

Abstract
Identification of mutations at familial loci for amyotrophic lateral sclerosis (ALS) has provided novel insights into the aetiology of this rapidly progressing fatal neurodegenerative disease. However, genome-wide association studies (GWAS) of the more common (∼90%) sporadic form have been less successful with the exception of the replicated locus at 9p21.2. To identify new loci associated with disease susceptibility, we have established the largest association study in ALS to date and undertaken a GWAS meta-analytical study combining 3959 newly genotyped Italian individuals (1982 cases and 1977 controls) collected by SLAGEN (Italian Consortium for the Genetics of ALS) together with samples from Netherlands, USA, UK, Sweden, Belgium, France, Ireland and Italy collected by ALSGEN (the International Consortium on Amyotrophic Lateral Sclerosis Genetics). We analysed a total of 13 225 individuals, 6100 cases and 7125 controls for almost 7 million single-nucleotide polymorphisms (SNPs). We identified a novel locus with genome-wide significance at 17q11.2 (rs34517613 with P = 1.11 × 10−8; OR 0.82) that was validated when combined with genotype data from a replication cohort (P = 8.62 × 10−9; OR 0.833) of 4656 individuals. Furthermore, we confirmed the previously reported association at 9p21.2 (rs3849943 with P = 7.69 × 10−9; OR 1.16). Finally, we estimated the contribution of common variation to heritability of sporadic ALS as ∼12% using a linear mixed model accounting for all SNPs. Our results provide an insight into the genetic structure of sporadic ALS, confirming that common variation contributes to risk and that sufficiently powered studies can identify novel susceptibility loci.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a late-onset progressive neurodegenerative disorder mainly affecting motor neurones. ALS is the most common adult onset motor neurone disease with prevalence of 5 per 100 000 and with family history, age and male gender as the major risk factors (1–4). While familial ALS is well characterized with several causative genes identified to date, the genetic architecture of the more common sporadic form is poorly understood. Previous genome-wide association studies (GWAS) have identified several loci associated with ALS risk such as DPP6, ITPR2, FGGY and UNC13a (5–11) that have failed to be replicated in independent populations (12–15). One exception is a locus on chromosome 9p21 (15) that has been reliably replicated (16). At this locus, an expanded hexanucleotide repeat in the C9orf72 gene has been recently identified as the causative mutation in a large proportion of familial (23–47%) as well as sporadic (∼5%) ALS cases (17–20). This finding indicates that well-powered studies can identify novel loci associated with ALS susceptibility. To discover further loci, we designed a large GWAS meta-analysis with sufficient power to detect risk alleles with small effect sizes as observed in other neurodegenerative and complex diseases. There is evidence for a strong genetic component in sporadic ALS with heritability estimated to be 0.61 (95% CI: 0.38–0.78) in a combined study of 171 ALS twin pairs collected by three independent studies from Britain and Sweden (21), so we sought to estimate heritability derived from unrelated individuals and explained by common variation using GWAS data. Phenotype variation in complex traits is owing to interactions of genetic and environmental factors; thus, the quantification of the genetic variance is relevant in the study of multifactorial diseases. Heritability, the proportion of phenotype explained by genetic variance, is typically estimated in closely related individuals such as twin pairs, but this can inflate estimates as a consequence of epistatic interactions or shared environment (22,23). In contrast, heritability explained by associated SNPs identified in GWAS as passing-accepted thresholds of significance (typically P ≤ 5 × 10−8) explains only a small fraction of the genetic variation in most complex diseases. The difference between the phenotypic variance explained by GWAS results and those estimated in family studies is referred to as the ‘missing heritability problem’ and possibly is explained by incomplete linkage disequilibrium (LD) between genotyped SNPs and causal variants and/or by the presence of gene-by-gene or gene-by-environment interactions (22,23). We have estimated heritability of ALS considering all SNPs simultaneously regardless of their association with ALS phenotype and compared this with heritability from twin studies.
RESULTS
Association analyses
We analysed genotype raw data from eight independent studies including 3959 newly genotyped Italian individuals (1982 cases and 1977 controls) and 11 611 individuals (5195 cases and 6416 controls) from previously published studies. Full description of the sample size included in this study is reported in Table 1 and Supplementary Material, Table S1.
