

Abstract
Autism spectrum disorder (ASD) consists of a group of complex developmental disabilities characterized by impaired social interactions, deficits in communication and repetitive behavior. Multiple lines of evidence implicate mitochondrial dysfunction in ASD. In postmortem BA21 temporal cortex, a region that exhibits synaptic pathology in ASD, we found that compared to controls, ASD patients exhibited altered protein levels of mitochondria respiratory chain protein complexes, decreased Complex I and IV activities, decreased mitochondrial antioxidant enzyme SOD2, and greater oxidative DNA damage. Mitochondrial membrane mass was higher in ASD brain, as indicated by higher protein levels of mitochondrial membrane proteins Tom20, Tim23 and porin. No differences were observed in either mitochondrial DNA or levels of the mitochondrial gene transcription factor TFAM or cofactor PGC1α, indicating that a mechanism other than alterations in mitochondrial genome or mitochondrial biogenesis underlies these mitochondrial abnormalities. We further identified higher levels of the mitochondrial fission proteins (Fis1 and Drp1) and decreased levels of the fusion proteins (Mfn1, Mfn2 and Opa1) in ASD patients, indicating altered mitochondrial dynamics in ASD brain. Many of these changes were evident in cortical pyramidal neurons, and were observed in ASD children but were less pronounced or absent in adult patients. Together, these findings provide evidence that mitochondrial function and intracellular redox status are compromised in pyramidal neurons in ASD brain and that mitochondrial dysfunction occurs during early childhood when ASD symptoms appear.
Keywords: Autism, Mitochondria, Temporal cortex, Development
Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental condition that encompasses a range of cognitive and behavioral phenotypes including autism, Asperger’s syndrome, and pervasive developmental disorder characterized by impaired social interaction, communication, and language, and repetitive and stereotyped behaviors (American Psychiatric Association, 1994). Pathological findings suggest that altered neurodevelopment during early postnatal life is crucial to ASD pathogenesis, and may result in excessive neurons and/or overgrowth of axons (Courchesne et al., 2007) and dendritic spines (Hutsler and Zhang, 2010). The etiology and biological basis of ASD pathology are unknown in most cases and both genetic predisposition and environmental factors are suggested to participate (Rossignol and Frye, 2012b).
Impaired mitochondrial function has been implicated in normal aging and neuropsychiatric disorders, including Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), mood disorder and schizophrenia (Lin and Beal, 2006; Manji et al., 2012; Schon and Manfredi, 2003; Trushina and McMurray, 2007). Mitochondria produce energy via oxidative phosphorylation (OXPHOS) through the respiratory chain, which generates more than 95% of neuronal ATP under normal physiological conditions (Benard et al., 2007). The respiratory chain involves electron transport chain (ETC.) proteins NADH-ubiquinone oxidoreductase (Complex I), succinate ubiquinone oxidoreductase (Complex II), ubiquinone cytochrome c oxidoreductase (Complex III), cytochrome c oxidase (Complex IV), and the ATP synthase (Complex V). Inefficient electron transfer through ETC. complexes causes brain pathology due to loss of energy, while defects of these enzymes, particularly Complexes I, II and III, cause the respiratory chain to leak electrons that react with oxygen to form toxic reactive radical species.
Recent evidence suggests that mitochondrial dysfunction could participate in the development and clinical features of ASD (Rossignol and Frye, 2012a). Studies have identified features associated with the biochemical endophenotype of mitochondrial energy deficiency, including abnormal plasma biomarkers that relate to mitochondrial dysfunction, such as plasma lactic acid, pyruvate, carnitine and amino acids (Weissman et al., 2008), and depressed ETC. complex function (Giulivi et al., 2010) with reduced mitochondrial membrane potential (James et al., 2009) in ASD lymphoblastoid cell lines. Complex I deficiency is the most common mitochondrial defect identified in ASD and has been found in association with Complex III and Complex IV deficiencies (Haas, 2010). Evidence for low pyruvate dehydrogenase complex (PDHC) activities, a higher rate of mitochondrial hydrogen peroxide production and mitochondrial DNA (mtDNA) overreplication and/or deletions, has been identified in a subset of ASD children (Giulivi et al., 2010).
In postmortem human brain, Chauhan et al. (2011) reported decreased expression of mitochondrial respiratory chain complexes in cerebellum, temporal lobe, and frontal lobe of ASD children. ASD patients showed significantly lower levels of Complexes III and V in the cerebellum, of Complex I in the frontal cortex, and of Complexes II, III and V in the temporal cortex. Anitha et al. (2012) identified downregulation of the expression of mitochondrial ETC. genes in anterior cingulate gyrus, motor cortex and thalamus of autism patients, compared to matched controls. More recently, oxidative damage to DNA (Rose et al., 2012) and proteins (Sajdel-Sulkowska et al., 2011) and inflammation have been found to be associated with low glutathione redox status (Rose et al., 2012) in cerebellum and temporal cortex of autism brain.
Here, we confirm findings of altered respiratory chain proteins in ASD brain, and identify novel features that further characterize abnormal mitochondrial function in ASD. To do so, we measured mitochondrial proteins in ASD brain in a larger cohort of postmortem brain tissue samples, analyzing BA21 in the lateral temporal lobe, a site involved in auditory processing, language and social perception implicated in ASD-associated behaviors (Bigler et al., 2007; Jou et al., 2010). In addition to confirming a decrease in protein expression and Complex I and IV enzyme activities in the temporal cortex from ASD cases, we identified decreased protein levels of the mitochondrial antioxidant enzyme SOD2 and increased oxidative mtDNA damage in ASD patients aged 2–9 years. We also identified increased mitochondrial mass in ASD brain, as indicated by increased protein levels of mitochondrial membrane proteins Tom20, Tim23 and porin. Altered mitochondrial dynamics were evidenced by increased mitochondrial fission proteins (Fis1 and Drp1) and decreased fusion proteins (Mfn1, Mfn2 and Opa1) in ASD patients. We did not identify any significant changes in mtDNA sequence, mtDNA copy number or levels of the mitochondrial gene transcription factor TFAM and cofactor PGC1α, suggesting that a mechanism other than an altered mitochondrial genome or gene expression underlies the mitochondrial abnormalities observed in ASD. Our findings provide further evidence for compromised mitochondrial function and intracellular redox status in ASD brain.
Methods and materials
Brain tissue
Frozen and fixed temporal lobe sections of ASD (n=20, age range 3–60 years) and control subjects (n=25, age range 2–65 years) were obtained from the Autism Tissue Portal, the Harvard Brain Bank and the NICHD Brain and Tissue Bank for developmental disorders at the University of Maryland. All frozen brain samples were kept at −80 °C and fixed brain sections in 10% formalin. Brain samples were divided into three age groups: 2–9 y, 13–20 y and 46–67 y. The case information and sample size within each age group in patients and controls were summarized in Table 1. Donors with autism fit the diagnostic criteria of DSM-IV and were confirmed by the Autism Diagnostic Interview—Revised. All controls are age-, PMI- and gender-matched subjects without known neurologic disease. The postmortem intervals did not exceed 32 h (ASD patients, range 3–32 h; controls, range 5–30 h). This study was approved by the Columbia University Medical School Institutional review board.
Table 1.
