticodon swap associated with a muscle disease. Nat Genet. 1993;4:284–288. doi: 10.1038/ng0793-284. [DOI] [PubMed] [Google Scholar]
- 11.Mitomap. MITOMAP: a human mitochondrial genome database. 2003 http://www.mitomap.org.
- 12.Sarzi E, Brown MD, Lebon S, et al. A novel mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet. 2007;143A:33–41. doi: 10.1002/ajmg.a.31565. [DOI] [PubMed] [Google Scholar]
- 13.Leshinsky-Silver E, Lev D, Malinger G, et al. Leigh disease presenting in utero due to a novel missense mutation in the mitochondrial DNA-ND3. Mol Genet Metab. 2010;100:65–70. doi: 10.1016/j.ymgme.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Taylor RW, Singh-Kler R, Hayes CM, Smith PEM, Turnbull DM. Progressive mitochondrial disease resulting from a novel missense mutation in the mitochondrial DNA ND3 gene. Ann Neurol. 2001;50:104–107. doi: 10.1002/ana.1084. [DOI] [PubMed] [Google Scholar]
- 15.McFarland R, Kirby DM, Fowler KJ, et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Ann Neurol. 2004;55:58–64. doi: 10.1002/ana.10787. [DOI] [PubMed] [Google Scholar]
- 16.Bugiani M, Invernizzi F, Alberio S, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Crimi M, Papadimitriou A, Galbiati S, et al. A new mitochondrial DNA mutation in ND3 gene causing severe Legh syndrome with early lethality. Pediatr Res. 2004;55:842–846. doi: 10.1203/01.PDR.0000117844.73436.68. [DOI] [PubMed] [Google Scholar]
- 18.Leshinsky-Silver E, Lev D, Tzofi-Berman Z, et al. Fulminant neurological deterioration in a neonate with Leigh syndrome due to maternally transmitted missense mutation in the mitochondrial ND3 gene. Biochem Biophys Res Commun. 2005;334:582–587. doi: 10.1016/j.bbrc.2005.06.134. [DOI] [PubMed] [Google Scholar]
- 19.Lebon S, Chol M, Benit P, et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet. 2003;40:896–899. doi: 10.1136/jmg.40.12.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chae JH, Lee JS, Kim KJ, et al. A novel ND3 mitochondrial DNA mutation in three Korean children with basal ganglia lesions and complex I deficiency. Pediatr Res. 2007;61(5 pt 1):622–624. doi: 10.1203/pdr.0b013e3180459f2d. [DOI] [PubMed] [Google Scholar]
- 21.Ferlin T, Landrieu P, Rambaud C, et al. Segregation of the G8993 mutant mitochondrial DNA through generations and embryonic tissues in a family at risk of Leigh syndrome. J Pediatr. 1997;131:447–449. doi: 10.1016/s0022-3476(97)80074-1. [DOI] [PubMed] [Google Scholar]
- 22.Harding AE, Holt IJ, Sweeney MG, Brockington M, Davis MB. Prenatal diagnosis of mitochondrial DNA 8993 T>G disease. Am J Hum Genet. 1992;50:629–633. [PMC free article] [PubMed] [Google Scholar]
- 23.Leshinsky-Silver E, Perach M, Basilevsky E, et al. Prenatal exclusion of Leigh syndrome due to T8993C mutation in mitochondrial DNA. Prenat Diagn. 2003;23:31–33. doi: 10.1002/pd.516. [DOI] [PubMed] [Google Scholar]
- 24.Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2010;40:1484–1488. doi: 10.1038/ng.258. [DOI] [PubMed] [Google Scholar]