Table 1.
Sample size and genotype platforms
| Source | Sample ancestry | Sample size (N) | Cases (N) | Control (N) | Genotyping Illumina arrays | SNPs before imputation | SNPs after imputation |
|---|---|---|---|---|---|---|---|
| SLAGEN Consortium | Italian | 3959 | 1982 | 1977 | 660K | 657366 | 8391895 |
| UMC Utrecht | Dutch | 911 | 461 | 450 | 317K | 317503 | 8328981 |
| Utrecht, Umeå, Leuven | Dutch, Swedish, Belgian | 2806 | 1364 | 1442 | 370K | 317503 | 8372645 |
| MGH, King's College, Evry | Northern American, British, French | 3916 | 1710 | 2206 | 370K | 307790 | 8370626 |
| Beaumont Hospital, Dublin | Irish | 432 | 221 | 211 | 550K | 561466 | 8268635 |
| NIH publicly available, Coriell biobank | Northern American | 547 | 276 | 271 | 550K | 555352 | 8325506 |
| MND DNA bank and RADIANT study | British | 2252 | 663 | 1589 | 610K | 471994 | 8372429 |
| NIH publicly available | Italian | 747 | 500 | 247 | 550K | 500002 | 8312642 |
Independent studies analysed in the combined international meta-analysis were genotyped on different type of Illumina platforms. Italian data were collected by the SLAGEN Consortium (Italy) and the Dutch, Swedish, French, Belgian, Irish and North American, and Italian raw data were collected by the International ALS-GWAS Consortium (ALSAGEN). British cases were collected by UK National MND Bank samples and British controls by the RADIANT study (DeCC, BACCs) and NIH publicly available data from Coriell biobank (www.coriell.org).
Raw data for each study were assessed for quality control (QC) separately following the same criteria (see Supplementary Material and Table S2 for full discussion). Population structure of the individual cohorts was studied by means of principal components analysis (PCA) with EIGENSTRAT software (24,25), and outliers were identified by the projection of the first 10 principal components (Supplementary Material, Methods and Fig. S1).
To achieve the maximal coverage, we imputed genome-wide filtered originally genotyped data for each study (see Materials and methods). Overall, the average number of inferred genotypes was 8 342 920 SNPs varying proportionally to the original genotyping platform. After filtering (Supplementary Material, Methods), the eight datasets included genotype data for almost 7 million SNPs in 13 225 individuals (6100 cases and 7125 controls) (Supplementary Material, Table S2). Cleaned imputed genotypes were tested for association with ALS separately using SNPTESTv2.4.0 (26), and the logistic regression analyses were adjusted by the appropriate principal component axes estimated in the individual cohorts (Supplementary Material, Table S3). After genomic inflation control, we combined the logistic regression analysis results of each study in a meta-analysis using the program METAL (27). We adopted the ‘standard error’ analysis scheme, which combines effect size estimates (β-coefficients) across the studies weighted according to the inverse of the corresponding standard errors. As a result of the meta-analysis, we observed two loci reaching genome-wide threshold of significance, rs3849943 with P = 7.69×10−9 (OR 1.16, 95% CI: 1.10–1.22; average posterior probability (APP) 0.999; minor allele frequency (MAF) cases 0.268, controls 0.238) on chromosome 9p21 and SNP rs34517613 with P = 1.11 × 10−8 (OR 0.82, 95% CI: 0.76–0.87; APP 0.9279; MAF cases 0.108, controls 0.129) on chromosome 17q11.2.
A third locus, rs1788776, at 18q11.2 was very close to genome-wide significance threshold (P = 7.67 × 10−8, OR 0.87, 95% CI: 0.76–0.87; APP 0.9716; MAF cases 0.392, controls 0.362) (Table 2, Figs. 1 and 2, Supplementary Material, Fig. S2). With the exception of the locus on 9p21, loci previously found to be associated with ALS risk (5–11) did not reach genome-wide significance in our meta-analysis (Supplementary Material, Table S4) although SNP rs12608932 (UNC13A) at 19p13.3 had not complete coverage across datasets as it failed the QC threshold in the British dataset (Supplementary Material, Fig. S3). Additionally, examination of these previously associated loci in the Italian cohort as an independent replication study only found evidence for association for the 9p21 locus, and for the ITPR2 locus while significant, direction of effect is the opposite to that reported in the original study (8) (Supplementary Material, Fig S3). Only the ITPR2 locus showed significant evidence for heterogeneity across the different cohorts of European ancestry analysed here (Supplementary Material, Table S5). Further larger studies are needed to confirm the role of these loci in ALS susceptibility.