Demographic data for post mortem frozen brains.
| Variable | Children
|
Adolescence
|
Adults
|
||||
|---|---|---|---|---|---|---|---|
| Control (n=7) | Autism (n=8) | Control (n=9) | Autism (n=5) | Control (n=9) | Autism (n=7) | ||
| Age (yrs) | Range | 2–8.3 | 2–9 | 13.2–20 | 14.2–20 | 51–60 | 46–67.3 |
| Means±SD | 4.6±2.1 | 6±2.3 | 17.8±1.6 | 17.0±2.5 | 55.2±3.1 | 55.3±6.7 | |
| Postmortem interval (hs) | Range | 12–25 | 3–32 | 9–30 | 9–28 | 16–29 | 15–24 |
| Means±SD | 16.9±3.6 | 18.4±10.8 | 22.2±7.3 | 20.9±7.6 | 22.0±2.9c | 23.9±4.9 | |
| Gender | Male | 4 | 6 | 9 | 5 | 9 | 7 |
| Female | 3 | 2 | 0 | 0 | 0 | 0 | |
| Cause of death | Cancer | 0 | 2 | 0 | 0 | 3 | 2 |
| Cardiac | 0 | 0 | 1 | 0 | 4 | 3 | |
| Accident | 2 | 1 | 4 | 1 | 0 | 0 | |
| Asphyxiationa | 5 | 4 | 1 | 2 | 0 | 0 | |
| Seizure | 0 | 0 | 0 | 1 | 0 | 0 | |
| Otherb | 0 | 1 | 3 | 1 | 2 | 2 | |
| Length of storage (ms) | Range | 26.0–127.0 | 13.3–120 | 35.0–107.0 | 45.9–127.0 | N/A | 12.7–198.0 |
| Means±SD | 75.5±34.9 | 47.7±38.7 | 68.6±29.0 | 70.5±35.8 | N/A | 73.4±71.5 | |
| Seizure/epilepsy history (n) | 0 | 1 | 0 | 2 | 0 | 3 | |
Abbreviations: yrs, years; hs, hours; ms, months.
Includes drowning and choking on food/drugs.
Includes diabetes, pneumonica, natural, etc.
Compared to childhood controls, p<0.05.
Western blot analysis
Frozen brain tissue samples were homogenized in 1× RIPA buffer supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Sigma). The suspensions were centrifuged at 13,000 g at 4 °C for 30 min. Supernatants were collected and assayed for total protein using the Bradford method (BioRad). Samples were aliquoted into labeled vials and stored at −80 °C until use. Fifty microgram of total protein of each sample was mixed with NuPAGE sample buffer and separated in 4–12% NuPAGE Bis–Tris gel (Invitrogen) and transferred to Whatman PROTRAN Nitrocellulose Transfer Membranes (0.25 μm). For immunoblotting, membranes were washed with 1× Tris-buffered saline with 0.1% Tween 20 (TBS-T), blocked with 5% dry milk in 1× TBS-T at room temperature (RT) for 1 h. The membranes were incubated overnight with the primary antibodies for respiratory chain proteins Complex I NDUFS3 subunit (Mitoscience, MS110), Complex II 30KD subunit (Mitoscience, MS203), Complex III UQCR2 subunit (Mitoscience, MS304), Complex IV subunit COX1 (Mitoscience, MS404), COX2 (Mitoscience, MS405) and COX4 (Mitoscience, MS408), Complex V subunit α ATP5A (Mitoscience, MS507), fusion proteins Mfn1 (Santa Crutz, sc50330), Mfn2 (Abcam ab56889) and Opa1(BD Bioscience, BP612606), fission proteins Fis1 (Proteintech, 10586-1-AP) and Drp1(Santa Crutz, sc32898), mitochondria outer membrane proteins porin (Abcam, ab15895) and Tom20 (Proteintech, 11802-1-AP), inner membrane protein Tim23 (Proteintech, 11123-AP), matrix protein cytochrome c (Abcam, ab13575), mitochondrial gene transcription factors PGC1alpha (Abcam, ab54481) and TFAM (Abcam, ab119684), actin (Sigma, A5441), and catalase (Mitoscience, MS721). Following the primary antibody exposure, the membranes were washed with 1× TBS-T buffer three times at 10 min intervals, then incubated for 1 h at RT with appropriate secondary antibodies, followed by three additional washes at 10 min intervals. Protein bands were visualized using ECL or Supersignal Chemiluminescent reagents (Pierce). The densities of immunoreactive bands were quantified using ImageJ and presented as mean±SEM. A reference standard sample was used on each gel to normalize every other band on the same blot, and then compare across multiple blots because every band in the dataset is normalized to the same standard.
MitoChip assay
Samples were prepared for MitoChip analysis as previously described (van Eijsden et al., 2006). Briefly, long-range PCR was performed using three PCR primer sets that can amplify the entire mtDNA, using 100 ng of input DNA for each reaction. The cycling conditions for all reactions were: 1) 95 °C for 2 min; 2) 95 °C for 15 s; 3) 68 °C for 7 min; 4) repeat step 2 for 29 times; 5) final extension for 12 min. As a control for PCR amplification and subsequent hybridization, a 7.5-kb plasmid DNA (Tag IQ-EX template) was amplified concomitantly with the test samples, using forward and reverse primers included in the CustomSeq™ kit (Affymetrix). The procedures for sample pooling, DNA fragmentation, and labeling were identical for both first- and second-generation MitoChip assays. Pre-hybridization, hybridization, washing, and scanning of the MitoChip were performed as described in the Affymetrix CustomSeq™ resequencing protocol. The analysis of microarray data was performed using resequencing analysis (RA) Software (Genechip Sequencing Analysis Software Version 4.1.0, Affymetrix Inc., 2005).
mtDNA quantification
mtDNA content was assessed with the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems) in 100 ng total DNA extracted from autopsy samples. We measured mtDNA using the 12S ribosomal TaqMan mitochondrial assay labeled with 6FAM fluorochrome and the primers 5′-CCA CGG GAA ACA GCA GTG ATT-3′ and 5′-CTA TTG ACT TGG GTT AAT CGT GTG A-3′. The nuclear single copy gene RNAseP was measured with the kit PDARs RNAseP (Applied Biosystems) and a VIC labeled probe. We used calibration curves to quantitate the number of mtDNA versus nDNA copies.
Detection of mtDNA deletions
For the detection of mtDNA deletions, we used long-range PCR amplification with Takara LA Taq Kit (Fisher Scientific). A 7.305 kb fragment was amplified by PCR reaction in a thermal cycler (GenAmp PCR System 9700; Applied Biosystem) using a primer set (XBA 8286F 5′-CTC TAG AGC CCA CTG TAA AGC TAA CTT AGC-3′ and MBO 15591-B 5′ GGG ACG GAT CGG AGA ATT GTG TTA GGC G-3′). The conditions for the PCR reactions were: initial denaturation at 94 °C for 2 min, followed by 30 cycles: 15 s at 94 °C, 8 min at 68 °C and 13 min at 68 °C and 4 °C hold. Products were separated on 1% agarose gel with 0.01% ethidium bromide and visualized by ultraviolet light trans-illuminator.
Mitochondrial Complex I and Complex IV dipstick assay
Activities of Complexes I and IV were determined with the Mitochondrial Dipstick Assay kit, performed according to the manufacturer’s instructions (Abcam ab109876, ab109720). Fifteen micrograms of proteins was allowed to wick up laterally through the dipstick membrane, the dipsticks were transferred into the appropriate (Complex I or Complex IV) enzyme substrate buffer and enzyme activities were calculated by measuring the optical density of precipitating, colorimetric enzyme reaction products using the ImageJ® program. Amount of Complexes I and IV was quantified using MetaPath Mito Disease 4 Plex Dipstick Array (Abcam 109879) and presented by the optical density of corresponding protein band. All measurements were done in duplicates, and Complex I and IV catalytic activities were normalized to total protein amount and amount of Complex proteins, respectively.
