

Abstract
Mitochondrial diseases involve the respiratory chain, which is under the dual control of nuclear and mitochondrial DNA (mtDNA). The complexity of mitochondrial genetics provides one explanation for the clinical heterogeneity of mitochondrial diseases, but our understanding of disease pathogenesis remains limited. Classification of Mendelian mitochondrial encephalomyopathies has been laborious, but whole-exome sequencing studies have revealed unexpected molecular aetiologies for both typical and atypical mitochondrial disease phenotypes. Mendelian mitochondrial defects can affect five components of mitochondrial biology: subunits of respiratory chain complexes (direct hits); mitochondrial assembly proteins; mtDNA translation; phospholipid composition of the inner mitochondrial membrane; or mitochondrial dynamics. A sixth category—defects of mtDNA maintenance—combines features of Mendelian and mitochondrial genetics. Genetic defects in mitochondrial dynamics are especially important in neurology as they cause optic atrophy, hereditary spastic paraplegia, and Charcot–Marie–Tooth disease. Therapy is inadequate and mostly palliative, but promising new avenues are being identified. Here, we review current knowledge on the genetics and pathogenesis of the six categories of mitochondrial disorders outlined above, focusing on their salient clinical manifestations and highlighting novel clinical entities. An outline of diagnostic clues for the various forms of mitochondrial disease, as well as potential therapeutic strategies, is also discussed.
Introduction
Over the past 50 years, since the first description of a patient with bona fide mitochondrial disease,1 extraordinary progress has been made—and continues—in the field of mitochondrial neurology. Although initially studied mainly by neuromuscular disease specialists, mitochondrial diseases were soon recognized to be more than just myopathies and, given the frequent involvement of the brain in these disorders, they are often termed mitochondrial encephalomyopathies.2 Strictly speaking, mitochondrial diseases encompass defects in any of the multiple metabolic pathways that are contained within the mitochondrion (Figure 1). In this Review, however, we focus on defects that involve the mitochondrial respiratory chain—the ‘business end’ of energy metabolism and site of oxidative phosphorylation (OXPHOS), where most cellular ATP is generated. The convention of considering mitochondrial encephalomyopathies as defects of the mitochondrial respiratory chain is justified given the biochemical complexity of the terminal mitochondrial bioenergetic pathway, its unique dual genetic control (with mitochondrial DNA [mtDNA] and nuclear DNA [nDNA] working in concert; Box 1), and the extraordinary clinical and genetic heterogeneity of the diseases related to respiratory chain dysfunction. We focus on the salient clinical manifestations of the various defects of mtDNA and nDNA, highlighting novel clinical entities of practical importance (that is, described in more than a single patient) and conceptually notable observations. The diagnostic ‘threads’ that can lead neurologists towards a correct diagnosis are outlined, together with a discussion of the potential therapeutic options for the various mitochondrial diseases. With further research and the use of novel technologies, currently unsuspected pathogenic underpinnings of mitochondrial disorders should be revealed, hopefully leading to improved therapeutic options for patients with these devastating disorders.
Figure 1.

The mitochondrial respiratory chain. Electrons derived from cellular dehydrogenases in the Krebs cycle and in β-oxidation spirals are passed ‘horizontally’ along four protein complexes and two small carriers (the electron transport chain) that are embedded in the MIM. The electrons travel from complex I (an NADH dehydrogenase) and complex II (a succinate dehydrogenase) to CoQ10 (a small mobile electron carrier also known as ubiquinone), then to complex III (ubiquinone oxidoreductase), cytochrome c (another small mobile electron carrier) and complex IV (cytochrome c oxidase), ultimately producing water. Concomitant with this horizontal flow of electrons, ‘vertical’ vectorial transport of dehydrogenase-derived protons from the matrix across the MIM into the intermembrane space takes place. This process creates an electrochemical proton gradient across the MIM that is used to drive complex V (F0F1-ATP synthase), a rotary motor that converts ADP to ATP. Conventionally, the five complexes comprise the oxidative phosphorylation system. The biosynthetic pathway of CoQ10, beginning with acetyl-CoA, is shown on the right; mutated biosynthetic genes that cause deficiency are show in bold text. Abbreviations: ANT, adenine nucleotide translocator; CACT, carnitine–primary CoQ10 acylcarnitine translocator; CoA, coenzyme A; CoQ10, coenzyme Q10; CPT, carnitine palmitoyltransferase; DIC, dicarboxylate carrier; ETF, electron-transfer flavoprotein; ETF-DH, ETF-dehydrogenase; FAD, flavin adenine dinucleotide; IMS, intermembrane space; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; PDHC, pyruvate dehydrogenase complex; TCA, tricarboxylic acid. Image first printed in Molecular Neurology.155
Box 1. Mitochondrial DNA.
The concept of endosymbiosis was proposed at the beginning of the 20th century,2 but was popularized in 1967 by Lynn Sagan Margulis.154 Endosymbiosis occurred about two billion years ago, when early eukaryotic cells were invaded by bacteria that had adapted to an increasingly oxygen-rich atmosphere and were on the path to becoming the permanent endosymbionts that we call mitochondria. A corollary of endosymbiosis is that all eukaryotic cells still contain, in addition to their original nuclear DNA (nDNA), a genetic ‘relic’ of the prokaryotic invaders, namely mitochondrial DNA (mtDNA). In the course of evolution, mtDNA has lost much of its autonomy—most of its genes have actually been transferred to the nuclear genome—and has essentially become the ‘slave’ of nDNA. Human mtDNA is a 16,569-bp circular, double-stranded molecule that contains 37 genes encoding two ribosomal RNAs, 22 transfer RNAs, and 13 polypeptides (all subunits of the respiratory chain complexes). Human mtDNA contains no introns and the genes are closely apposed, with some showing partial overlap. Some mRNAs have no termination codons as they are created post-transcriptionally by polyadenylation—a nucleus-encoded function. Of the approximately 85 subunits of the respiratory chain, only 13 are encoded by mtDNA: seven subunits of complex I (ND1–ND6); one subunit of complex III (cytochrome b); three subunits of complex IV (COX I, COX II and COX III); and two subunits of complex V (ATPase 6 and ATPase 8). Complex II is entirely encoded by nDNA and is, therefore, a good marker of mitochondrial abundance.
Disease classification
At the molecular level, mitochondrial encephalomyopathies are broadly classified as either those due to mutations in mtDNA, which cause maternally inherited or sporadic disorders, or disorders due to mutations in nDNA, which show a Mendelian inheritance pattern. A less obvious but more practical way of categorizing these heterogeneous disorders is to divide them into three groups: mtDNA defects; nDNA defects that directly or indirectly affect the respiratory chain; and defects of mtDNA maintenance (impairment of intergenomic communication). Separate classification of defects of mtDNA maintenance is useful, as these disorders directly involve the mitochondrial genome, often manifesting as multiple mtDNA deletions or mtDNA depletion. Furthermore, although unequivocally Mendelian with regard to inheritance, disorders that involve defects of mtDNA maintenance share much of the clinical heterogeneity of primary mtDNA-related diseases because the polyploid mtDNA is involved in both groups. Below, in order to organize a heterogeneous group of mitochondrial disorders, we outline what is known about disorders associated with mtDNA defects, describe defects of mtDNA maintenance separately, and divide nDNA-associated disorders into five major groups on the basis of functionally distinct molecular defects: mutations in genes encoding subunits of the respiratory chain (direct hits); mutations in genes encoding ancillary—usually assembly—proteins (indirect hits); mutations in genes affecting mtDNA translation; mutations in genes controlling the phospholipid composition of the mitochondrial inner membrane (MIM); and mutations in genes involved in mitochondrial dynamics.
Disorders of mtDNA defects
The molecular era of mitochondrial encephalomyopathies began in 1988 with the identification of the first mtDNA mutations.3,4 Since then, over 260 pathogenic mutations and 120 large-scale rearrangements (single mtDNA deletions) have been identified in the mtDNA molecule (Figure 2), together with many more putatively nonpathogenic, ‘neutral’ polymorphisms.5 The abundance of mtDNA point mutations is often attributed to the proximity of the ‘naked’ (that is, histone-free) mtDNA to the respiratory chain and its byproducts, namely reactive oxygen species (ROS), as well as to the low repair capacity of mtDNA. This view is rather simplistic, however, as mtDNA is not naked but is packaged within DNA–protein assemblies (nucleoids),6 and has robust DNA base-excision repair mechanisms.7 Most primary mtDNA single deletions occur in regions flanked by short repeated sequences,8 and can develop during repair of damaged mtDNA.9 The high rate of mtDNA repair and mutation leads to a correspondingly high prevalence of mtDNA-related diseases. In the north east of England, for example, one in 10,000 people are clinically affected by mtDNA-related disorders, and one in 6,000 individuals are considered at risk.10 Screening of umbilical cord blood from newborns for the 10 most common pathogenic mtDNA point mutations reveals that around one in 200 infants harbours a mutation in these genes.11 A supersensitive next-generation sequencing approach has revealed low-level variations (0.2–2.0% heteroplasmy [the coexistence of mutated and wild-type mtDNA variant within a cell]) in blood and skeletal muscle of clinically unaffected individuals (a phenomenon termed ‘universal heteroplasmy’).12 The possibility exists, therefore, that silent hereditary mtDNA mutations could expand over time and manifest as seemingly de novo mitochondrial disease.
Figure 2.

The mitochondrial morbidity map. Schematic map of the 16,569-bp mtDNA, in which coloured sections represent protein-coding genes: seven subunits of complex I (ND; pink sections); one subunit of complex III (cyt b; light blue section); three subunits of cytochrome c oxidase (CO; purple sections); two subunits of ATP synthase (A6 and A8; yellow sections): 12 S and 16 S ribosomal RNA (green sections); and 22 transfer RNAs identified by three-letter codes for the corresponding amino acids (blue sections). Diseases due to mutations in genes that impair protein synthesis are indicated as blue circles. Mutated genes that encode respiratory chain proteins are indicated as pink circles. Numbers in circles represent number of mutations reported at the given site. Abbreviations: Cyt b, cytochrome b; FBSN, familial bilateral striatal necrosis; LHON, Leber hereditary optic neuropathy; LS, Leigh syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonus epilepsy with ragged-red fibres; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia and retinitis pigmentosa; ND, NADH-dehydrogenase (complex I); PEO, progressive external ophthalmoplegia.