Table 2.
ALS-GWAS meta-analysis results
| Chromosome | SNP | Position | A1 | A2 | APP | Info | MAF_ALS | MAF_CONT | OR | P-value | Direction | Low 95% CI | Up 95% CI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 9 | rs3849943 | 27533382 | C | T | 1 | 0.999 | 0.268 | 0.238 | 1.166 | 7.69E−09 | +−−+++++ | 1.107 | 1.228 |
| 9 | rs3849941 | 27528948 | A | G | 0.993 | 0.986 | 0.266 | 0.236 | 1.166 | 9.54E−09 | −++−−−−− | 1.106 | 1.229 |
| 9 | rs2477523 | 27526894 | T | A | 0.997 | 0.992 | 0.269 | 0.239 | 1.163 | 1.06E−08 | −++−−−−− | 1.104 | 1.225 |
| 9 | rs2453555 | 27553868 | G | A | 0.995 | 0.988 | 0.27 | 0.239 | 1.163 | 1.07E−08 | −++−−−−− | 1.104 | 1.225 |
| 17 | rs34517613 | 23634379 | C | T | 0.922 | 0.734 | 0.108 | 0.129 | 0.822 | 1.11E−08 | ++++++++ | 0.769 | 0.879 |
| 17 | rs12937920 | 23614784 | C | T | 0.951 | 0.854 | 0.125 | 0.149 | 0.83 | 1.24E−07 | ++++++++ | 0.775 | 0.889 |
| 17 | rs9909055 | 23620255 | C | T | 1 | 1 | 0.217 | 0.249 | 0.857 | 1.98E−07 | ++++++++ | 0.809 | 0.908 |
| 17 | rs16964203 | 23606837 | A | C | 0.978 | 0.954 | 0.224 | 0.257 | 0.86 | 2.55E−07 | −−−−−−−− | 0.812 | 0.911 |
| 17 | rs2292633 | 23599507 | G | A | 0.956 | 0.906 | 0.221 | 0.254 | 0.86 | 2.82E−07 | ++++++++ | 0.812 | 0.911 |
| 17 | rs35714695 | 23743915 | G | A | 0.973 | 0.934 | 0.169 | 0.192 | 0.845 | 3.21E−07 | ++++++++ | 0.792 | 0.901 |
| 17 | rs739439 | 23747949 | C | T | 0.994 | 0.984 | 0.169 | 0.193 | 0.842 | 3.97E−07 | ++++++++ | 0.788 | 0.900 |
| 17 | rs9900311 | 23619534 | G | A | 0.997 | 0.992 | 0.219 | 0.252 | 0.858 | 4.58E−07 | ++++++++ | 0.808 | 0.911 |
| 18 | rs1788776 | 19498035 | G | A | 0.971 | 0.947 | 0.392 | 0.362 | 0.872 | 7.67E−08 | +++++−++ | 0.830 | 0.917 |
| 18 | rs1629335 | 19475137 | C | G | 0.956 | 0.927 | 0.383 | 0.353 | 0.874 | 1.00E−07 | −−−−−−−− | 0.832 | 0.918 |
| 18 | rs62093304 | 19489968 | T | C | 0.979 | 0.96 | 0.389 | 0.36 | 0.875 | 2.09E−07 | −−−−−+−− | 0.832 | 0.920 |
| 18 | rs12957850 | 19487958 | G | T | 0.988 | 0.976 | 0.408 | 0.382 | 0.878 | 2.64E−07 | +++++−++ | 0.836 | 0.923 |
| 18 | rs1652373 | 19482645 | G | T | 1 | 1 | 0.409 | 0.382 | 0.877 | 2.66E−07 | +++++− | 0.834 | 0.922 |
| 18 | rs12606211 | 19499478 | C | T | 0.979 | 0.961 | 0.408 | 0.381 | 0.88 | 3.29E−07 | +++++−++ | 0.838 | 0.924 |
| 18 | rs1711468 | 19498537 | C | A | 0.97 | 0.945 | 0.41 | 0.384 | 0.882 | 4.16E−07 | +++++−++ | 0.840 | 0.926 |
Summary of the most significant associated SNPs in the international ALS-GWAS meta-analysis from imputed genotypes. Meta-analysis (METAL) was performed weighting by the β-coefficient estimates and the inverse of their corresponding standard errors. Effect size of reference allele A1 is expressed with positive and negative symbols; symbols ‘+’ and ‘−’ indicate the direction of the effect size (beta values from regression), where a plus symbol means that increasing frequency of allele A1 is correlated with increasing trait values and vice versa.