Immunohistochemistry
Immunohistochemistry (IHC) was performed in children with ASD (n=8) and age-matched controls (n=7), according to general protocols. One cubic centimeter brain sections were fixed in 10% neutral buffered formalin and were embedded in paraffin blocks. Seven-micron thick consecutive sections were prepared, deparaffinized and rehydrated. Antigen retrieval was performed in a steamer for 40 min with Trilogy buffer (Cell Marque). Sections were then incubated with primary antibodies against mitochondrial membrane proteins porin (Abcam), mtCOX2 (Abcam), SOD2 (Abcam), fission proteins Fis1 (Proteintech) and Drp1 (Santa Crutz), fusion proteins Opa1 (BD Bioscience), Mfn1(Santa Crutz) and Mfn2 (Abcam) at 4 °C overnight, followed by an appropriate species of secondary antibodies conjugated to Alexa 488 (Invitrogen) for 1 h at room temperature. Negative control sections were held in PBS without primary antibody during the primary incubation. Background immunoreactivity with anti-rabbit and anti-mouse secondary antibodies alone was negligible.
8-OHdG immunostaining
Antigen retrieved brain sections were treated with proteinase K (20 μg/ml) at 37 °C for 45 min. The slides were blocked for endogenous peroxidase activity with peroxidase block (DAKO), washed 5 min in buffer, and incubated with serum free protein block (DAKO) for 1 h. The sections were incubated with anti-8-OHdG antibody (Abcam, 1:5000) at 37 °C for 1 h, followed by Alexa-488 conjugated secondary antibody at RT for 1 h. The images were acquired using Olympus BX-61 epifluorescent microscope and Image J software was used to quantify the staining intensity from 30 randomly selected pyramidal neurons per case.
Imaging
To image porin, COX2 and SOD2 positive mitochondria, we used a Leica Multiphoton imaging system with a 63× objective and zoom factors 2 (porin, SOD2) and 3 (COX2). The fluorescent intensity in the soma of individual neuron was quantified using ImageJ. For mitochondrial fusion and fission proteins as well as 8-OHdG staining, we used an Olympus BX 61 epifluorescent microscope equipped with Metamorph imaging software. Three fields (each field 438.6 μm by 330.2 μm) of layer V of temporal cortex were selected and imaged at 20× magnification. In each field, 20 neurons sectioned near their equator, based on a section plane that included the nucleolus, were selected and outlined manually. The fluorescent intensity was obtained for each neuronal soma, averaged and corrected for background by subtracting the fluorescent intensity of the negative control sections. All images were acquired under identical conditions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Normal distribution of the data was determined using the Kolmogorov–Smirnov test. Differences in the normalized intensity value among the ASD and control groups was performed with the non-parametric Kruskal–Wallis test followed by Dunn’s post-test, because some datasets are not normally distributed. Correlations were analyzed using Spearman’s correlation test and nonlinear regression. Results are expressed as mean±SEM. p<0.05 was considered statistically significant.
Results
Demographic variables in controls and patients
ASD is typically diagnosed at the age of 2–4 y, and a fraction of children outgrow severe disabilities by adolescence and recover following years of behavioral intervention (Helt et al., 2008). Based on this development trajectory, we examined brain samples from patients and controls at different ages: childhood (age 2–9 y), adolescence (age 13–20 y) and later adulthood (age 46–67 y), with sample age ranges largely determined by tissue availability. Demographic and clinical characteristics of patients and controls are detailed in Table 1. Within each age group, controls were matched with patients for age, post-mortem interval (PMI), gender and length of tissue storage. With the exception of PMI, which was different only between controls aged <10 y and controls>45 y (p=0.038), all pairwise comparisons of the factors on Table 1 were not different.
Mitochondrial respiratory chain protein levels in temporal lobe BA21 of ASD patients
Levels of mitochondrial respiratory chain proteins were measured in BA21 by Western blotting (Fig. 1A), using a panel of MitoSciences mouse monoclonal antibodies (see Methods and materials) that recognize subunits that are labile when the complexes are not assembled. The relative densities of the bands of different protein complexes were normalized to the level of β-actin, and the distribution of the data is plotted in Fig. 1B. We observed significantly lower levels of Complex I subunit NDUFS3 in ASD patients. When the samples were stratified by age, significantly lower levels of Complex I subunit NDUFS3 were only detected in the <10 yr group. There was no significant difference in Complex II 30KD subunit levels between ASD patients and controls at any age group. Levels of Complex III UQCR2 subunit decreased significantly in patients aged <10 y and >45 y. Protein levels of Complex IV were detected using antibodies against subunits 1 (COX1), 2 (COX2) and 4 (COX4): COX 1 level was significantly reduced in ASD patients aged <10 y but not in patients aged 10–20 y and >45 y, COX2 level was decreased in ASD patients in the two younger groups, while COX4 protein level remained the same between patients and controls among all age groups. Complex V subunit α ATP5A was significantly reduced in ASD patients in the age group<10 y, but not in the other groups.
Fig. 1.

Mitochondrial respiratory chain protein levels in BA21 ASD postmortem brain. (A) Representative images for Western blots of respiratory chain protein Complexes I–V in ASD cases and age-matched controls. C, control; A, ASD; R, a protein sample of temporal lobe brain tissue used as a reference for comparison of band intensities of samples loaded in different gels; (B) scatterplots of the relative density of the bands of different respiratory chain complexes in ASD and control brains, normalized to actin. Compared to age-, PMI-matched controls, *p<0.05, **p<0.01.
In summary, we observed significant decreases in protein levels of Complex I, III, IV, and V subunits in ASD brain: the mean level of these subunits was lower in ASD patients than in controls in <10 y age group, while Complex III was also reduced in patients >45 y. Complex II was not significantly different in ASD patients. These results indicate a decreased expression of mitochondrial respiratory chain complexes in ASD brains, which implies a defect in mitochondrial oxidative phosphorylation.
Western blot analysis of mitochondrial mass
To determine whether the decreased levels of respiratory chain protein subunits resulted from a loss of mitochondria, we measured overall mitochondrial mass in ASD brain by immunoblot against the mitochondria outer membrane proteins porin, Tom20, mitochondrial inner membrane protein Tim23, and intermembrane mitochondria protein cytochrome c (Arthur et al., 2009; Brinckmann et al., 2010). Contrary to our expectations (Fig. 2), the protein levels of porin, Tom20 and Tim23 were significantly higher in ASD brain in the age group<10 y, whereas the levels of cytochrome c were comparable in ASD and control samples. We observed significantly higher levels of Tom20 and Tim23 in ASD children than in control children, while in the group>45 y, Tom20 was higher in control than in ASD cases.
Fig. 2.

Levels of mitochondrial membrane proteins Tom20, Porin and Tim23 and intermembrane space protein cytochrome c (Cyto c) in BA21 ASD postmortem brain. (A) Representative images for Western blots of Tom20, Tim23, porin and Cyto c in ASD patients and age-matched controls. C, control; A, ASD, R, reference sample; (B) scatterplots for Tom20, Tim23 and porin relative intensity in ASD patients and controls, normalized to actin. Compared to age-, PMI-matched controls, *p<0.05, **p<0.01.
Mitochondrial DNA analysis in temporal lobe of young ASD patients
As we observed a decrease in protein levels of Complex IV subunits COX1 and COX2, which are encoded by mitochondrial DNA (mtDNA), we examined the sequence and amount of brain mtDNA in childhood cases, including 8 ASD children and 7 controls. Aside from a synonymous mutation (m.7064T>C), MitoChip sequence analysis (Affymetrix GeneChip® Human Mitochondrial Resequencing Array) showed that the ASD patients possessed only known polymorphisms (G5460A, G5178A, G9055A, G6150A, T6253C). Long-range PCR amplification ruled out the presence of large-scale deletions and duplications, even in small amounts (Fig. 3A). Real-time PCR analysis moreover excluded changes in mtDNA quantity (Fig. 3B). There is thus no evidence for changes in mitochondrial DNA sequences or copy numbers that are abnormal in these ASD patients, although the DNA exhibits higher levels of oxidative damage (see below).