Lumping or splitting?
From the beginning of the molecular era,13 controversy prevailed between ‘lumpers’, who considered mtDNA-related diseases as a clinical ‘swamp’ of heterogeneous multisystemic disorders, and ‘splitters’, who identified and assigned acronyms to several well-defined syndromes, including Kearns–Sayre syndrome (KSS); chronic progressive external ophthalmoplegia (CPEO); Pearson syndrome (PS); mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS); myoclonus epilepsy with ragged-red fibres (MERRF); Leber hereditary optic neuropathy (LHON); neuropathy, ataxia and retinitis pigmentosa (NARP); and maternally inherited Leigh syndrome (MILS). 25 years later, the debate of ‘lumping’ versus ‘splitting’ remains unresolved: although many multisystem disorders do not fit any known syndromic definition and many overlapping syndromes have been described, the validity of the above defined syndromes has been largely confirmed, and each one has been associated with one or a few predominant mtDNA mutations.5,14
The signs and symptoms that characterize six exemplary mtDNA-related diseases have been identified (Table 1). Two of these diseases, KSS and PS, are due to large-scale deletions. Another two, MELAS and MERRF, are caused by transfer RNA (tRNA) mutations that globally affect mitochondrial protein synthesis, whereas the remaining two diseases, NARP and MILS, are attributable to mutations in a protein-coding gene. The complexity of mitochondrial genetics provides some explanation for the clinical heterogeneity of diseases due to mutations in these genes. For example, the degree of heteroplasmy of the same mutation in the gene encoding ATPase 6 (m.8993T>G) is clearly related to the severity and age at onset of NARP, a disorder of young adults that shows ~70% mutation load, and MILS, a devastating neurodegenerative disease with onset in infancy or early childhood in which the mutation load is >90%.15
Table 1.
Signs and symptoms of six key mitochondrial diseases
| Tissue or factor | Sign or symptom | Δ-mtDNA-associated disease
|
tRNA-associated disease
|
ATPase 6-associated disease
|
|||
|---|---|---|---|---|---|---|---|
| KSS | Pearson | MERRF | MELAS | NARP | MILS | ||
| CNS | Seizures | − | − | + | + | − | + |
| Ataxia | + | − | + | + | + | +/− | |
| Myoclonus | − | − | + | +/− | − | − | |
| Psychomotor retardation | − | − | − | − | − | + | |
| Psychomotor regression | + | − | +/− | + | − | + | |
| Hemiparesis/hemianopia | − | − | − | + | − | − | |
| Cortical blindness | − | − | − | + | − | − | |
| Migraine-like headaches | − | − | − | + | − | − | |
| Dystonia | − | − | − | + | − | + | |
|
| |||||||
| PNS | Peripheral neuropathy | +/− | − | +/− | +/− | + | − |
|
| |||||||
| Muscle | Weakness | + | − | + | + | + | + |
| Ophthalmoplegia | + | +/− | − | +/− | − | − | |
| Ptosis | + | − | − | +/− | − | − | |
|
| |||||||
| Eye | Pigmentory retinopathy | + | − | − | − | + | +/− |
| Optic atrophy | − | − | − | − | +/− | +/− | |
| Cataracts | − | − | − | − | − | − | |
|
| |||||||
| Blood | Sideroblastic anaemia | +/− | + | − | − | − | − |
|
| |||||||
| Endocrine | Diabetes mellitus | +/− | − | − | +/− | − | − |
| Short stature | + | − | + | + | − | − | |
| Hypoparathyroidism | +/− | − | − | − | − | − | |
|
| |||||||
| Heart | Conduction block | + | − | − | +/− | − | − |
| Cardiomyopathy | +/− | − | − | +/− | − | +/− | |
|
| |||||||
| Gastrointestinal | Exocrine pancreatic dysfunction | +/− | + | − | − | − | − |
| Intestinal pseudo-obstruction | − | − | − | − | − | − | |
|
| |||||||
| ENT | Sensorineural hearing loss | − | − | + | + | +/− | − |
|
| |||||||
| Kidney | Fanconi syndrome | +/− | +/− | − | +/− | − | − |
|
| |||||||
| Laboratory testing | Lactic acidosis | + | + | + | + | − | +/− |
| Muscle biopsy (ragged-red bres) | + | +/− | + | + | − | − | |
|
| |||||||
| Inheritance | Maternal | − | +/− | + | +/− | + | + |
| Sporadic | + | − | − | − | − | − | |
Abbreviations: ENT, ear, nose and throat; KSS, Kearns–Sayre syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonus epilepsy with ragged-red fibres; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia and retinitis pigmentosa.
Pathogenesis: terra incognita
One conundrum that lies at the heart of mitochondrial genetics is how mutations in tRNA can cause syndromes as diverse as MELAS and MERRF (Table 1). Unless different tRNAs—such as tRNALeu(UUR) in MELAS and tRNALys in MERRF—have subtly different functions,16 one would expect pathogenic mutations in any tRNA to impair protein synthesis and ATP production to a similar extent. Even more puzzling for neurologists is the tendency for different mtDNA mutations to manifest in certain brain areas or structures, such as the subpial arterioles in MELAS, the choroid plexus in KSS, and the olivocerebellar pathway in MERRF.17,18
Immunohistochemical studies of these structures to compare the abundance of mtDNA-encoded versus nDNA-encoded respiratory chain subunits support the concept that affected brain areas harbour high loads of the relevant mutation.19 Such selective tissue vulnerability is well illustrated in studies of the optic nerve.20 The axons of the retinal ganglion cells of the inner retina converge to become the optic nerve, a structure that is unmyelinated before crossing the lamina cribrosa, and heavily myelinated thereafter. Prelaminar axons are highly dependent on OXPHOS energy, probably for signal transmission and to sustain electrical potential, whereas the myelinated post-laminar axons exhibit much less energetically ‘costly’ saltatory conduction, except at the nodes of Ranvier, which are rich in mitochondria. This asymmetric distribution of mitochondria and energy demand probably explains the particular vulnerability of the retinal ganglion cells and their unmyelinated neurons to LHON-associated mutations in complex I genes.20
One major obstacle to functional studies of primary mtDNA mutations is the lack of animal models, in part owing to the difficulty of introducing mutant mtDNA into mitochondria of mammalian cells in a heritable fashion. An alternative approach has been to use cybrid cells—established human cell lines that are ‘emptied’ of their own mtDNA then repopulated with exogenous mitochondria harbouring various proportions of mutant genomes.5,21 This technique has been used to largely confirm the hypothesis that single deletions and pathogenic point mutations in mtDNA impair respiration and protein synthesis, and decrease ATP production.22 For all major mutations, the threshold required to cause disease, as assessed in vitro, seem to be both high and steep: for the m.3243A>G mutation that is associated with MELAS, the threshold is around 90%.23 These data cannot be extrapolated to the in vivo situation, however, as demonstrated in oligosymptomatic carriers of the m.3243A>G mutation, who had mutation loads below this threshold but nevertheless had abnormal 31P-magnetic resonance spectroscopy (MRS) findings24 and bicycle ergometry (a method to test muscle performance),25 as well as abnormal lactate peaks in both cerebrospinal fluid (CSF) and brain parenchyma, as measured by 1H-MRS.26
The problem of homoplasmy
Pathogenic mtDNA mutations are usually heteroplasmic, whereas neutral polymorphisms are homoplasmic. Many exceptions to this rule exist, however, and awareness of the importance of pathogenic homoplasmic mutations is increasing. In fact, the first point mutation associated with a human disease (namely, an m.11778G>A mutation in the ND4L gene in LHON) was homoplasmic,4 as are the other two common LHON-associated mutations (m.3460G>A in ND1 and m.14484T>C in ND6). The latest puzzling clinical phenotype associated with—but not yet explained by—a homoplasmic mutation is a severe but reversible infantile mitochondrial myopathy characterized biochemically and histochemically by profound, though not exclusive, deficiency of cytochrome c oxidase (COX) in muscle.27 This homoplasmic mutation (m.14674T>C in tRNAGlu) was initially considered a neutral polymorphism, but was subsequently found in 17 patients from 12 families of various ethnic origins,28 and the same mutation (or a T>G mutation at the same site) was confirmed in eight Japanese patients.29 The pathogenicity of the mutation was established using high-resolution northern blot analysis of muscle biopsies during the symptomatic phase and through biochemical studies of cybrid cell lines, but the mutation by itself does not explain the tissue specificity or reversibility of the biochemical and clinical phenotypes. A role for a developmentally regulated nuclear modifier factor in this disorder has been postulated28 but not yet identified.
At least three factors can influence the phenotypic expression of homoplasmic mtDNA mutations: mtDNA haplotype, nDNA background and epigenetic factors. All three factors are illustrated in LHON—one of the most prevalent mtDNA-related diseases, with an estimated frequency of 15 in 100,000. In support of the importance of the mtDNA haplotype, strong evidence indicates that the penetrance of LHON is increased by an association with the European mtDNA haplogroup J.30 The fact that this disease predominantly affects men has been attributed to X-linked modifier loci, although no such gene has been clearly identified.30 A more likely explanation for this sex-related difference seems to be epigenetic factors, namely a protective effect of oestrogens in women. Indeed, the beneficial effect of oestrogens in disease prevention has been clearly documented in LHON cybrid cells.31
Disorders of nDNA defects
Since the first report of nDNA defects in 1995,32 identification of mutations in nuclear genes that affect the mitochondrial respiratory chain has occurred at an escalating pace (Box 2). These advances have been made possible by rapidly evolving technologies, from linkage analysis and homozygosity mapping, through to monochromosomal transfer, gene complementation, sequencing of candidate genes and, finally, to genome-wide (whole-exome) sequencing or screening of the portion of the genome that encodes the approximately 1,700 mitochondrial proteins in a process termed mito-exome sequencing.33 The advantages of these approaches are both practical (enabling diagnosis of puzzling cases and providing information to inform genetic counselling) and heuristic (leading to discovery of novel mutant genes and novel disease mechanisms).34
Box 2. Disorders due to mutations in nDNA.