Figure 1.
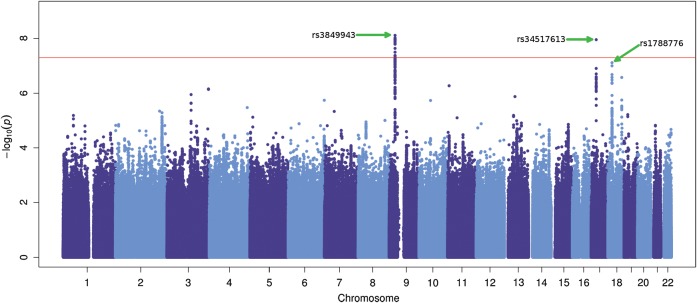
Manhattan plot of the international ALS meta-analysis. Scatter plot of chromosome position (x-axis) against –log10 GWAS meta-analysis P-values (y-axis) from imputed data. The threshold of genome-wide significance (P = 5 × 10−8) is indicated as a horizontal red line. At locus 9p21.2, 18 SNPs close to Corf72 gene lie above the red line (most significant SNP is rs3849943 with P = 7.69 × 10−9). Locus 17q11.2 shows SNP rs34517613 (P = 1.11 × 10−8) to be significantly associated. SNP rs1788776 at 18q11.2 is very close to the threshold of significance with P = 7.67 × 10−8. Manhattan plot was produced using ggplot2 package in R.
Figure 2.
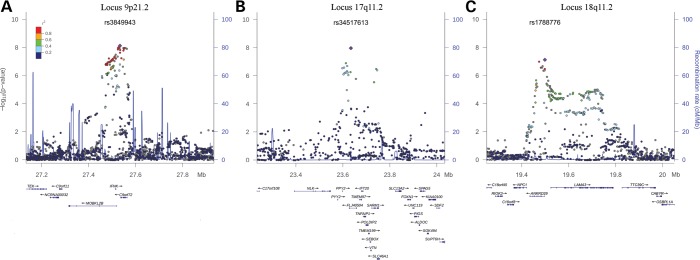
Regional association plots of the associated loci. LD structure of the three regions associated with ALS in the international GWAS meta-analysis. For each plot, the −log10 P-values (left y-axis) of SNPs are shown according to their chromosomal positions (x-axis); the genetic recombination rates are shown on the right y-axis. −log10 P-values are shown for both genotyped and imputed SNPs distributed in a 0.8-megabase genomic region. The top SNP of each region is indicated as a diamond, and SNPs colour reflects LD correlation (r2). (A) locus at 9p21, (B) 17q11.2 and (C) 18q11.2. LD plots were generated by LocusZoom v1.1.
As inclusion of non-confounding covariates such as gender and age can significantly reduce power to identify truly associated variants particularly when the disease prevalence is low (<1%), we have not included non-confounding covariates in logistic regression analyses presented here (28). In confirmation of our approach, the most significant SNPs (P ≤ 1 × 10−5) mapping in the two candidate loci at 17q and 18q showed no gender-by-SNP interaction in multi regression analysis (see Materials and methods).
For these loci, we searched for secondary signals by performing logistic regression conditioning upon the most associated SNPs under an additive model. There was no evidence of independent SNPs in any of these regions (Supplementary Material, Table S6). As the international meta-analysis included samples from Northern and Southern Europe, we tested the most associated SNPs for genetic heterogeneity quantifying the effect of the degree of variation between studies (Supplementary Material, Methods). We observed no significant heterogeneity in the distribution of allele frequencies between European populations for these SNPs (Supplementary Material, Table S7).