Fig. 3.

mt DNA analysis in BA21 temporal lobe of young ASD patients. (A) Agarose gel electrophoresis shows no deletion in ASD patients. M, HindIII digested Lambda DNA marker, C (+) positive control for mtDNA deletion, C (−) normal control; 1 to 8, ASD patients. (B) Mitochondrial DNA copy number, represented as the number of mitochondria DNA versus nuclear DNA.
Levels of PGC1α or TFAM proteins in temporal lobe of ASD brain
Changes in mitochondria biogenesis might underlie decreased ETC. levels or increased mitochondrial content in ASD. Proteins that regulate mitochondrial biogenesis and activate mitochondria gene expression include PGC1α and TFAM (Kim et al., 2010). In neurodegenerative disorders including HD and PD, the levels of both proteins are decreased, implicating them in the pathogenesis (Taherzadeh-Fard et al., 2011; Tsunemi and La Spada, 2012). In contrast, we found no significant difference in the levels of PGC1α or TFAM between ASD and control samples (Fig. 4), suggesting that an alternative mechanism leads to mitochondrial abnormalities in ASD temporal cortex. However, there appears to be an age-related decrease in PGC1α between young and adolescent groups in both control and ASD.
Fig. 4.
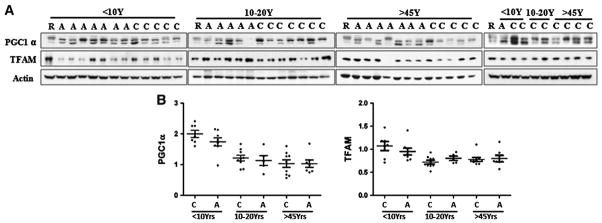
Levels of mitochondrial gene transcription factor TFAM and cofactor PGC1α in BA21 ASD postmortem brain. (A) Representative images for Western blots of TFAM and PGC1α in ASD patients and age-matched controls. C control; A, ASD, R, reference sample; (B) scatterplots for the relative density of the bands of PGC1α and TFAM, normalized to actin. No significant difference in levels of PGC1α and TFAM was found between ASD and controls in any age group.
Mitochondrial Complex I/IV activities in young ASD temporal lobe
When normalized to the amount of total protein, dipstick assay shows decreased Complex I and Complex IV activities in ASD patient brain (Fig. 5A). By quantifying the amount of Complexes I and IV, we found a significant decrease in the quantity of Complexes I and IV in ASD brain (Fig. 5B), consistent with our results determined by Western blot (Fig. 1). However, while normalized to the quantity of Complexes I and IV, no differences in Complex I and IV enzyme activities per Complex I and IV protein complex units were detected between ASD and controls (Fig. 5C). These results imply that the decrease in Complex I and Complex IV activities is associated with a decrease in the quantity of these two complexes.
Fig. 5.

Mitochondrial Complex I and IV enzyme activities in BA21 temporal lobe of young ASD patients. (A) Representative images for dispstick assay of Complex I and IV enzyme activities, quantity of Complex I and IV multisubunit complexes. Note that the antibodies stripped to the dipstick nitrocellulose membrane are capable of capturing the multisubunit complex in its native form when the sample is extracted with the detergent included in the kits. (B) Decreased mitochondrial Complex I and IV enzyme activities in ASD temporal lobe. The enzyme activities were normalized to the amount of total proteins and represented as Complex I and IV enzyme activities per μg total protein. Compared to age-, PMI-matched controls, **p<0.01; (C) decreased mitochondrial Complex I and IV quantities in ASD temporal lobe. The amount of complexes was presented by the optical density of Complex I and Complex IV bands. Compared to age-, PMI-matched controls, **p<0.01; (D) Complex I and IV enzyme activities normalized to the amount of complexes. No differences were detected in enzyme activity per Complex I or IV unit between ASD and controls.
Analysis of antioxidant enzymes catalase and mitochondria SOD2
Mitochondria are both the major source of reactive oxygen species (ROS) production and the major target of ROS damage. In mitochondria, superoxide is converted to the less reactive metabolite, hydrogen peroxide, by the mitochondrial matrix enzyme manganese superoxide dismutase (MnSOD, SOD2) (Weydert and Cullen, 2010). Hydrogen peroxide levels are in turn regulated by glutathione peroxidase and by peroxisomal catalase activity. Remaining hydrogen peroxide can react with iron to produce highly reactive hydroxyl radicals. We analyzed the protein levels of the two antioxidant enzymes, catalase and mitochondrial superoxide dismutase SOD2 (Fig. 6A & B). While catalase was not changed in the temporal lobe of ASD patients at any age, SOD2 was significantly lower in ASD children, but no significant difference in SOD2 protein levels was detected between adolescent and adult ASD patients.
Fig. 6.
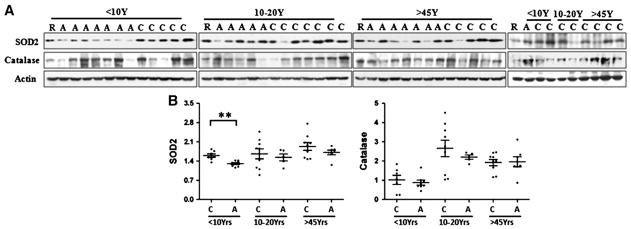
Levels of antioxidant enzyme SOD2 and catalase in BA21 ASD postmortem brain. (A) Representative images for Western blots of SOD2 and catalase in ASD cases and age-matched controls. C, control; A, ASD, R, reference sample; (B) scatterplots for the relative density of the bands of SOD2 and catalase, normalized to actin. Compared to age-, PMI-matched controls, **p<0.01.
8-OHG hydroxyguanosine immunolabel
The low level of SOD2 protein in childhood ASD temporal cortex suggests that there may be oxidative damage to DNA. Oxidatively modified nucleoside 8-hydroxy-2′-deoxyguanosine (8-OHdG) is widely used as a biomarker of oxidative DNA damage (de Souza-Pinto et al., 2008). We determined 8-OHdG levels in temporal cortex of ASD children and control subjects. We observed a low level of the 8-OHdG label in the nucleolus and cytoplasm of pyramidal neurons in controls, which may reflect basal DNA oxidation due to postmortem fixation. In contrast, there was intense cytoplasmic staining in pyramidal neurons of ASD patients (Fig. 7A & B), suggesting that these primary projection neurons are particularly sensitive to oxidative DNA damage. Negative controls incubated in secondary antibody alone were unlabeled (not shown). There was no correlation between 8-OHdG signal and PMI or other factors in tissue handling or cause of mortality (Fig. 7C and see below). Thus, the data revealed oxidative mtDNA damage in pyramidal neurons in temporal cortex of young ASD patients.
Fig. 7.

Levels of 8-OHdG in BA21 ASD postmortem brain. (A) Representative images for immunohistochemistry of 8-OHdG in temporal lobe layer V pyramidal neurons. Pyramidal neurons were identified by a characteristic triangular morphology with a prominent apical dendrite pointing toward pial surface. Samples from 3 controls (C1, 2 yrs; C2, 4 yrs; C3, 5 yrs) and 3 ASD patients (A1, 3 yrs; A2: 3 yrs; A3, 7 yrs) were presented. Pyramidal neurons from boxed areas in C2 and A1 were shown in CP and AP. Scale bar, 20 μm. (B) Quantification of fluorescent intensity of cytoplasmic 8-OHdG per pyramidal neuron. Control, n=7; ASD, n=8. **, compared to controls, p<0.01. (C) Correlation analysis and non-linear regression of 8-OHdG fluorescent intensity with confounding factors (r, Spearman’s correlation coefficient; p, significance p value; R2, coefficient of determination).