Mutations in respiratory chain subunits (‘direct hits’)
Leigh syndrome
Mutations in respiratory chain ancillary proteins (‘indirect hits’)
Leigh syndrome with cytochrome c oxidase deficiency (SURF1)
Leigh syndrome and cardiopathy (SCO2, COX10, COX14, COX15, COA5, FAM36A, TACO1)
Leigh syndrome and hepatopathy (SCO1)
GRACILE syndrome
Defects of mitochondrial protein importation: Mohr–Tranebjaerg deafness–dystonia syndrome (TIMM8A); spastic paraplegia-13 (HSP60)
Defects of mtRNA translation
Fatal neonatal lactic acidosis (MRPS16, MRPS22, RMND1)
Spastic paraplegia (SPG7); dominant hereditary ataxia (AFG3L2)
Infantile hepatocerebral syndrome (GFM1); infantile encephalomyopathy (TUFM)
Myopathy, lactic acidosis, sideroblastic anaemia (PUS1); reversible hepatopathy (TRMU)
Leukoencephalopathy, brainstem and spinal cord involvement, lactic acidosis (DARS2)
Late-onset Leigh syndrome with COX deficiency (TACO1)
Defects of the MIM lipid milieu
Barth syndrome (TAZ)
Sengers syndrome (AGK)
Megaconial encephalomyopathy (CHKB)
MEGDEL (SERAC1)
Childhood myoglobinuria (LPIN1)
Defects of mitochondrial dynamics
DOA; DOA-plus (OPA1)
Charcot–Marie–Tooth type 2A (MFN2)
Charcot–Marie–Tooth type 4A (GDAP1)
Fatal infantile encephalomyopathy (DRP1, MFF)
Representative implicated genes are indicated in parentheses. Abbreviations: DOA, dominant optic atrophy; GRACILE, growth retardation, aminoaciduria, cholestasis, iron overload, and early death; MEGDEL, 3-methylglutaconic aciduria with sensorineural deafness, encephalopathy, and Leigh-like syndrome.
Direct hits
‘Direct hits’ refers to pathogenic mutations that directly affect nDNA-encoded respiratory chain subunits. Such mutations have been identified in all five complexes of the respiratory chain, but are most abundant in the gigantic complex I, affecting at least 16 of the 45 structural subunits. Conversely, mutations of nDNA-encoded complex IV respiratory chain subunits are surprisingly rare: only two of the 11 nDNA subunits (namely, COX subunit 6B1 and COX subunit 7B) harbour deleterious mutations.35,36
In keeping with the all-or-nothing effect of Mendelian mutations (most of which are recessive), as opposed to the variegated effect of heteroplasmic mtDNA mutations, disorders due to direct hits to the mitochondrial respiratory chain generally manifest at or soon after birth and are severe, often being lethal in infancy.
The clinical picture of disorders caused by nDNA mutations are remarkably homogeneous and to a large extent correspond with Leigh syndrome, a disorder that reflects the detrimental effects of energy shortage on the developing nervous system. Leigh syndrome is defined neuropathologically (or neuroradiologically) by bilateral symmetrical lesions throughout the nervous system, particularly in the basal ganglia, thalamus, brainstem and cerebellar roof nuclei. Neuronal loss, proportionate loss of myelin, reactive astrocytosis, and proliferation of cerebral microvessels are evident at the microscopic level. Clinically, children with Leigh syndrome have psychomotor retardation or regression, respiratory abnormalities, hypotonia, failure to thrive, seizures, dystonia and blindness.
Indirect hits
Even when all nDNA-encoded subunits of the various complexes are expressed correctly, they must be translated, imported into mitochondria, and directed to the MIM. At this site they assemble with their mtDNA and nDNA-encoded counterparts, acquire prosthetic groups, multimerize (if necessary), and further assemble into supercomplexes. Mutations in genes involved in these pathways are referred to as ‘indirect hits’, as they indirectly affect the mitochondrial respiratory chain.
In 1998, the search for the molecular basis of COX-deficient Leigh syndrome led to simultaneous discovery by two groups of the first mutant mitochondrial assembly gene, SURF1.37,38 More recently, mutant assembly factors have been found through whole-genome sequencing of DNA from patients with specific respiratory chain complex deficiencies, such as in individuals with deficiencies of complex I.39 The clinical manifestations of indirect hits that affect complex I40,41 tend to be more heterogeneous than those associated with direct hits to this complex, although considerable overlap exists between the two groups. All described patients with indirect hit mutations had encephalopathy that clinically resembles Leigh syndrome, but often with leukodystrophy rather than grey matter involvement.42 Cardiomyopathy was more common with indirect hit mutations than with direct hit mutations and, at times, was the dominating feature of the former. Although onset is in infancy or early childhood, and early death is common, the course can be prolonged and, in some cases, shows signs of fluctuation.42
The first assembly defect in complex III was identified in 2002 in Finnish infants with an extremely severe syndrome named GRACILE—an acronym summarizing the main symptoms and signs: growth retardation, aminoaciduria, cholestasis, iron overload, and early death (before 5 months of age).43 The protein that is mutated in this disorder—mitochondrial chaperone BCS1—is needed for insertion of the Rieske iron–sulphur (FeS) subunit into complex III. FeS clusters are prosthetic groups of complex I, complex II, complex III and the Krebs cycle enzyme aconitase. BCS1 is one of a series of FeS assembly proteins (including frataxin, the protein altered in Friedreich ataxia) that is associated with diverse generalized or tissue-specific disorders with multiple respiratory chain defects (Box 2).
Mutations in the assembly protein SURF1 are among the most common causes of Leigh syndrome, and SURF1 should be sequenced in all children with COX-deficient Leigh syndrome. Mutations in at least seven more COX assembly factors (SCO1, SCO2, COX10, COX14,44 COX15, COA5,45 FAM36A46 and TACO147) have been associated with human diseases. Mutations in any of these genes can cause encephalopathy, but each shows preferential involvement of one tissue other than the brain. For example, mutations in SCO2, COX10, COX15 and COA5 cause severe cardiomyopathy, whereas mutations in SCO1 cause hepatopathy.48
A relatively new group of disorders that are attributable to deficiency of coenzyme Q10 (CoQ10, also known as ubiquinone) can also be included in the ‘indirect hits’ group. Notably, however, the situation in this disorder is the converse of that described above for complex I, III, and IV deficiencies: instead of one mutant protein disrupting the assembly of a multimeric complex, mutations in a cascade of biosynthetic enzymes result in deficiency of one relatively simple component of the respiratory chain (Figure 1). CoQ10 is an integral part of the respirasome, where it shuttles electrons from complex I and complex II to complex III, acts as an antioxidant, and regulates apoptosis.49 The effects of CoQ10 deficiency were first described in 1989 in two sisters with the clinical triad of mitochondrial and lipid storage myopathy, recurrent muscle breakdown with myoglobinuria, and encephalopathy (seizures, ataxia and mental retardation).50 Molecular defects in CoQ10 biosynthetic genes (PDSS2 and COQ2), however, were not reported until 2006.51,52 Mutations in the same and other biosynthetic genes (PDSS1, COQ6, ADCK3 and COQ9) followed,53–57 and five clinical syndromes are now attributed to primary CoQ10 deficiency. In clinical practice, it is important to consider primary CoQ10 deficiency in the differential diagnosis of juvenile ataxias and infantile encephalomyopathies, as most patients with these disorders benefit from CoQ10 supplementation.58
A so far unique, ‘toxic’ indirect hit was documented in children with ethylmalonic encephalopathy—an early-onset disorder with microangiopathy, chronic diarrhoea, greatly elevated levels of ethylmalonic acid and short-chain acylcarnitines in body fluids, and apparently isolated COX deficiency in skeletal muscle.59 Studies of patients with this disorder and Ethe1-null mice (an animal model of the disease) revealed that the affected gene product, ETHE1, is a matrix sulphur dioxygenase. Dysfunction of ETHE1 results in excessive accumulation of hydrogen sulphide, which is a ‘gasotransmitter’ (gaseous signalling molecule) and—at high concentrations—a powerful COX inhibitor.60
A prerequisite for the assembly of any respiratory chain complex is the import of nDNA-encoded subunits from the cytoplasm into mitochondria, so it seems reasonable to add one more category to the indirect hits group—namely, defects of mitochondrial protein importation. Surprisingly, considering the complexity of the mitochondrial importation machinery,61 only a handful of diseases belong to this category (Box 2).
Defects of mtRNA translation
The mitochondrial genome is transcribed into nine monocistronic and two dicistronic mRNAs. Translation of these mRNAs into the 13 mtDNA-encoded respiratory chain subunits is effected by mitoribosomes, which consist of one large subunit (48 proteins) and one small subunit (29 proteins). The translation process comprises four phases, each requiring multiple ancillary factors.62 During the initiation phase, the start site of the mRNA is selected and the initiator tRNA (fMet-tRNA) is base-paired to the mRNA. During the elongation phase, the mRNA codons are read sequentially and the amino acids are incorporated by aminoacyl-tRNA synthetases into the growing peptide chain. In the termination phase, the complete polypeptide is released, and the ribosome complex is again made available in the recycling phase. The translation machinery requires translation initiation factors (IF2 and IF3), elongation factors (EF-Tu, EF-Ts and EF-G1) and release factors (eRF1 and ICT1), plus several translational activators (TACO1 and LRP130) and specific tRNA base modifiers (TRMU and PUS1).