As additional evidence, we tested the candidate loci at 17q and 18q in a novel independent replication cohort of 2074 cases and 2556 controls collected in Italy, Netherlands and Germany (see full description in the Supplementary Material, Methods and Table S8). For each locus, we selected two variants, the most associated in the GWAS meta-analysis (imputed data) and combined joint analysis (original genotype data) (Supplementary Material, Table S9). We found suggestive evidence for association in the independent replica analysis for SNP rs34517613 at 17q (P = 0.055; OR 0.89, 95% CI: 1.00–0.78) and when combined in meta-analysis with the full GWAS data, the association identified in the discovery sample became more significant (Pcombined = 8.62 × 10−9; OR 0.833, 95% CI: 0.891–0.778). We found no additional evidence for association at the 18q11.2 locus (Supplementary Material, Table S10).
The locus at 17q11.2 is gene rich with a number of plausible candidates (Fig. 2). In an effort to refine associations at this locus and at 18q11.2, we searched for non-synonymous SNPs in LD (r2 ≥ 0.4) with our two lead signals to identify possible functional variants that explained the associations with ALS risk. No non-synonymous variants were found in LD at the 17q11.2 locus; however, three SNPs, rs739439 (r2 = 0.416) in the 3′ untranslated region, the intronic SNPs rs35714695 (r2 = 0.457) and rs34660379 (r2 = 0.412) within the SARM1 (sterile alpha and TIR-containing motif 1) gene were in LD (Table 2, Fig. 2).
At the suggestive 18q11.2 locus, the lead SNP rs1788776 was only in strong LD with SNPs in the ANKRD29 gene (Fig. 2), an ankyrin repeat domain-containing protein of undetermined function, these included missense SNPs (rs12956232, r2 = 0.78; rs1788758, r2 = 0.78; rs12960692, r2 = 0.74; rs11662113, r2 = 0.53) (Table 2, Fig. 2).
Next, we looked for cis eQTL (expression quantitative trait loci) in LD with our lead SNPs. All SNPs in LD (r2 ≥ 0.2) with rs34517613 and rs1788776 were analysed for association with cis eQTL using several publicly available eQTL databases (Supplementary Material), but none reached the P-value threshold for significance. This should not be considered conclusive as expression data from central nervous system tissues are still rather limited across eQTL databases and further studies should be carried out.
Contribution of C9orf72 to chromosome 9p21 association signal
As chromosome 9p21 was found to be strongly associated in our meta-analysis, we further investigated the contribution of ALS sporadic cases that also carry the expanded hexanucleotide repeat in the C9orf72 gene to this association. Information about carriers of the pathologic expansion in C9orf72 gene was available for 2287 of 6100 (37.3%) ALS cases from the Italian, Dutch and British studies included in the meta-analysis. The frequency of the expanded repeats carriers in the sporadic cases we have analysed progressively decreased as you proceed from Northern to Southern Europe ranging from 8 to 4.7%, respectively, in the British and Italian patients (see Materials and methods, Supplementary Material, Table S10). Our results confirm data previously reported (17–20). We performed a meta-analysis of this sample subset (2287 sporadic cases and 4162 controls) including and excluding cases (n = 144) with the expanded hexanucleotide repeat. Significant association of SNP rs3849943 decreased from P = 7.72 × 10−5 to P = 0.052 when the carriers were excluded from the logistic regression analysis, confirming that the association of 9p21 locus was largely dependent on carriers of the C9orf72 expansion (Supplementary Material, Fig. S4).