An increase in compromised mitochondria in pyramidal neurons
Our findings of decreased mitochondrial ETC. proteins and increased oxidative stress together with increased mitochondria mass in the temporal cortex of ASD children suggested that there might be an increase in compromised mitochondria in ASD brain. To address this, we immunolabeled COX2 and SOD2 to visualize functional mitochondria and porin (Arthur et al., 2009; Keeney et al., 2006) to evaluate mitochondrial mass in fixed brain sections from the ASD and control children. Consistent with the results from Western blot, we observed a significantly lower COX2 and SOD2 fluorescence and significantly higher porin immunofluorescence in pyramidal neurons in ASD temporal cortex (Fig. 8 A & B). We normalized protein levels of respiratory chain complexes and SOD2 to total mitochondrial protein content indicated by the level of Tom20 (Fig. 8C). The protein level per mitochondrial protein unit was reduced by 40% for Complex I, 22% for Complex II, 45% for Complex III, 30% for Complex IV, 45% for Complex V, and 43% in SOD2, consistent with decreased functional proteins in mitochondria of ASD pyramidal neurons. We observed similar results when normalizing levels of these functional proteins to porin protein (not shown). Together, these data suggest that compromised mitochondria accumulate in pyramidal neurons in temporal cortex of younger ASD subjects.
Fig. 8.

An increase in damaged mitochondria in BA21 ASD postmortem brain. Layer V pyramidal neurons (10–20 neurons per case) were immunolabeled for porin, SOD2, and COX2. Scale bar, 10 μm. (A) Representative images for immunohistochemistry of porin, SOD2, COX2 in temporal lobe layer V pyramidal neurons. (B) Quantification of porin, SOD2 and COX2 immunofluorescent intensity in pyramidal neurons in temporal lobe layer V from ASD patients and controls. Compared to control subjects, ASD patients showed a decrease in SOD2, COX2 intensity, but an increase in porin fluorescence. Control, n=7; ASD, n=8. Compared to controls, *p<0.05, **p<0.01. (C) Protein levels of mitochondria respiratory chain proteins and SOD2, normalized to the level of mitochondrial outer membrane protein Tom20. Control, n=7; ASD, n=8. Compared to controls, *p<0.05, **p<0.01.
Analysis of mitochondrial fission and fusion proteins in ASD temporal cortex
Mitochondrial dynamics are regulated by the fission proteins Drp1, and Fis1 and fusion proteins Mfn1, Mfn2 and Opa1 (Detmer and Chan, 2007), and are associated with mitochondrial morphology and size, distribution, function, and turnover. Impaired mitochondrial dynamics are increasingly implicated in neurodegenerative disorders including PD, HD and AD (Chen and Chan, 2009). Our results revealed significant differences in the levels of Drp1, Fis1, Mfn1/2 and Opa1 between ASD and controls (Fig. 9A & B): Drp1 level was increased by 14% in ASD cases<10 y and 18% in patients over 45. Fis1 levels were significantly increased by 15% in ASD patients aged <10 y. In contrast, the levels of fusion proteins were lower by 25% for Mfn1, 14% for Mfn2 and 14% for Opa1 in ASD patients under 10 y. Consistent with the changes revealed by immunoblot analysis, lower immunoreactivity in pyramidal neurons was noted for Mfn1 and Opa1 whereas immunolabel for Drp1 and Fis1 was higher in ASD patients under 10 y. In summary, mitochondrial fission proteins are elevated and fusion proteins decreased in ASD children (Fig. 9C).
Fig. 9.

Mitochondrial fission and fusion protein expression in BA21 ASD postmortem brain tissue. Representative immunoblot (A) and quantification (B) revealed that in ASD temporal lobe, levels of fission protein Drp1 and Fis1 were significantly higher, while levels of fusion proteins Mfn1, Mfn2 and Opa1 were significantly lower in young ASD cases. Compared with age-matched controls, *p<0.05, **p<0.01. The greatest differences in protein levels of Mfn1/2, Opa1, Drp1 and Fis1 were in cases under the age 10, with 86–100% of patients showing lower/higher levels than the lower/higher range for controls. (C) Representative images for immunohistochemistry of Mfn1/2, Opa1, Drp1 and Fis1 in temporal lobe layer V pyramidal neurons in childhood ASD patients and controls. Compared to controls, ASD neurons displayed a decrease in Mfn1/2, Opa1 intensity, but an increase in Fis1 and Drp1 fluorescence. Control, n=7; ASD, n=8. Scale bar, 50 μm.
Correlation analysis of mitochondrial protein levels with confounding factors
Comparison of protein expression in postmortem brain tissue is complicated by numerous confounding factors, including age, gender, postmortem interval, cause of death and other variables. Thus, we performed correlation analysis between these factors and the levels of mitochondrial proteins. As shown in Table 2, there was no significant correlation between the levels of mitochondrial respiratory chain proteins and seizure history, gender or cause of death. The length of storage slightly affected the levels of Complex I protein (coefficient of determination R2=0.029) and Fis1 (R2=0.0003). Seizure history was significantly correlated with protein levels of fusion proteins Mfn2 (R2=0.032) and Opa1 (R2=0.067); the lower Mfn2 and Opa1 levels in childhood ASD brains remained significant even after the single case with seizure history was removed. For adolescent and adult groups, the significantly lower Mfn1, Mfn2 and Opa1 may be related to seizure history, as more patients manifested seizure symptoms, although it should be noted that seizures are very common in ASD patients (Tuchman and Rapin, 2002). The protein level of Complex IV COX1 subunit increased with PMI, while levels of Complex V, Mfn2 and Opa1 were negatively correlated with PMI (R2=0.111, 0.089, 0.017, respectively). The levels of most mitochondrial proteins fell slightly with age (R2: Complex IV COX4, 0.146; Complex V, 0.111; cytochrome c, 0.101; PGC1α, 0.072; TFAM, 0.019; Mfn1, 0.026; Mfn2, 0.089; Opa1, 0.061), while SOD2 (R2: 0.027) and catalase (R2: 0.062) increased with age. In summary, there are changes with aging in mitochondrial proteins that appear to be common to all individuals. However, the changes in mitochondrial proteins we found between ASD patients and controls were not the result of differences in PMI, cause of death, gender or storage time in any age group.
Table 2.
Correlation between expression levels of mitochondrial proteins and confounding factors.