Mutations in genes encoding mRNA polyadenylation factors, ribosomal proteins, ribosomal protein assembly factors, aminoacyl-tRNA synthetases, elongation factors, release factors, translational activators, and tRNA modifiers have been associated with a rapidly expanding group of human diseases (Box 2). The discovery of mtRNA translation defects was prompted by frequent clinical observations of patients, usually infants, with severe neurological involvement (often manifesting as Leigh syndrome, leukodystrophy or recessive ataxia), hepatocerebral syndrome or cardiomyopathy, and lactic acidosis. The biochemical hallmark of these disorders—and the clue to their mitochondrial aetiology—was the combined deficiency of multiple respiratory chain enzymes without evidence of altered mtDNA maintenance (no mtDNA depletion or multiple deletions).63,64 Homozygosity or candidate gene mapping, integrative genomics, and whole-exome sequencing uncovered the molecular genetic basis of these disorders.62,65,66
Notably, at least one condition involving impaired mtRNA translation—namely, acute liver failure of infancy due to mutations in the tRNA-modifying gene TRMU—is potentially reversible.67 This situation is reminiscent of the aforementioned reversible COX-deficient myopathy associated with a homoplasmic mutation in tRNAGlu. Interestingly, TRMU protein modifies three tRNAs, one of which is tRNAGlu, and one patient with reversible COX-deficient myopathy harboured a mutation in TRMU rather than in tRNAGlu.68
Our understanding of disorders of mtRNA translation is clearly a work in progress, as shown by the recent discovery in two laboratories of mutations in a gene (RMND1) that has previously been associated not with mtDNA translation, but with a possible role in assembly of ribosomal proteins.69,70
Defects of the MIM lipid milieu
The phospholipid component of the MIM, in which the respiratory chain resides, provides much more than a passive cell scaffold. Alterations of the lipid milieu are increasingly being associated with mitochondrial encephalomyopathies.
Cardiolipin—a dimeric molecule composed of two phosphatidylglycerol moieties connected by a glycerol group71—is the signature mitochondrial phospholipid and a major component of the MIM, where it is synthesized. Cardiolipin contains four acyl chains, the composition of which depends on the deacylation–reacylation activity of tafazzin, a phospholipid–lysophospholipid transacylase that confers cell-type and tissue specificity.72,73 Cardiolipin deficiency was first documented in the early 2000s in cultured fibroblasts from patients with Barth syndrome—an X-linked mitochondrial myopathy and cardiopathy, with neutropenia and stunted growth.74,75 Since then, four new disease entities characterized by deficiencies in different phospholipids have been identified in rapid succession (Box 2). Similarly to Barth syndrome, Sengers syndrome primarily affects heart and muscle, with the distinctive additional clinical feature of congenital cataracts. Despite convincing evidence that ADP–ATP translocase 1 (ANT1) was missing from muscle of patients with this condition, no mutation was found in the corresponding gene, and whole-exome sequencing subsequently revealed mutations in the acylglycerol kinase gene.76 Acylglycerol kinase catalyses the phosphorylation of diacylglycerol and monoacylglycerol to form phosphatidic acid or lysophosphatidic acid. Loss of either of these two phospholipids presumably prevents assembly of ANT1 in the MIM, but the mechanism is currently unknown.76 Notably, as phosphatidic acid is a precursor of cardiolipin, it thereby provides a point of convergence in Sengers syndrome and Barth syndrome, which could explain some of the clinical similarities between the two disorders.
The mitochondria-associated endoplasmic reticulum (ER) membrane (MAM) is a close physical and functional association between the ER and mitochondria. These membranes consist of specialized detergent-insoluble lipid raft domains that are rich in cholesterol and sphingolipids, and function as platforms for membrane proteins (Figure 3).77 MAMs are involved in multiple functions, including lipid transport, cholesterol metabolism, calcium signalling, energy metabolism, apoptosis and mitochondrial dynamics. Although the MAM is a recently described subcellular structure—the structure, although not with the acronym MAM, was first described in 199078—diseases related to MAM dysfunction are already being recognized.79–82
Figure 3.
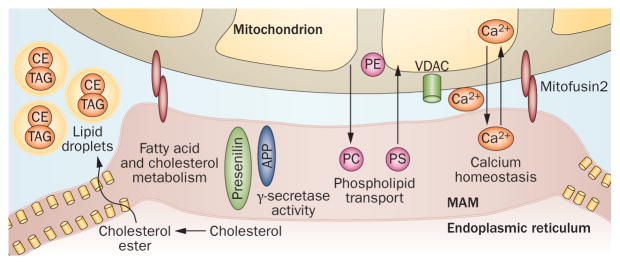
Structure and function of the MAM. The ER communicates with mitochondria via the MAM, a specialized subregion of the ER with the characteristics of a lipid raft. The MAM is the regulatory site in the cell for phospholipid metabolism, cholesterol ester and fatty acid metabolism, lipid droplet formation, calcium homeostasis, and mitochondrial dynamics. Abbreviations: APP, amyloid precursor protein; CE, cholesteryl ester; ER, endoplasmic reticulum; MAM, mitochondria-associated ER membrane; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; TAG, triacylglycerol, VDAC, voltage-dependent anion-selective channel. Original image courtesy of E. Area-Gomez, Columbia University, NY, USA.
In 1998, a new condition characterized by congenital myopathy and mental retardation was reported.83 The unusual muscle morphology, which included giant mitochondria that were displaced to the periphery of the fibres, was later observed in spontaneously mutant dystrophic mice that harboured changes in the gene encoding choline kinase-β (Chkb).84 Sequencing of CHKB in 15 patients with the disorder revealed 11 pathogenic mutations.85 CHKB catalyses the first step in the biosynthesis of phosphatidylcholine—a phospholipid that is formed through a biosynthetic pathway within the MAM. The relationship between phospholipid abnormality and mitochondrial dysfunction could, therefore, be mediated through the MAM. Several proteins involved in mitochondrial dynamics are also integral parts of the MAM,86 raising the possibility that MAM dysfunction might explain both the increased size and the intracellular displacement of mitochondria in patients with CHKB mutations.81
The role of the MAM in the aetiology of mitochondrial diseases that are caused by altered MIM phospholipid composition was confirmed in another disorder, termed MEGDEL, which is characterized by 3-methylglutaconic aciduria type IV,87 deafness, and Leigh syndrome-like encephalopathy. In this case, whole-exome sequencing revealed mutations in SERAC1. The SERAC1 protein is located in the MAM and controls exchange of phospholipids between the ER and mitochondria.88 Analysis of fibroblasts from patients with MEGDEL revealed alteration in the distribution of phosphatidylglycerol species and in the composition of cardiolipin subspecies. Quantitative or qualitative alterations in cardiolipin could, therefore, be a common denominator in the pathogenesis of disorders other than Barth syndrome.
Defects of mitochondrial dynamics
In keeping with their bacterial origin, mitochondria constantly move, fuse and divide, often forming tubular networks that favour a balanced distribution of energy throughout the cell.89 Interference with mitochondrial motility, fusion or fission results in disease (Box 2), and the nervous system is particularly susceptible as it is highly dependent on oxidative energy, and mitochondria must travel long distances along central axons and peripheral nerves.
Mitochondrial fusion requires coordinated action of pro-fusion GTPases in the mitochondrial outer membrane (MOM), namely mitofusins MFN1 and MFN2, and in the MIM (such as OPA1), together with MIM scaffolding proteins (prohibitin 2 and stomatin-like protein 2). Mitochondrial fission requires cytosolic dynamin-related protein 1 (DRP1; also a GTPase), which is recruited to the MOM together with two partners: mitochondrial fission 1 (FIS1) and mitochondrial fission factor (MFF).90 Once recruited to the MOM, DRP1 forms a spiral that wraps around the mitochondrion like a noose and severs the mitochondrial membranes using GTP hydrolysis.
Mutations in fusion proteins, such as MFN2 or GDAP1, result in Charcot–Marie–Tooth neuropathies (types 2A and 4A).91–93 However, mutations in the MFN2 gene have also been associated with a multisystem disorder that includes mitochondrial myopathy with multiple mtDNA deletions and depletion—that is, a defect of mtDNA maintenance (discussed below).94,95 Disorders of mitochondrial fission are less common than disorders of mitochondrial fusion, but mutations in DRP1 have been reported in an infant with mitochondrial and peroxisomal abnormalities, lactic acidosis, and a rapidly fatal encephalopathy with microcephaly, abnormal brain development and optic atrophy.96 A second patient with defective mitochondrial fission was identified through homozygosity mapping and mito-exome sequencing in the child of a consanguineous family with developmental delay, microcephaly and optic atrophy.97 This infant harboured a homozygous mutation in MFF, and both his clinical presentation and the morphological features of his cultured fibroblasts (excessive tubular pattern of mitochondria and peroxisomes) were similar to those observed in the aforementioned patient with a DRP1 mutation. The role of altered mitochondrial dynamics in neurological diseases is beyond the scope of this Review, but is addressed elsewhere.98–100
Defects of mtDNA maintenance
Mitochondria in eukaryotic cells have lost much of their original independent function, with the nucleus performing the role of mtDNA maintenance, including replication and integrity. When the dialogue between the two genomes becomes impaired, the resulting diseases are characterized by mtDNA depletion, multiple mtDNA deletions, and site-specific mtDNA point mutations.101
Initially, a simplistic concept was proposed whereby mtDNA depletion and mtDNA multiple deletions were distinct conditions with characteristic and distinguishable phenotypes. Following the advent of whole-genome sequencing, however, it is now recognized that the two conditions often coexist, and that mutations in the same genes can predominantly cause either mtDNA depletion or multiple mtDNA deletions (Table 2). Impairment of mtDNA maintenance can be attributable to defects in the replication machinery or in the intramitochondrial pool of deoxynucleoside triphosphates (dNTPs), the DNA building blocks.
Table 2.
Defects of mtDNA maintenance
| Mutated gene | mtDNA depletion | Multiple mtDNA deletions |
|---|---|---|
| TK2 | Infantile or adult myopathy SMA phenocopy | Adult autosomal recessive PEO |
| DGUOK | Infantile hepato-cerebral syndrome | Adult myopathy±PEO |
| PEO1 | Hepato-cerebral syndrome Infantile-onset spinocerebellar ataxia | Adult autosomal dominant PEO-plus |
| SUCLA2 | Infantile encephalomyopathy | — |
| SUCLG1 | Infantile encephalomyopathy Methylmalonic aciduria | — |
| RRM2B | Infantile encephalomyopathy | Adult autosomal dominant or autosomal recessive PEO-plus |
| MPV17 | Infantile hepatocerebral syndrome Navajo neurohepatopathy | Adult autosomal recessive PEO-plus |
| TYMP | Mitochondrial neurogastrointestinal encephalomyopathy | Mitochondrial neurogastrointestinal encephalomyopathy |
| POLG | Hepato-cerebral syndrome (Alpers syndrome) | Adult autosomal dominant or autosomal recessive PEO-plus; SANDO; MIRAS |
| POLG2 | — | Adult autosomal dominant PEO |
| ANT1 | — | Adult autosomal dominant PEO-plus |
| OPA1 | — | DOA; PEO-plus |
| MFN2 | — | DOA-plus |
| GFER | — | Congenital cataract, encephalomyopathy |
Abbreviations: DOA, dominant optic atrophy; MIRAS, mitochondrial recessive ataxia syndrome; mtDNA, mitochondrial DNA; PEO, progressive external ophthalmoplegia; SANDO, sensory ataxic neuropathy, dysarthria and ophthalmoparesis; SMA, spinal muscular atrophy.