Heritability estimation
We used the Genome-wide Complex Trait Analysis (GCTA) method (29) to quantify additive genetic variance (narrow sense heritability) explained by all common SNPs regardless of their association with disease. GCTA uses a linear mixed-effect model that includes a random effect to estimate the polygenic component of trait variation (29). We performed GCTA analysis on 13 225 individuals (6100 ALS cases and 7125 controls) whose pair wise relationship was estimated to be <0.05 (PI_HAT) and used a restricted maximum likelihood (REML) algorithm to estimate the variance explained by imputed SNPs data. Analyses were carried out separately in the eight cohorts using a more stringent quality control threshold for imputed SNPs (MAF >0.01, posterior probability >0.9 and information measure >0.8) (see Materials and methods, Supplementary Material, Table S2) under the assumption of an ALS prevalence of 5 per 100 000. Heritability estimates were similar (∼11.5%) for all cohorts (Supplementary Material, Table S12) and were combined in meta-analysis modelled for random effects and weighted by sample size. The summary heritability was 0.119 (95% CI: 0.110–0.127) (Fig. 3).
Figure 3.

Heritability estimate across ALS-GWA studies. Heritability estimated in eight independent ALS cohorts. Forest plot shows heritability value and confidence interval (95%) calculated in each study at the reported prevalence of 5/100 000. Black boxes indicate the single studies, and box sizes are proportional to the sample (N). Grey bars specify lower and upper limit of 95% confidence interval.
Exclusion of the chromosome 9p21 locus did not affect heritability estimates. As ALS is a disease of aging and age is increasing in the human population, an increase in the prevalence of ALS can be predicted. Consequentially, the true prevalence of ALS may be higher than that used in our analysis. We have therefore calculated heritability over a range of prevalences, but for all prevalences, estimation of heritability attributable to common variation does not exceed 0.25 (Supplementary Material, Fig. S5).
DISCUSSION
In this study, we present the largest GWAS meta-analysis to date in ALS. Overall, we analysed 13 225 individuals with sufficient power to capture allelic association with small effects and low MAF. The novel associated SNP rs34517613 at 17q11.2 was confirmed when genotype data from the independent replication cohort were combined in meta-analysis with full GWAS data. We have also replicated the 9p21 locus at genome-wide significance; this locus was also identified by linkage studies of familial ALS patients and by analogy it may be that there is a corresponding familial locus for the 17q11.2 locus identified here, though we are not aware of any reports.
The lead SNP at this locus was in LD with three SNPs in the SARM1 gene. Interestingly, SARM1 orthologues, dSarm in Drosophila melanogaster and Sarm1 in mice, have been recently found to play a direct role in an axonal self-destruction pathway, a mechanism known as Wallerian degeneration, which shares morphological similarities with the axon dying-back degeneration observed in ALS and in other neurodegenerative pathologies (30,31). As axonal degeneration is an early feature of ALS progression, a mouse model for familial ALS, SOD1G93A, was crossed with the WldS mouse, a spontaneous mutant with phenotype of prolonged survival of injured axons because of a chromosomal rearrangement that disrupts two genes, Ube4b and Nmnat1. In the SOD1G93A/WldS, the progression of axonopathies was attenuated, suggesting that the Wallerian pathway could be involved in the axon loss observed in ALS (32,33).
We estimated the heritability of ALS owing to common variation as ∼12% based on a prevalence of 5 per 100 000 (1,2), although the true prevalence of ALS may be higher as consequence of increasing age in the human population as discussed earlier. We have also only considered autosomal SNPs and additional variance may be encoded on the X chromosome. Twin studies estimate heritability owing to all genetic variation including rare monogenic forms of the disease, whereas we have estimated heritability as a consequence of common polymorphisms either directly typed or imputed. The difference between heritability estimated from twin studies and from analysis of common SNP polygenic variation suggests a substantial role for variation not captured by genome-wide association studies.
Our estimate for the heritability of ALS explained by common variation is lower than that for other late-onset neurodegenerative diseases. Using similar approaches, heritability for late-onset Parkinson's disease was 0.31 (34), 0.24 for late-onset Alzheimer's disease and 0.30 for multiple sclerosis (35). These differences in heritability owing to common variants are reflected in the difficulty in identifying genome-wide significant loci for ALS compared with these diseases. For example, a two-stage GWA study of Parkinson's disease with a stage 1 sample size of ∼6000 individuals identified two strong association signals (36), and a study of Alzheimer's disease with a similar-sized stage 1 sample identified two loci additional to the well-replicated APOE locus (37). In comparison with these studies, our combined sample size is >13 000 individuals, suggesting that the genetic architecture of ALS may be different from other more common neurodegenerative diseases. A recent study of ALS in a Han Chinese population identified two loci of genome-wide significance in a combined sample of ∼5 000 (38); strikingly these loci were not replicated in populations of European ancestry, nor in the combined meta-analysis reported here. This suggests there may be considerable heterogeneity in ALS risk loci across different ethnicities; in support of this, the expanded repeat in the C9orf72 gene has been reported to be infrequent in Asian populations (39,40).