| Storage time | Seizure history | Age at death | PMI | Gender | Cause of death | ||
|---|---|---|---|---|---|---|---|
| Complex I | r (p) | −0.350 (0.044) | 0.015 (0.922) | −0.082 (0.594) | 0.120 (0.435) | −0.229 (0.411) | −0.200 (0.193) |
| Complex II | r (p) | −0.002 (0.993) | 0.150 (0.322) | −0.110 (0.480) | −0.160 (0.305) | −0.131 (0.641) | 0.220 (0.153) |
| Complex III | r (p) | −0.023 (0.899) | 0.076 (0.622) | −0.006 (0.967) | 0.098 (0.527) | −0.197 (0.483) | 0.023 (0.883) |
| Complex IV (COX1) | r (p) | −0.240 (0.163) | 0.038 (0.806) | 0.170 (0.256) | 0.430 (0.003) | −0.164 (0.560) | −0.120 (0.430) |
| Complex IV (COX2) | r (p) | −0.170 (0.349) | 0.096 (0.532) | −0.077 (0.615) | −0.140 (0.352) | −0.098 (0.727) | −0.038 (0.803) |
| Complex IV (COX4) | r (p) | −0.110 (0.549) | 0.280 (0.061) | −0.390 (0.007) | −0.270 (0.071) | 0.229 (0.411) | 0.180 (0.242) |
| Complex V | r (p) | −0.006 (0.975) | 0.150 (0.330) | −0.470 (0.001) | −0.370 (0.014) | −0.066 (0.817) | 0.110 (0.473) |
| Tom20 | r (p) | 0.079 (0.656) | 0.058 (0.705) | −0.077 (0.615) | 0.069 (0.658) | 0.131 (0.641) | 0.110 (0.491) |
| Tim23 | r (p) | 0.065 (0.714) | −0.190 (0.220) | 0.260 (0.086) | 0.190 (0.216) | 0.247 (0.375) | 0.093 (0.542) |
| Porin | r (p) | −0.130 (0.474) | 0.073 (0.634) | −0.250 (0.099) | 0.210 (0.180) | 0.148 (0.599) | −0.150 (0.331) |
| Cyto c | r (p) | 0.150 (0.397) | 0.120 (0.449) | −0.490 (0.001) | −0.180 (0.255) | 0.196 (0.483) | −0.034 (0.824) |
| SOD2 | r (p) | −0.140 (0.417) | 0.038 (0.806) | 0.420 (0.004) | 0.120 (0.435) | −0.147 (0.600) | −0.029 (0.848) |
| Catalase | r (p) | −0.076 (0.668) | −0.190 (0.208) | 0.450 (0.002) | 0.130 (0.394) | −0.229 (0.411) | 0.097 (0.528) |
| PGC1α | r (p) | −0.150 (0.396) | 0.073 (0.634) | −0.610 (0.001) | −0.180 (0.236) | −0.263 (0.344) | −0.210 (0.169) |
| TFAM | r (p) | −0.260 (0.130) | 0.078 (0.610) | −0.370 (0.013) | 0.180 (0.242) | 0.410 (0.129) | −0.254 (0.09) |
| Drp1 | r (p) | 0.062 (0.728) | −0.150 (0.322) | 0.074 (0.631) | 0.260 (0.084) | 0.000 (1.000) | 0.061 (0.689) |
| Fis1 | r (p) | 0.047 (0.000) | 0.130 (0.410) | −0.007 (0.962) | 0.100 (0.501) | 0.246 (0.377) | −0.075 (0.623) |
| Mfn1 | r (p) | −0.210 (0.237) | 0.270 (0.071) | −0.350 (0.020) | −0.079 (0.611) | −0.164 (0.560) | 0.190 (0.212) |
| Mfn2 | r (p) | −0.120 (0.503) | 0.350 (0.017) | −0.630 (0.000) | −0.400 (0.008) | −0.181 (0.520) | 0.067 (0.663) |
| Opa1 | r (p) | −0.110 (0.529) | 0.420 (0.004) | −0.440 (0.002) | −0.410 (0.006) | 0.033 (0.908) | 0.160 (0.282) |
r, Spearman’s correlation coefficient; p, significance p value.
Discussion
This study demonstrates mitochondrial defects within the temporal cortex of ASD children by demonstrating changes in proteins affecting mitochondrial OXPHOS, Complex I and IV enzyme activities, antioxidant defense, dynamics and content. Perhaps most importantly, the data indicate that many of the differences between ASD patients and control subjects occur predominantly in childhood, when ASD symptoms are initially expressed, and that several of these features are present in cortical pyramidal neurons, the primary neuron of the cortex. We examined BA21 cortex, which is widely implicated in ASD as it participates in brain networks involved in social-communicative processes including language (Gillberg and Coleman, 2000), social and speech perception (Redcay, 2008), and intention and comprehension (Allison et al., 2000), all of which are impaired in ASD (American Psychiatric Association, 1994). Abnormalities in ASD temporal cortex have been confirmed by functional imaging and pathological studies, including disturbed gene transcription profiles (Garbett et al., 2008; Voineagu et al., 2011), increased dendritic spine densities in excitatory pyramidal neurons (Hutsler and Zhang, 2010), and reduced functional specificity (Shih et al., 2011). Our identification of alterations in the mitochondrial respiratory chain protein levels and enzyme activities, mitochondrial antioxidant enzyme SOD2, mitochondrial fission and fusion proteins, and localized oxidative stress in pyramidal neurons in temporal cortex of ASD children indicates mitochondria dysfunction in this cortical region and may represent a pathological correlate of ASD behavioral deficits.
Mitochondrial defects and oxidative stress in temporal lobe of ASD brain
In postmortem brain tissue from a smaller cohort of patients and controls, Chauhan et al. (2011) identified a significant reduction in respiratory chain protein expression in ASD, with lower levels of Complexes II, III and IV in temporal cortex of ASD children. We also find that levels of each respiratory chain protein except Complex II were decreased in ASD children aged <10 y, and further that level of Complex IV COX2 subunit was decreased in ASD patients aged 10–20 y, whereas patients>45 y showed significantly low levels of Complex III. The most severe respiratory chain damage was apparent in the ASD children, as patients<10 y showed lower levels of Complex I, III, IV, and V proteins and decreased Complex I and IV enzyme activities. Severe respiratory chain abnormalities at a younger age may result in mitochondria dysfunction and abnormal energy metabolism and contribute to the onset of ASD symptoms: nevertheless, <30% of adult patients also demonstrated decreased levels of mitochondrial ETC. complexes, and so it may be that mitochondrial abnormalities occur throughout life for some patients.
ETC. proteins are encoded both by mtDNA genes and by nuclear DNA (ntDNA) genes (DiMauro and Schon, 2008) and so the pattern of biochemical changes in the brain of ASD children could be due to mutations in mtDNA or decreased mtDNA abundance (mtDNA depletion) (Pons et al., 2004). However, our detailed molecular analysis of mtDNA in the brain of eight ASD children ruled out direct mtDNA involvement in these patients. The ETC. abnormalities may rather be secondary to environmental stressors, including aberrant Ca++ signaling, alterations in immune function, toxic compounds, or oxidative stress (James et al., 2009).
Respiratory chain complex defects compromise ATP synthesis and accelerate the generation of free radicals including superoxide anions, hydrogen peroxide, and hydroxyl radicals that cause oxidative damage to proteins and DNA (Henchcliffe and Beal, 2008). Consistently, we observed extensive staining of oxidatively modified mtDNA in pyramidal neurons in temporal cortex from young ASD patients. These results support the conclusion of Chauhan et al. (2011), who found that the level of lipid hydroperoxide (LOOH), a product of fatty acid oxidation, was significantly increased in the cerebellum and temporal cortex in patients aged 4–10 years but not in those aged 14–39 years.
Several changes in ASD tissue, such as increased peroxidation (Rossignol and Frye, 2012a) and membrane abnormalities (Chauhan and Chauhan, 2006) are consistent with effects of reactive oxygen species (ROS). Antioxidant enzymes work in combination to protect cells from ROS damage (Andersen, 2004). We found that the level of the mitochondrial antioxidant enzyme SOD2 was significantly lower in the temporal lobe of ASD patients under the age of 10 y, suggesting that oxidative stress could be specific to ASD children. Our results are consistent with Meguid et al., who found low SOD protein level in plasma of ASD patients (Meguid et al., 2011), and Yorbik et al., who reported that SOD activity is lower in ASD blood than in controls (Yorbik et al., 2002). We did not however observe any change in catalase protein level in ASD patients at any age, suggesting that the oxidative stress may originate from mitochondrial rather than peroxisomal dysfunction. A diminished capacity to counteract ROS renders mitochondria more vulnerable to oxidative damage, which can lead to a vicious cycle of increased ROS generation (James et al., 2009).