Defects in replication machinery
The replicative machinery (replisome) includes the catalytic subunit of polymerase (encoded by the POLG gene), the accessory subunit (encoded by POLG2), and the replicative helicases Twinkle (encoded by PEO1)102 and DNA2.103 Study of the POLG gene provides the ultimate example of how a single mutant gene can cause either mtDNA depletion or multiple mtDNA deletions and result in diverse syndromes. Depending largely on which of the three POLG domains (polymerase, exo-nuclease or linker region) harbours the mutation(s), the clinical phenotype ranges from a severe hepatocerebral disorder of infancy or childhood (known as Alpers syndrome), through to adult-onset autosomal dominant or autosomal recessive progressive external ophthalmoplegia (AD-PEO or AR-PEO, respectively), to parkinsonism and other clinical phenotypes, including sensory ataxic neuropathy, dysarthria and ophthalmoparesis (SANDO), or mitochondrial recessive ataxia syndrome.102,104 Mutations in PEO1 usually cause adult-onset AD-PEO with multiple mtDNA deletions, but can also cause Alpers-like autosomal recessive hepatocerebral syndrome with mtDNA liver depletion, or infantile-onset spinocerebellar ataxia (IOSCA)—a disease that is prevalent in the Finnish population and is characterized by mtDNA depletion in the brain.105
Defects involving the dNTP pool
An adequate and balanced pool of the four dNTPs (dATP, dGTP, dCTP and dTTP) is necessary to provide the precursors of mtDNA replication. Defects in six enzymes that control the delicate balance of the dNTP pool have been associated primarily with mtDNA depletion,106 and result in four major syndromes (Table 2): myopathy (caused by mutations in TK2); hepatoencephalopathy (associated with mutations in DGUOK); encephalomyopathy (linked to mutations in SUCLA2, SUCLG1 or RRM2B); and mitochondrial neurogastrointestinal encephalomyopathy (MNGIE; caused by mutations in TYMP). The function of a seventh protein, MPV17, which is localized to the MIM, has not yet been clarified, but mutations in MPV17 cause severe mtDNA depletion associated with either encephalohepatopathy or Navajo neurohepatopathy, a disease that is endemic in the Navajo population of southwestern USA.107
Transgenic mice have been generated for all of the disorders of mtDNA maintenance, and are providing valuable information on pathogenesis and therapeutic approaches. In Tk2-knockout mice, for example, the CNS is as severely affected as skeletal muscle, thus reproducing the frequent spinal muscular atrophy phenocopy that is manifest in patients with TK2 deficiency.108
Other defects
An additional gene, OPA1, has been shown to be associated with myopathy and PEO.109–113 This finding was unexpected because mutations in OPA1 were initially associated with purely ophthalmological conditions, namely dominant optic atrophy (DOA) or Kjer disease,114,115 and the gene product, OPA1, is a MIM protein associated with mitochondrial fusion rather than with mitochondrial maintenance. However, a syndromic disorder termed DOA-plus has emerged, and is characterized by the generally sequential appearance of optic atrophy with visual failure, sensorineural deafness, ataxia, myopathy, axonal sensory–motor polyneuropathy, and PEO.109–113 Muscle biopsy in these patients shows scattered ragged-blue, COX-negative fibres, and long-range PCR reveals multiple mtDNA deletions. Interestingly, the proportion of COX-negative fibres is much higher in patients with DOA-plus than in those with nonsyndromic DOA. OPA1 is now known to be involved in both mitochondrial membrane dynamics and mtDNA maintenance; in fact, a specific OPA1 isoform facilitates tethering of nucleoids to the MIM—a function that is of crucial importance for mtDNA replication and distribution.116 In addition, OPA1 mutations lead to impairment of the OXPHOS system, as documented in studies of primary skin fibroblasts,117 muscle histo-chemistry, and 31P-MRS studies of muscle, which showed reduction of phosphorylation potential and impairment of ATP production.118
The heuristic value of deep-sequencing of DNA in puzzling cases is illustrated by the discovery of the first identified mitochondrial exonuclease, mitochondrial genome maintenance exonuclease 1 (encoded by C20orf72, renamed MGME1), which is involved in mtDNA replication. MGME1 mutations are responsible for a childhood-onset or adult-onset syndrome characterized by PEO, emaciation and respiratory failure, and associated with both mtDNA depletion and multiple mtDNA deletions in muscle.119
Diagnosis: Ariadne’s threads
Patients with suspected mitochondrial disease represent a considerable diagnostic challenge. Greek mythology tells us how Theseus, after killing the Minotaur in the Labyrinth of Crete, was helped out of that maze by Ariadne, who gave him a skein of wool that he unwound on his way in and rewound on his way out. Are there ‘Ariadne’s threads’ that can direct the modern-day neurologist, if not to the correct diagnosis, at least to the likely disease category?
Family history
Maternal inheritance of a disorder is indicative of an mtDNA-related disease, but can be obscured owing to heteroplasmy. A family history must, therefore, be taken meticulously, acknowledging ‘soft signs’ in the maternal lineage, such as short stature, migraines, hearing loss, diabetes, exercise intolerance, and psychiatric disorders (especially depression). We should not forget, however, that disorders due to single mtDNA deletions (such as KSS, PS and CPEO) are almost always sporadic,120 including diseases due to de novo mutations, such as most changes affecting the mitochondrial DNA cyto-chrome b gene (MTCYB).121 Parental consanguinity suggests autosomal recessive inheritance, and homo-zygosity mapping has helped to identify several mutated nuclear genes.
Multisystem disorders
Multisystem disorders are indicative of both mtDNA-related and nDNA-related disease. However, some syndromes are so typically associated with mtDNA mutations (KSS, PS, MELAS, MERRF, NARP, MILS and LHON) or with mutations in specific nuclear genes (MNGIE, SANDO, Alpers syndrome and DOA) that they provide useful shortcuts to the molecular diagnosis, as sequencing can be targeted to these common disease-associated genes. By contrast, a diagnosis of Leigh syndrome indicates a mitochondrial disease, but provides little information about which genome is involved or in which subgroup of mitochondrial disorders the patient should be categorized. Application of whole-exome sequencing in patients with Leigh syndrome of unknown cause has already revealed novel mutated genes in this disorder.
Three organs—namely, the heart, liver and kidney—are often involved in mitochondrial diseases, usually together with the brain, but occasionally in the absence of encephalopathy. Hypertrophic cardiomyopathy, often rapidly fatal but sometimes chronic as in Barth syndrome, can be the presenting and predominant feature. Liver involvement is typically associated with mtDNA depletion or defects of mtDNA translation (‘hepatocerebral syndromes’), but can occasionally be seen as part of direct or indirect hits to respiratory chain complexes. Nephropathy typically presents as proximal tubulopathy with Fanconi syndrome (a disorder that is characterized by failed reabsorption and excessive urinary excretion of glucose, amino acids, uric acid, phosphate and bicarbonate), but presentation as glomerulonephrosis with albuminuria should raise suspicion of CoQ10 deficiency.
PEO
If diagnoses of myasthenia gravis and oculopharyngeal muscular dystrophy are excluded, PEO is a strong clue to mitochondrial dysfunction. The extreme vulnerability of extraocular muscles to mitochondrial dysfunction correlates with their dependence on oxidative metabolism. PEO is commonly associated with primary mtDNA large-scale rearrangements (such as in KSS and CPEO) and with nDNA-related defects of mtDNA maintenance, which also result in mtDNA deletions or depletion. Sporadic cases of PEO suggest primary mtDNA mutations, whereas autosomal dominant or recessive inheritance suggests disorders of mtDNA maintenance. Maternal inheritance is rare, but reports exist of PEO due to mutations in mtDNA tRNA genes, including the common MELAS mutation m.3243A>G.122
Onset
Onset varies widely between mtDNA-related diseases, even within members of the same family, probably owing to varying degrees of heteroplasmy.15 In most Mendelian disorders, with the exception of some defects of mtDNA maintenance (especially those with multiple mtDNA deletions), onset is in infancy or early childhood and often follows several months of normal development. Prenatal manifestations of mitochondrial disorders are uncommon, as shown by the rare occurrence of arthrogryposis and dysmorphisms. In a few instances, foetal cardiopathy is revealed on foetal bradycardia analysis or on echocardiography. Adult onset is frequent in disorders of intergenomic communications (most notably multiple mtDNA deletions with PEO), probably owing to the gradual accumulation of mtDNA defects.
Laboratory tests
Lactic acidosis is a hallmark of all mitochondrial diseases, but is neither invariably present nor necessarily severe. Increased lactate in the CSF, detectable by 1H-MRS, is a frequent finding in patients with these conditions.
Serum creatine kinase is only mildly elevated in most mitochondrial disorders. In mtDNA depletion myopathy due to TK2 deficiency, however, increased serum levels of creatine kinase serves as a diagnostic red flag.
In 2011, fibroblast growth factor 21 (FGF-21) was introduced as a valuable new serum or plasma biomarker for detection of mitochondrial disorders, especially those involving skeletal muscle.123 Although the diagnostic accuracy of FGF-21 is better than that of all conventional biomarkers, including lactate, the test has not yet been widely used, possibly because it requires ELISA, which is not routinely available, or simply owing to reluctance from the clinical community to adopt this new assay.