In conclusion, we have identified a novel locus for sporadic ALS risk at 17q11.2, but not replicated the suggestive evidence for a second locus at 18q11.2 and confirmed the association at 9p21 with the expanded hexanucleotide repeat in the C9orf72 gene. Furthermore, we provide evidence from our heritability estimates that further common variation affecting ALS risk remains to be detected by current GWAS platforms and with larger cohorts but that denser genome-wide assays and next generation sequencing technologies are required to detect rarer variation.
MATERIALS AND METHODS
Participating individuals
The discovery sample consisted of a novel Italian cohort collected by the Italian Consortium for the Genetics of ALS (SLAGEN) and seven independent published studies collected by ALSGEN, The International Consortium on Amyotrophic Lateral Sclerosis Genetics (5–11,15,16). All patients fulfilled the El Escorial revised criteria for ALS (41). Written informed consent according to the Declaration of Helsinki was obtained from all patients and healthy subjects participating to this study. Local ethical committees for each participating institution approved these studies. All samples across the eight datasets were of European ancestry, and a full description of demographic details is reported in the Supplementary Material, Table S1. All patients with family history of ALS or carrying Mendelian risk genes were excluded from analyses. Screening for the expanded hexanucleotide repeat in the C9orf72 gene were performed subsequently; information of mutation carries was available for a subset of cases included in the Italian, Dutch and British cohorts (see Supplementary Material for full description).
A replication sample of 4630 individuals (2074 cases and 2556 controls) was collected across Italy, The Netherlands and Germany using the same diagnostic criteria (see Supplementary Material, Table S8).
Genotyping procedures
Samples from the individual cohorts were genotyped on different Illumina beadchips (Illumina, CA, USA) as shown in Table 1. Additional SNPs were genotyped in the replication phase by PCR-based KASP technique by the KBioscience (UK) facility for the Italian samples (622 cases and 971 controls) or by TaqMan_2013 PCR method for the Dutch (877 cases and 1226 controls) and German samples (575 cases and 359 controls) (Supplementary Material). Cross-validation between methodologies showed 100% concordance.
Statistical analysis
Quality control and meta-analysis of imputed data
Before imputation analysis QC of samples and markers was performed separately in the eight studies using the PLINK software package (42). A detailed description of QC procedure is reported in the Supplementary Material and Table S2. Genotype data from British cases and controls were merged and analysed as single cohort as fully described in the Supplementary Material and Table S2.
Ancestry differences between individuals within each cohort were detected by PCA. Principal component axes were generated by genotypes of a genome-wide subset of LD-independent SNPs using EIGENSTRAT software (24,25), and outliers identified by the first 10 principal components (PCs) were removed. The number of significant principal components was estimated by Tracy–Widom distribution using the program TW statistic (25) and included as covariates in the logistic regression analyses of each study for population stratification control. Scatter plots of PC1 and PC2 showed no evidence of substructure between cases and controls within the single cohorts (Supplementary Material, Fig. S1). As an example, Supplementary Material, Figure S5 shows the population structure of the novel Italian cohort by the projection of the first two PC axes.
After QC, original genotype data of each study were tested for genomic inflation and lambda estimates resulted to be minimal (λ(gc) < 1.02). Individual datasets were imputed genome wide separately using the IMPUTE.v2 program (43,44), which employs combined reference panels of known phased haplotypes provided by HapMap 3 (Feb 2009), 1000 Genomes Project (Mar 2010) (NCBI build 36 coordinates) and the study's sample genotypes (Supplementary Material). In each cohort, imputed genotypes were tested for association with ALS status by logistic regression analysis (SNPTESTv2.4.0) including the specific PCs as confounder covariates to control population stratification. For each test, statistic spurious associations and genomic inflation were controlled by plotting the observed quantiles versus the expected ones in Q–Q plots and by factor λ(gc) estimate (Supplementary Material, Fig. S7 and Table S3) (R package software).