A cumulative respiratory chain deficiency and oxidative DNA damage over time may impair mitochondrial function, and could influence the onset or severity of ASD and co-morbid symptoms. Patients may develop compensatory mechanisms that sustain adequate energy production despite chronic mitochondrial respiratory chain deficiency and oxidative stress (Celotto et al., 2011), through increases in glycolytic flux, ketogenesis and Kreb’s cycle activity, increases in mtDNA copy numbers to maintain normal levels of mitochondrial transcripts (Cannon et al., 2011), or through age-related increases in DNA repair (Humphreys et al., 2007). A higher mitochondrial maximal respiratory rate reported for ASD patients has been suggested to provide a compensatory response to the depression in complex I function (Benzecry et al., 2009; Holtzman, 2008). Such compensatory mechanisms may explain why mitochondrial dysfunction was less severe in older ASD cases.
Disturbed mitochondrial homeostasis in temporal lobe of ASD brain
Our results indicate that mitochondrial mass, as measured by levels of mitochondrial membrane proteins porin, Tom20, and Tim23, is increased in primary neurons of ASD temporal cortex. This effect on mitochondria mass could be caused by an imbalance between mitochondrial biogenesis and degradation. Mitochondria biogenesis is regulated by the crosstalk between nuclear and mitochondrial genomes and is coordinated by nuclear co-activators including PGC1α and the nuclear respiratory factors NRF1 and NRF2 (Finck and Kelly, 2006). PGC1α co-activates NRF1 and NRF2, which in turn coordinate the expression of mitochondria transcription factor A (TFAM), thus increasing the expression of mtDNA. Additional nuclear receptors, including PPARα, PPARγ, thyroid hormone receptors, retinoid receptors and glucocorticoid receptors, regulate mitochondria biogenesis through effects on PGC1α (Kelly and Scarpulla, 2004). Levels of PGC1α and TFAM are decreased in neurodegenerative conditions that have been associated with mitochondria dysfunction and oxidative stress (Ekstrand et al., 2007; Finck and Kelly, 2006; Kelly and Scarpulla, 2004). In contrast to reports in these neurodegenerative disorders, however, we did not find any increase in mitochondrial transcription factors in ASD, and so the accumulation of mitochondria and the decrease in ETC./SOD2 protein levels may not occur at the transcriptional level.
Mitochondria degradation provides an alternative mechanism for mitochondrial quality control and is mediated by molecular chaperones that can selectively remove excess and damaged proteins from mitochondrial outer membrane (Luce et al., 2010). Injury of mitochondria can impair fusion, and as a stress response, it can activate fission-dependent fragmentation that helps to promote sequestration by a degradation of mitochondria by macroautophagy, a process known as mitophagy (Twig et al., 2008). Mitophagy targets damaged mitochondria or subsets of mitochondria producing excessive reactive oxygen species to autophagosomes for removal via ubiquitin-mediated recognition and selective targeting (Kraft et al., 2010). Numerous ubiquitin genes have been implicated in ASD pathogenesis (Glessner et al., 2009). One of these genes, PARK2, encodes parkin, a cytosolic protein that moves into uncoupled mitochondria to assist in their destruction (Scheuerle and Wilson, 2011). Defects in parkin-mediated mitophagy have been implicated in both PD and AD (Khandelwal et al., 2011; Narendra et al., 2008). We have identified an increase in compromised mitochondria in ASD temporal cortex, which is unlikely to be due to altered levels of gene transcription. These findings suggest that mitophagy may be impaired in our ASD tissue. Alternatively, a decrease in mitochondrial respiratory chain proteins and SOD2 could be caused by mitochondrial protein importing defects (Pfanner et al., 2004; Yamamoto et al., 2011). The increase in Tom20 protein level however suggests that there may be an enhanced import for the mitochondrial protein presequences. We further detected increased Tim23 protein level, although it could be that translocation of other proteins by the Tim23 complex may be impaired in a membrane potential dependent manner (Pfanner et al., 2004).
Our data demonstrate significant changes in the expression of mitochondrial fission and fusion proteins in temporal cortex in ASD, which could lead to changes in the morphology, number and function of mitochondria (Han et al., 2011; Knott and Bossy-Wetzel, 2008). The reduced levels of fusion proteins Mfn1/2 and Opa1 and increased levels of fission proteins Fis1 and Drp1 are expected to cause fragmentation of mitochondria and perinuclear accumulation of mitochondria, which might leave more distal areas of neurons (dendrites and axons) devoid of the organelles (Wang et al., 2009). While increased fission via overexpression of fission proteins Fis1 and Drp1 isolates compromised segments of mitochondria and would normally promote mitophagy (Twig et al., 2008), this process requires a constitutively active basal level of macroautophagy (Gilkerson et al., 2012) that may be disturbed in the disorder.
Implication of mitochondrial dysfunction in ASD synaptic pathology
The current postmortem study was performed in patients who were already diagnosed with ASD, and does not distinguish between causes and consequences. Altered mitochondrial protein levels and the increase in compromised mitochondria could be secondary to other metabolic or genetic abnormalities identified in autism, e.g., environmental triggers such as toxin and metabolites derivates, seizures, medications (Rossignol and Frye, 2012a), or primary carnitine deficiency identified in some nonsyndromic autism cases (Celestino-Soper et al., 2012). However, the resulting mitochondrial dysfunction could be detrimental to synapse formation, as trafficking of mitochondria into dendritic protrusions correlates with the development and morphological plasticity of spines (Li et al., 2004). Such mitochondrial dysfunction could further lead to reduced synaptic neurotransmitter release (Anderson et al., 2008; Whittaker et al., 2011), which has been implicated in the etiology of ASD and hyperexcitability of neocortical circuits (Chao et al., 2010). The collapse of mitochondria membrane potential may further impair the clearance of intracellular calcium in presynaptic terminals, which can interfere with synaptic function and structure (Billups and Forsythe, 2002; Verstreken et al., 2005). If present during critical developmental periods, mitochondrial dysfunction could promote synaptic calcium deregulation and disruption of normal neuronal networks that have been postulated to underlie cognitive attention deficits in ASD. Consistently, we observed a significant increase in compromised mitochondria and oxidative stress in patients aged 2–10, the critical developmental window during which improper synaptogenesis develops in ASD, particularly in temporal cortex, a region with an altered gene transcription profile (Garbett et al., 2008; Voineagu et al., 2011) and higher than normal dendritic spine density in pyramidal neurons.
While treatment with mitochondrial cofactor supplementation, including antioxidants, co-enzyme Q10, carnitine and B-vitamins may improve mitochondrial function and behavior in some children with ASD (Weissman et al., 2008), systematic studies documenting the efficacy of these treatments for mitochondrial dysfunction in children with ASD are lacking. Mitochondrial abnormalities have been identified in MECP2 mutant (Grosser et al., 2012; Kriaucionis et al., 2006), TSC mutant (Goto et al., 2011) ASD mouse and Drosophila models (Yao et al., 2011). Further characterization of the mechanisms underlying mitochondrial dysfunction in these model systems may lead to an increased understanding of ASD brain pathology and provide therapeutic directions.
Acknowledgments
Funding
This study was supported by the Simons Foundation. Additional support for the Sulzer laboratory is from the Parkinson’s Disease and JPB Foundations. The DiMauro laboratory is funded by NICHD grant HD32062 and the Marriott Mitochondrial Disorder Clinical Research Fund (MMDCRF).
We are grateful to Dr. Daniel Geshwind for his critical review of the manuscript. We also thank the Autism Tissue Portal, Harvard Brain Bank and Maryland NICHD Brain & Tissue Bank for kindly providing us brain tissues for the present study.
Footnotes
Conflict of interest statement
None.