Muscle biopsy
Despite its invasive nature, muscle biopsy remains the gold standard for diagnosis of mitochondrial diseases, especially those due to primary mtDNA mutations.14 Systematic analysis of frozen, cross-sectioned muscle with histochemical reactions for COX, succinate dehydrogenase (SDH) or both provides valuable diagnostic clues.
The SDH stain reflects the activity of complex II of the respiratory chain—the only complex that is entirely encoded by nDNA. Consequently, this stain is unaffected by deleterious mutations of mtDNA and provides an excellent marker of mitochondrial abundance. By contrast, the three catalytic subunits of COX are encoded by mtDNA, and the COX stain is an excellent index of mtDNA function. As well as mutations in COX I, COX II or COX III, any mtDNA mutation that impairs mitochondrial protein synthesis (that is, large-scale deletions or point mutations in tRNA or ribosomal RNA genes) results in a decrease in or lack of COX staining.
Importantly, heteroplasmic mtDNA mutations are unevenly distributed along syncytial muscle fibres, such that adjacent segmental sections can have widely varying amounts of mutant mtDNAs. Cross sections of the muscle biopsy from patients harbouring pathogenic mtDNA mutations, therefore, reveal a mosaic pattern of COX-negative and COX-positive fibres (Figure 4).
Figure 4.

Muscle biopsy in patients with mitochondrial disease. a | In a patient with MERRF (harbouring a mitochondrial DNA point mutation that impairs mitochondrial protein synthesis), two ragged-red fibres are evident with the modified Gomori trichrome stain. b–d | Using the SDH stain, the same fibres appear ‘ragged blue’ (part b) but are COX-negative (part c), and stain blue with the combined COX and SDH stain (COX-deficient fibres stain a lighter blue; part d). Abbreviations: MERRF, myoclonus epilepsy with ragged-red fibres; SDH, succinate dehydrogenase. Image first printed in Molecular Neurology.155
Mitochondria proliferate in response to energy failure, so COX deficiency is often accompanied by excess mitochondria, showing as hyperintense SDH stain (these fibres are termed ‘ragged-blue’ by analogy with the ‘ragged-red’ fibres revealed with the modified Gomori trichrome stain; Figure 4). A mosaic pattern of normally stained fibres mixed with ragged-blue COX-negative fibres in a muscle biopsy is a robust clue to the diagnosis of an mtDNA-related disease affecting mitochondrial protein synthesis (or, more rarely, affecting one of the three mtDNA-encoded COX subunits). A mosaic pattern of ragged-blue but COX-positive fibres suggests a mutation in an mtDNA gene that encodes a respiratory chain subunit other than COX I, II, or III (for example, a gene encoding a complex I subunit or cytochrome b).124
Heteroplasmy explains not only the mosaic distribution of COX-negative fibres, but also the varying degrees of ‘COX negativity’, as some fibres have a deficiency rather than a lack of staining. To help reveal the COX-deficient fibres, the combined COX–SDH stain is extremely effective: when the two stains are superimposed in normal fibres, the brown COX stain prevails and overshadows the blue SDH stain. However, even a small decrease in COX activity allows the SDH stain to shine through, making the COX-deficient fibres appear blue. This technique has also been used to reveal COX-deficient motor neurons in patients with amyotrophic lateral sclerosis,125 MELAS,17 and other mtDNA-related diseases.126
In Mendelian disorders such as COX-deficient Leigh syndrome, muscle histochemistry shows diffuse COX deficiency,127 which is an important diagnostic clue (Figure 4). Conversely, owing to the overlapping features of Mendelian and mitochondrial genetics, muscle biopsies from patients with defects of mtDNA maintenance—and especially from cases of PEO with multiple mtDNA deletions—show a mosaic pattern of COX-negative ragged-blue fibres. This morphological pattern, together with a family history suggestive of Mendelian inheritance, should prompt a search for mutations in mtDNA maintenance genes, even in clinically atypical cases.119,128
Ultrastructural studies of muscle are usually only confirmatory in that they show mitochondrial proliferation. However, the presence of paracrystalline inclusions, consisting of crystals of creatine kinase, reinforces the diagnosis of mitochondrial diseases; the presence of giant displaced mitochondria suggests impaired mitochondrial dynamics;81 and abnormal cristae are typical of Barth syndrome.129
Biochemistry
Biochemical analysis is usually conducted in frozen muscle tissue or cultured fibroblasts. A detailed, step-by-step procedure for the measurement of the activities of complexes I–IV is available,130 and should facilitate comparison of data among laboratories.
Individual respiratory complex deficiencies are expected to occur in patients with mutations in mtDNA protein-coding genes and in individuals with mutations in genes encoding single respiratory chain subunits (direct hits) or assembly genes for specific complexes (indirect hits). As the respiratory chain is organized in supercomplexes, however, a severe defect in one complex is likely to be accompanied by less-severe defects in one or more additional complexes. For example, mutations in the MTCYB gene cause defects not only of complex III but also of complex I.121
Impaired biochemical activities of all complexes that contain subunits encoded by mtDNA suggest either mutations in mtDNA genes controlling protein synthesis globally (tRNA or ribosomal RNA genes, single deletions) or mutations in nDNA genes controlling mtDNA maintenance (multiple deletions, depletion, defects of mtDNA translation, defects of the MIM lipid milieu).
Particular biochemical patterns exist that are suggestive of specific problems. For example, combined defects of complexes II and III are often associated with primary CoQ10 deficiency, whereas combined defects of complexes I, II and III may be attributable to defective assembly of the FeS cluster proteins, particularly if they are accompanied by aconitase deficiency.131
Therapeutic approaches
Mitochondrial diseases are defined here as defects of the respiratory chain, but no panacea for respiratory chain dysfunction and no universal therapy for mitochondrial disorders currently exist. Nevertheless, pharmacological treatments (for example, antiepileptic drugs) and surgical remedies (such as blepharoplasty) are useful in prolonging and improving the lives of patients with mitochondrial disease. Several strategies aimed at curing (or even preventing) mitochondrial diseases have shown promise in vitro or in animals, or have produced encouraging preliminary results in humans, and are briefly reviewed below.
Enhancement of respiratory chain function
The most obvious therapeutic approach to mitochondrial disorders is to enhance respiratory chain function, thereby mitigating both energy crisis (ATP deficit) and oxidative stress (toxic build-up of ROS). The compound used widely in patients with mitochondrial disease is CoQ10, owing to the pivotal role of this molecule in electron transport, its antioxidant properties, and its safety even at high doses. With the exception of some patients with primary CoQ10 deficiency who show a dramatic improvement following CoQ10 supplementation,132 however, the effects of CoQ10 are largely modest and subjective.133 Two synthetic analogues of CoQ10— idebenone and parabenzoquinone—seem more promising. Idebenone, a short-chain benzoquinone, has shown positive results in two studies of patients with LHON.134,135 The second compound, a parabenzoquinone named EPI-743, has so far been tested only in open-label studies: this compound reversed vision loss in four of five patients with LHON,136 produced clinical improvement in 12 children with various mitochondrial disorders,137 and arrested or reversed disease progression in 13 children with genetically proven Leigh syndrome.138
Elimination of noxious compounds
The second logical therapeutic approach to mitochondrial disease is to eliminate the noxious compounds that accumulate in these disorders. To decrease brain lactate in patients with MELAS, we used dichloroacetate, which keeps pyruvate dehydrogenase in the active form and favours lactate oxidation. However, this agent had unacceptable neurotoxicity, leading to treatment discontinuation.139 A more promising ‘detoxifying therapy’ seems to be allogeneic haematopoetic stem-cell transplantation (AHSCT) aimed at restoring sufficient thymidine phosphorylase activity in patients with MNGIE to normalize circulating, toxic levels of thymidine and deoxyuridine.132 As of 2010, five of 11 patients with MNGIE who had undergone AHSCT were alive, and had normal blood thymidine phosphorylase activity and virtually undetectable levels of thymidine and deoxyuridine. A safety study of AHSCT in patients with MNGIE is under way.
Shifting of heteroplasmy
For mtDNA-related disorders, an obvious but challenging goal is to shift heteroplasmy to lower the mutation load to subthreshold levels. Several strategies have been attempted in cybrid cell lines, often with good results. When deprived of glucose and exposed to ketogenic media, cybrids harbouring single mtDNA deletions shifted their heteroplasmy level and recovered mitochondrial function, probably through selective mitochondrial autophagy (mitophagy).140 A genetic approach to heteroplasmic shifting involves use of restriction endonucleases to eliminate specific pathogenic mutations.141 Dormant stem cells in skeletal muscle (satellite cells) have fewer mtDNA mutations than do adult muscle fibres, and muscle regeneration after induced myonecrosis was used to shift heteroplasmy in a patient with strabismus.142
Alteration of mitochondrial dynamics
Mitochondrial dynamics could be exploited therapeutically in two opposing ways. Mitochondrial fission could be enhanced to favour mitophagy, a natural ‘quality control’ function that sequesters and eliminates dysfunctional mitochondria, perhaps sensing their abnormally low membrane potential.140,143 Alternatively, enhancement of mitochondrial fusion and ‘networking’ would allow complementation of ‘bad’ and ‘good’ mitochondria and normalization of overall mitochondrial function.144
If mitochondrial proliferation is a futile compensatory attempt, a potential therapeutic approach could be to try to improve on nature’s strategy by enhancing mitochondrial biogenesis through activation of the transcriptional co-activator PGC-1a. The advantage over disease-induced mitochondrial proliferation, which favours mutated mtDNAs, is that pharmacological upregulation of mitochondrial biosynthesis increases numbers of all mtDNA molecules, allowing wild-type genomes to compensate for mutated ones. Encouraging results have been obtained in four mouse models of COX deficiency.145,146
Gene therapy
For nDNA-related mitochondrial disorders, ‘classic’ gene therapy is an option, but well-known issues are associated with this approach, including choice of appropriate viral or nonviral vectors, delivery to the affected tissues, and potential immunological reactions. Nonetheless, adeno-associated virus-mediated gene transfer has proven successful in two animal models: the Ant1 mutant mouse,147 and a mouse model of ethylmalonic encephalomyopathy.148
Cytoplasmic transfer
For mtDNA-related diseases, many of which are devastating and undiagnosable before birth, the ultimate goal is to prevent their occurrence altogether via cytoplasmic transfer. In this approach, the nucleus of an in vitro-fertilized oocyte from a carrier is transferred to an enucleated oocyte from a normal donor: the embryo will have the nDNA of the biological parents but the mtDNA of a normal mitochondrial donor. In nonhuman primate experiments using spindle–chromosomal complex transfer, the infants were healthy and devoid of maternal mtDNA.149 The same technique was applied to fertilized150 and unfertilized but parthenogenically activated151 human oocytes, and the cells were found to develop into normal blastocysts and contain exclusively donor mtDNA. Moreover, only donor mtDNA was detected after stem-cell lines from blastocysts were differentiated into neurons, cardiomyocytes and β-cells.151 Similar results were obtained in the UK after pronuclear transfer in abnormally fertilized human oocytes developed to the blastocyst stage.152 Despite some minor concerns,153 the stage seems set for approval of this technique for therapeutic application in the UK and the USA.
Conclusions
Mitochondrial disorders in neurology are either underdiagnosed (“what is this bizarre syndrome?”) or overdiagnosed (“this syndrome is so bizarre that it must be mitochondrial”). Here, we have outlined the complex genetic control of the mitochondrial respiratory chain, and described the typical clinical features that are associated with various genetic subgroups of mitochondrial disorders. These diagnostic clues can orient clinicians towards the correct molecular defect, which makes genetic counselling and prenatal diagnosis possible. Therapy for mitochondrial diseases is woefully inadequate and mostly palliative, but innovative therapeutic strategies developed in in vitro or animal models bode well for human application in the near future.
Key points.
Few neurological disorders are as phenotypically heterogeneous and diagnostically challenging as mitochondrial encephalomyopathies
The clinical heterogeneity of mitochondrial disorders can be explained by both the unique rules of mitochondrial genetics and the dependence of most mitochondrial functions on a wide variety of nuclear genes
Despite advances in understanding the molecular aetiology of mitochondrial diseases, their pathogenesis is still largely unknown
Through whole-exome or mito-exome sequencing, novel mutant genes and novel disease mechanisms of mitochondrial disease are being revealed
Promising areas of investigation for neurological mitochondria-associated disorders include defects in lipid composition of the mitochondrial membrane and defects of the mitochondria-associated membrane
Review criteria.
Most articles were selected from the extensive personal bibliographies of the authors. For recent references, up to April 2013, MEDLINE and PubMed were searched using keywords including “mitochondrial disease,” “mitochondrial dynamics,” “mtDNA” and “whole-exome sequencing”. Only articles in English were included.
Acknowledgments
The authors are supported by grants from the NIH (HD032062) and from the Marriott Mitochondrial Disorder Clinical Research Fund.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
S. DiMauro and V. Carelli researched data for the article. S. DiMauro wrote the article. All authors provided substantial contribution to discussion of content and to the review and/or editing of the manuscript before submission.
References
- 1.Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study. J Clin Invest. 1962;41:1776–1804. doi: 10.1172/JCI104637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shapira Y, Harel S, Russell A. Mitochondrial encephalomyopathies: a group of neuromuscular disorders with defects in oxidative metabolism. Isr J Med Sci. 1977;13:161–164. [PubMed] [Google Scholar]
- 3.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 5.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilkerson RW, Schon EA, Hernandez E, Davidson MM. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol. 2008;181:1117–1128. doi: 10.1083/jcb.200712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sykora P, Wilson DM, 3rd, Bohr W. A Repair of persistent strand breaks in the mitochondrial genome. Mech Ageing Dev. 2012;133:169–175. doi: 10.1016/j.mad.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schon EA, et al. A direct repeat is a hotspot for large-scale deletions of human mitochondrial DNA. Science. 1989;244:346–349. doi: 10.1126/science.2711184. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan KJ, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer AM, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 11.Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Payne BA, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowland LP. In: Mitochondrial Disorders in Neurology. Schapira AH, DiMauro S, editors. Butterworth-Heinemann; 1994. pp. 116–129. [Google Scholar]
- 14.Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 15.Tatuch Y, et al. Heteroplasmic mtDNA mutation (T>G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet. 1992;50:852–858. [PMC free article] [PubMed] [Google Scholar]
- 16.Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr. 1997;29:131–149. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- 17.Betts J, et al. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropath Appl Neurobiol. 2006;32:359–373. doi: 10.1111/j.1365-2990.2006.00731.x. [DOI] [PubMed] [Google Scholar]
- 18.Tanji K, Bonilla E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods Cell Biol. 2001;65:311–332. doi: 10.1016/s0091-679x(01)65019-2. [DOI] [PubMed] [Google Scholar]
- 19.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 20.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. doi: 10.1016/j.preteyeres.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 21.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 22.Pallotti F, et al. Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations. Biochem J. 2004;384:287–293. doi: 10.1042/BJ20040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chomyn A, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci USA. 1992;89:4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinnery PF, et al. Very low levels of the mtDNA A3243G mutation associated with mitochondrial dysfunction in vivo. Ann Neurol. 2000;47:3381–384. [PubMed] [Google Scholar]
- 25.Jeppesen TD, et al. Muscle phenotype and mutation load in 51 persons with the 3243A>G mtDNA mutation. Arch Neurol. 2006;63:1701–1706. doi: 10.1001/archneur.63.12.1701. [DOI] [PubMed] [Google Scholar]
- 26.Kaufmann P, et al. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology. 2004;62:1297–1302. doi: 10.1212/01.wnl.0000120557.83907.a8. [DOI] [PubMed] [Google Scholar]
- 27.DiMauro S, et al. Benign infantile mitochondrial myopathy due to reversible cytochrome c oxidase deficiency. Ann Neurol. 1983;14:226–234. doi: 10.1002/ana.410140209. [DOI] [PubMed] [Google Scholar]
- 28.Horvath R, et al. Molecular basis of infantile reversible cytochrome c oxidase deficiency. Brain. 2009;132:3165–3174. doi: 10.1093/brain/awp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mimaki M, et al. Reversible infantile respiratory chain deficiency: a clinical and molecular study. Ann Neurol. 2010;68:845–854. doi: 10.1002/ana.22111. [DOI] [PubMed] [Google Scholar]
- 30.Maresca A, La Morgia C, Caporali L, Valentino ML, Carelli V. The optic nerve: a “mito-window” on mitochondrial neurodegeneration. Mol Cell Neurosci. 2012;55:62–67. doi: 10.1016/j.mcn.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giordano C, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain. 2011;134:220–234. doi: 10.1093/brain/awq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourgeron T, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- 33.Calvo S, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra110. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coppola G, Geschwind DH. Genomic medicine enters the neurology clinic. Neurology. 2012;79:112–114. doi: 10.1212/WNL.0b013e31825f06d9. [DOI] [PubMed] [Google Scholar]
- 35.Massa V, et al. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet. 2008;82:1281–1289. doi: 10.1016/j.ajhg.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Indrieri A, et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet. 2012;91:942–949. doi: 10.1016/j.ajhg.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Z, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 38.Tiranti V, et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvo T, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system: what we have learned by studying its defects. Biochim Biophys Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 41.Tucker EJ, et al. Next-generation sequencing in molecular diagnosis: NUBPL mutations highlight the challenges of variant detection and interpretation. Hum Mut. 2012;33:411–418. doi: 10.1002/humu.21654. [DOI] [PubMed] [Google Scholar]
- 42.DiMauro S, Emmanuele V. In: Mitochondrial Disorders Caused by Nuclear Genes. Wong L-J, editor. Springer Science and Business Media; New York: 2013. pp. 3–25. [Google Scholar]
- 43.Visapää I, et al. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet. 2002;71:863–876. doi: 10.1086/342773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weraarpachai W, et al. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huigsloot M, et al. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488–493. doi: 10.1016/j.ajhg.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szklarczyk R, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet. 2012;22:656–667. doi: 10.1093/hmg/dds473. [DOI] [PubMed] [Google Scholar]
- 47.Weraarpachai W, et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 48.DiMauro S, Tanji K, Schon EA. The many faces of cytochrome c oxidase deficiency. Adv Exp Med Biol. 2012;748:341–357. doi: 10.1007/978-1-4614-3573-0_14. [DOI] [PubMed] [Google Scholar]
- 49.Trevisson E, DiMauro S, Navas P, Salviati L. Coenzyme Q deficiency in muscle. Curr Opin Neurol. 2011;24:440–456. doi: 10.1097/WCO.0b013e32834ab528. [DOI] [PubMed] [Google Scholar]
- 50.Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quinzii C, et al. A mutation in para-hydoxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez LC, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaproneyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mollet J, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heeringa SF, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mollet J, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagier-Tourenne C, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duncan AJ, et al. A nonsense mutation on COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84:558–566. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emmanuele V, et al. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch Neurol. 2012;69:978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tiranti V, et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tiranti V, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 61.Bolender N, Sickmann A, Wagner R, Meisinger C, Pfanner N. Multiple pathways for sorting mitochondrial precursor proteins. EMBO Rep. 2008;9:42–49. doi: 10.1038/sj.embor.7401126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta. 2012;1819:1035–1054. doi: 10.1016/j.bbagrm.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kemp JP, et al. Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain. 2011;134:183–195. doi: 10.1093/brain/awq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smits P, et al. Mutation in subdomain G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum Genet. 2011;19:275–279. doi: 10.1038/ejhg.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smits P, Smeitink J, van den Heuvel B. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chrzanowska-Lightowlers ZM, Horvath R, Lightowlers RN. 175th ENMC International Workshop: mitochondrial protein synthesis in health and disease, 25–27th June, 2010, Naarden, The Netherlands. Neuromusc Disord. 2011;21:142–147. doi: 10.1016/j.nmd.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 67.Schara U, et al. Acute liver failure with subsequent cirrhosis as the primary manifestation of TRMU mutations. J Inherit Metab Dis. 2011;34:197–201. doi: 10.1007/s10545-010-9250-z. [DOI] [PubMed] [Google Scholar]