References
- Allison T, et al. Social perception from visual cues: role of the STS region. Trends Cogn Sci. 2000;4:267–278. doi: 10.1016/s1364-6613(00)01501-1. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Press; Washington, D.C: 1994. Fourth Edition (DSM-IV) [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10:S18–S25. doi: 10.1038/nrn1434. (Suppl) [DOI] [PubMed] [Google Scholar]
- Anderson M, et al. Bridging from cells to cognition in autism pathophysiology: biological pathways to defective brain function and plasticity. Am J Biochem Biotechnol. 2008;4:167–176. [Google Scholar]
- Anitha M, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2012 doi: 10.1111/bpa.12002. http://dx.doi.org/10.1111/bpa.12002. [DOI] [PMC free article] [PubMed]
- Arthur CR, Morton SL, Dunham LD, Keeney PM, Bennett JPJ. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener. 2009;4:37. doi: 10.1186/1750-1326-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, et al. Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- Benzecry JM, et al. Are autistic spectrum disorders an expression of mitochondrial encephalopathies? Mitochondrion. 2009;9:62. [Google Scholar]
- Bigler ED, et al. Superior temporal gyrus, language function, and autism. Dev Neuropsychol. 2007;31:217–238. doi: 10.1080/87565640701190841. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckmann A, et al. Regionalized pathology correlates with augmentation of mtDNA copy numbers in a patient with myoclonic epilepsy with ragged-red fibers (MERRF-syndrome) PLoS One. 2010;5:e13513. doi: 10.1371/journal.pone.0013513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon MV, et al. Xenomitochondrial mice: investigation into mitochondrial compensatory mechanisms. Mitochondrion. 2011;11:33–39. doi: 10.1016/j.mito.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celestino-Soper PB, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci. 2012;109:7974–7981. doi: 10.1073/pnas.1120210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celotto AM, et al. Modes of metabolic compensation during mitochondrial disease using the Drosophila model of ATP6 dysfunction. PLoS One. 2011;6:e25823. doi: 10.1371/journal.pone.0025823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13:171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Chauhan A, et al. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem. 2011;117:209–220. doi: 10.1111/j.1471-4159.2011.07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, et al. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, et al. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair. 2008;7:1098–1109. doi: 10.1016/j.dnarep.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett K, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30:303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillberg C, Coleman M. The Biology of Autistic Syndromes. 3. Cambridge University Press; London: 2000. [Google Scholar]
- Giulivi C, et al. Mitochondrial dysfunction in autism. JAMA. 2010;304:2389–2396. doi: 10.1001/jama.2010.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto J, et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci. 2011;108:E1070–E1079. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser E, et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol Dis. 2012;48:102–114. doi: 10.1016/j.nbd.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Haas RH. Autism and mitochondrial disease. Dev Disabil Res Rev. 2010;16:144–153. doi: 10.1002/ddrr.112. [DOI] [PubMed] [Google Scholar]
- Han XJ, et al. Regulation of mitochondrial dynamics and neurodegenerative diseases. Acta Med Okayama. 2011;65:1–10. doi: 10.18926/AMO/43824. [DOI] [PubMed] [Google Scholar]
- Helt M, et al. Can children with autism recover? If so, how? Neuropsychol Rev. 2008;18:339–366. doi: 10.1007/s11065-008-9075-9. [DOI] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Holtzman D. Autistic spectrum disorders and mitochondrial encephalopathies. Acta Paediatr. 2008;97:859–860. doi: 10.1111/j.1651-2227.2008.00883.x. [DOI] [PubMed] [Google Scholar]
- Humphreys V, et al. Age-related increases in DNA repair and antioxidant protection: a comparison of the Boyd Orr Cohort of elderly subjects with a younger population sample. Age Ageing. 2007;36:521–526. doi: 10.1093/ageing/afm107. [DOI] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94. doi: 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- James SJ, et al. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009;23:2374–2383. doi: 10.1096/fj.08-128926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jou RJ, et al. Enlarged right superior temporal gyrus in children and adolescents with autism. Brain Res. 2010;1360:205–212. doi: 10.1016/j.brainres.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney PM, et al. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Khandelwal PJ, et al. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet. 2011;20:2091–2102. doi: 10.1093/hmg/ddr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet. 2010;19:3919–3935. doi: 10.1093/hmg/ddq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283–292. doi: 10.1196/annals.1427.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, et al. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, et al. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Luce K, et al. Mitochondrial protein quality control systems in aging and disease. Adv Exp Med Biol. 2010;694:108–125. doi: 10.1007/978-1-4419-7002-2_9. [DOI] [PubMed] [Google Scholar]
- Manji H, et al. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- Meguid NA, et al. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biol Trace Elem Res. 2011;143:58–65. doi: 10.1007/s12011-010-8840-9. [DOI] [PubMed] [Google Scholar]
- Narendra D, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N, et al. Assembling the mitochondrial outer membrane. Nat Struct Mol Biol. 2004;11:1044–1048. doi: 10.1038/nsmb852. [DOI] [PubMed] [Google Scholar]
- Pons R, et al. Mitochondrial DNA abnormalities and autistic spectrum disorders. J Pediatr. 2004;144:81–85. doi: 10.1016/j.jpeds.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Redcay E. The superior temporal sulcus performs a common function for social and speech perception: implications for the emergence of autism. Neurosci Biobehav Rev. 2008;32:123–142. doi: 10.1016/j.neubiorev.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Rose S, et al. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry. 2012;2:e134. doi: 10.1038/tp.2012.61. http://dx.doi.org/10.1038/tp.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012a;17:290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012b;17:389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdel-Sulkowska EM, et al. Brain region-specific changes in oxidative stress and neurotrophin levels in autism spectrum disorders (ASD) Cerebellum. 2011;10:43–48. doi: 10.1007/s12311-010-0223-4. [DOI] [PubMed] [Google Scholar]
- Scheuerle A, Wilson K. PARK2 copy number aberrations in two children presenting with autism spectrum disorder: further support of an association and possible evidence for a new microdeletion/microduplication syndrome. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:413–420. doi: 10.1002/ajmg.b.31176. [DOI] [PubMed] [Google Scholar]
- Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111:303–312. doi: 10.1172/JCI17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih P, et al. Functional differentiation of posterior superior temporal sulcus in autism: a functional connectivity magnetic resonance imaging study. Biol Psychiatry. 2011;70:270–277. doi: 10.1016/j.biopsych.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, et al. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol Neurodegener. 2011;6:32. doi: 10.1186/1750-1326-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, La Spada AR. PGC-1alpha at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol. 2012;97:142–151. doi: 10.1016/j.pneurobio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurol. 2002;1:352–358. doi: 10.1016/s1474-4422(02)00160-6. [DOI] [PubMed] [Google Scholar]
- Twig G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eijsden RG, et al. Chip-based mtDNA mutation screening enables fast and reliable genetic diagnosis of OXPHOS patients. Genet Med. 2006;8:620–627. doi: 10.1097/01.gim.0000237782.94878.05. [DOI] [PubMed] [Google Scholar]
- Verstreken P, et al. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JR, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS One. 2008;3:e3815. doi: 10.1371/journal.pone.0003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5:51–66. doi: 10.1038/nprot.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RG, et al. Impaired mitochondrial function abolishes gamma oscillations in the hippocampus through an effect on fast-spiking interneurons. Brain. 2011;134:e180–e181. doi: 10.1093/brain/awr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, et al. Dual role of the receptor Tom20 in specificity and efficiency of protein import into mitochondria. Proc Natl Acad Sci U S A. 2011;108:91–96. doi: 10.1073/pnas.1014918108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao A, et al. Drosophila FMRP regulates microtubule network formation and axonal transport of mitochondria. Hum Mol Genet. 2011;20:51–63. doi: 10.1093/hmg/ddq431. [DOI] [PubMed] [Google Scholar]
- Yorbik O, et al. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot Essent Fat Acids. 2002;67:341–343. doi: 10.1054/plef.2002.0439. [DOI] [PubMed] [Google Scholar]