- 68.Uusimaa J, et al. Reversible infantile respiratory chain deficiency is a unique, genetically heterogeneous mitochondrial disease. J Med Genet. 2011;48:660–668. doi: 10.1136/jmg.2011.089995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Janer A, et al. An RMND1 mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet. 2012;91:737–743. doi: 10.1016/j.ajhg.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garcia-Diaz B, et al. Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am J Hum Genet. 2012;91:729–736. doi: 10.1016/j.ajhg.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids. 2005;138:38–49. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 72.Schlame M, et al. The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat Chem Biol. 2012;8:862–869. doi: 10.1038/nchembio.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vreken P, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279:378–382. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 75.Schlame M, et al. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 76.Mayr JA, et al. Lack of mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am J Hum Genet. 2012;90:314–320. doi: 10.1016/j.ajhg.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper? Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- 79.Al-Saif A, Bohlega S, Al-Mohanna F. Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann Neurol. 2012;72:510–516. doi: 10.1002/ana.23641. [DOI] [PubMed] [Google Scholar]
- 80.Bellzil VV, Rouleau GA. Endoplasmic reticulum lipid rafts and upper motor neuron degeneration. Ann Neurol. 2012;72:479–480. doi: 10.1002/ana.23678. [DOI] [PubMed] [Google Scholar]
- 81.Gutierrez-Rios P, et al. Congenital megaconial myopathy due to a novel defect in the choline kinase beta (CHKB) gene. Arch Neurol. 2012;69:657–661. doi: 10.1001/archneurol.2011.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Area-Gomez E, et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishino I, et al. A new congenital muscular dystrophy with mitochondrial structural abnormalities. Muscle Nerve. 1998;21:40–47. doi: 10.1002/(sici)1097-4598(199801)21:1<40::aid-mus6>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 84.Sher RB, et al. A rostrocaudal muscular dystrophy caused by a defect in choline kinase beta, the first enzyme in phosphatidylcholine biosynthesis. J Biol Chem. 2006;281:4938–4948. doi: 10.1074/jbc.M512578200. [DOI] [PubMed] [Google Scholar]
- 85.Mitsuhashi S, et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidyl choline biosynthesis. Am J Hum Genet. 2011;88:845–851. doi: 10.1016/j.ajhg.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci. 2012;55:26–36. doi: 10.1016/j.mcn.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 87.Wortmann RL, et al. Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy. Brain. 2009;132:136–146. doi: 10.1093/brain/awn296. [DOI] [PubMed] [Google Scholar]
- 88.Wortmann RL, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Am J Hum Genet. 2012;44:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 89.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 90.Otera H, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Züchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 92.Baxter RV, et al. Ganglioside-induced differentiation-associated protein 1 is mutant in Charcot–Marie–Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–22. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- 93.Cuesta A, et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot–Marie–Tooth type 4A disease. Nat Genet. 2002;30:22–25. doi: 10.1038/ng798. [DOI] [PubMed] [Google Scholar]
- 94.Rouzier C, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 2012;136:23–34. doi: 10.1093/brain/awr323. [DOI] [PubMed] [Google Scholar]
- 95.Vielhaber S, et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013;125:245–256. doi: 10.1007/s00401-012-1036-y. [DOI] [PubMed] [Google Scholar]
- 96.Waterham HR, et al. A lethal defect of mitochondrial and peroxisomal fission. New Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 97.Shemseldin HE, et al. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet. 2012;49:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- 98.Milone M, Benarroch EE. Mitochondrial dynamics: general concepts and clinical implications. Neurology. 2012;78:1612–1619. doi: 10.1212/WNL.0b013e3182563c46. [DOI] [PubMed] [Google Scholar]
- 99.Schon EA, Przedborski S. Mitochondria in the next (neurode)generation. Neuron. 2011;70:1033–1053. doi: 10.1016/j.neuron.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen H, Chan DC. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hirano M, Lagier-Tourenne C, Valentino ML, Martí R, Nishigaki Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene. 2005;354:152–156. doi: 10.1016/j.gene.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 102.Copeland WC. Inherited mitochondrial diseases of DNA replication. Annu Rev Med. 2008;59:131–146. doi: 10.1146/annurev.med.59.053006.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ronchi D, et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet. 2013;92:293–300. doi: 10.1016/j.ajhg.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ylikallo E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2011;44:41–59. doi: 10.3109/07853890.2011.598547. [DOI] [PubMed] [Google Scholar]
- 105.Hakonen AH. Infantile-onset spinocerebellaer ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet. 2008;17:3822–3835. doi: 10.1093/hmg/ddn280. [DOI] [PubMed] [Google Scholar]
- 106.Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes—many genes, common mechanisms. Neuromusc Disord. 2010;20:429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 107.Karadimas CL, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Akman HO, et al. Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum Mol Genet. 2008;17:2433–2440. doi: 10.1093/hmg/ddn143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yu-Wai-Man P, Chinnery PF. Dysfunctional mitochondrial maintenance: what breaks the circle of life? Brain. 2012;135:9–11. doi: 10.1093/brain/awr352. [DOI] [PubMed] [Google Scholar]
- 110.Ranieri M, et al. Optic atrophy plus phenotype due to mutations in the OPA1 gene: two more Italian families. J Neurol Sci. 2012;315:146–149. doi: 10.1016/j.jns.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amati-Bonneau P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- 112.Hudson G, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- 113.Ferraris S, et al. Progressive external ophthalmoplegia and vision and hearing loss in a patient with mutations in POLG2 and OPA1. Arch Neurol. 2008;65:125–131. doi: 10.1001/archneurol.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Alexander C, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 115.Delettre C, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 116.Elachouri G, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011;12:12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Agier V, et al. Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta. 2012;1822:1570–1580. doi: 10.1016/j.bbadis.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 118.Lodi R, et al. Defective mitochondrial adenosine triphosphate production in skeletal muscle from patients with dominant optic atrophy due to OPA1 mutations. Arch Neurol. 2011;68:67–73. doi: 10.1001/archneurol.2010.228. [DOI] [PubMed] [Google Scholar]
- 119.Kornblum C, et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet. 2013;45:214–219. doi: 10.1038/ng.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chinnery PF, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–595. doi: 10.1016/S0140-6736(04)16851-7. [DOI] [PubMed] [Google Scholar]
- 121.Emmanuele V, et al. A novel mutation in the mitochondrial DNA cytochrome b gene (MTCYB) in a patient with MELAS syndrome. J Child Neurol. 2013;28:236–242. doi: 10.1177/0883073812445787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moraes CT, et al. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul Disord. 1993;3:43–50. doi: 10.1016/0960-8966(93)90040-q. [DOI] [PubMed] [Google Scholar]
- 123.Suomalainen A, et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Andreu AL, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. New Engl J Med. 1999;341:1037–1044. doi: 10.1056/NEJM199909303411404. [DOI] [PubMed] [Google Scholar]
- 125.Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 126.Borthwick GM, et al. Motor neuron disease in a patient with a mitochondrial tRNAIle mutation. Ann Neurol. 2006;59:570–574. doi: 10.1002/ana.20758. [DOI] [PubMed] [Google Scholar]
- 127.Sue CM, et al. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann Neurol. 2000;47:589–595. [PubMed] [Google Scholar]
- 128.Ronchi D, et al. Next-generation sequencing discloses DGUOK mutations in adult patients with mtDNA multiple deletions. Brain. 2012;135:3404–3415. doi: 10.1093/brain/aws258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Claypool SM, Boontheung P, McCafferty JM, Loo JA, Koehler CM. The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol Biol Cell. 2008;19:5143–5155. doi: 10.1091/mbc.E08-09-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 131.Haller RG, et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. J Clin Invest. 1991;88:1197–1206. doi: 10.1172/JCI115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hirano KI, Garone C, Quinzii C. CoQ10 deficiency and MNGIE: two treatable mitochondrial disorders. Biochim Biophys Acta. 2012;1820:625–631. doi: 10.1016/j.bbagen.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Glover EI, et al. A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve. 2010;42:739–748. doi: 10.1002/mus.21758. [DOI] [PubMed] [Google Scholar]
- 134.Carelli V, et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain. 2011;134:e188. doi: 10.1093/brain/awr180. [DOI] [PubMed] [Google Scholar]
- 135.Klopstock T, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–2686. doi: 10.1093/brain/awr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sadun AA, et al. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol. 2012;69:331–338. doi: 10.1001/archneurol.2011.2972. [DOI] [PubMed] [Google Scholar]
- 137.Enns GM, et al. Initial experience in the treatment of inherited mitochondrial diseases with EPI-743. Mol Genet Metab. 2012;105:91–102. doi: 10.1016/j.ymgme.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 138.Martinelli D, et al. EPI-743 reverses the progression of the pediatric mitochondrial disease—genetically defined Leigh symdrome. Mol Genet Metab. 2012;107:383–388. doi: 10.1016/j.ymgme.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 139.Kaufmann P, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 140.Gilkerson RW, et al. Mitochondrial autophagy in cells with mtDNA mutations results from the synergistic loss of transmembrane potential and mRORC1 inhibition. Hum Mol Genet. 2012;21:978–990. doi: 10.1093/hmg/ddr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tanaka M, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci. 2002;9:534–541. doi: 10.1159/000064726. [DOI] [PubMed] [Google Scholar]
- 142.Scott AB, Alexander DE, Miller JM. Bupivacaine injection of eye muscles to treat strabismus. Br J Ophthalmol. 2007;91:146–148. doi: 10.1136/bjo.2006.110619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen B, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–255. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 146.Viscomi C, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Flierl A, Chen YT, Coskun PE, Samulski RJ, Wallace DC. Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther. 2005;12:570–578. doi: 10.1038/sj.gt.3302443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Di Meo I, et al. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalomyopathy. EMBO Mol Med. 2012;4:1008–1014. doi: 10.1002/emmm.201201433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tachibana M, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tachibana M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Paull D, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Craven L, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Craven L, et al. Mitochondrial DNA disease: new options for prevention. Hum Mol Genet. 2011;20:R168–R174. doi: 10.1093/hmg/ddr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sagan Margulis L. On the origin of mitosing cells. J Theroet Biol. 1967;14:255–274. doi: 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- 155.DiMauro S, Schon EA. In: Mitochondrial Neurology. Waxman S, editor. Elsevier; 2007. pp. 535–551. [Google Scholar]