SNP statistic data were filtered for uncertainty of inferred genotypes according to posterior probability (APP) of >0.9 and statistical information of allele frequency (info) of >0.4 (26). The average number of SNPs filtered out in each dataset was 1.47 million (Supplementary Material, Table S2). Finally, filtered SNP tables were combined in a meta-analysis and analysed with the program METAL (27), applying the standard error scheme option that weights effect size estimates, or β-coefficients, using the inverse of the corresponding standard errors. The z-scores were calculated from the P-values of each study with the sign of the z-scores indicating the direction of allelic effect in that study (z > 0 for odds ratio >1). Meta-analysis was performed applying the fixed-effects model that does not account for study heterogeneity; therefore, heterogeneity was quantified by the Cochran's statistic (Q) and I2 (METAL). P-values were estimated by comparing the statistic with a X2 distribution (N—1 ‘degrees of freedom’, N = number of studies) (Supplementary Material and Table S5 and S7) (45,46). Meta-analysis was repeated stratifying by gender and testing for the gender-by-SNP interaction as described in Supplementary Material.
For the independent replication cohort, logistic regression analyses (SNPTESTv2.4.0) were performed separately in the Italian, Dutch and German samples and then combined in meta-analysis (METAL) applying standard error scheme as described earlier. Next, statistic tables of the three novel replication samples were combined with the discovery sample in a final meta-analysis (METAL).
Power calculation
The final meta-analysis cohort had 99.99% power to detect allelic association with an odds ratio (OR) of 1.2 and MAF of 0.25 at genome-wide significance (P = 5 × 10−8). For low-frequency variants with MAF ranging from 0.01 to 0.04 and of small effect (OR < 1.5), there was low power to capture variants with MAF 0.01, whereas for SNPs with MAF 0.02 power ranged from 60 to 98% for alleles with OR of 1.3 and 1.4, respectively (Supplementary Material, Fig. S8).
Screening of expanded repeats in C9orf72 gene
Hexanucleotide repeat expansion data for the C9orf72 gene were available for the Italian, Dutch and British cases (Supplementary Material, Table S10). As previously reported, carriers were defined as individuals with a range of the GGGGCC repeats of >23 units (17–20). We performed conditional logistic regression analysis on a subset of 2287 screened cases and 4162 controls including and excluding pathologic expansions carriers (n = 144).
Estimation of the genetic variance tagged by all SNPs
We used GCTA software (29) to estimate the proportion of ALS phenotypic variance explained by autosomal SNPs distributed genome wide. Genetic relationships between pairs of individuals were calculated separately in the eight cohorts where all samples were previously tested for cryptic relatedness and excluded if the proportion of IBD (identical-by-descent) estimate was >0.05 (Supplementary Material, Table S2). We estimated the genetic relationship matrix (GRM) including imputed genotypes filtered by MAF >0.01, posterior probability >0.9 and information measure >0.8. Filtered genotype data were submitted to GRM analysis, and GRMs output data were used in the REML analysis. In each dataset, REML analysis was carried out by fitting the specific principal components (Supplementary Material, Table S3) as covariates to control for possible population stratification. The variance estimate was transformed from the observed scale (V(1)/Vp) to a scale of liability (V(1)/Vp_L) by specifying a disease prevalence for ALS of 5 per 100 000 persons (1,2). The variance estimate of each cohort was combined by random effects meta-analysis weighted by sample size using the library rmeta in R toolset.
SUPPLEMENTARY MATERIAL
DATA AVAILABILITY
Data were accessed by formal request to the consortia members. The complete summary meta-analysis data (SNP, genomic position, odds ratio and P-value) are freely available at the ALSOD website (http://alsod.iop.kcl.ac.uk/) in a searchable format (47).
FUNDING
The SLAGEN Consortium was supported by funds from the Ministero della Salute Italiano (GWA-SLA, Ricerca Finalizzata 2007 grant number 31) and Fondazione Istituto Auxologico Italiano; I.F., C.G., B.C., J.F.P. and V.S. were supported by the Istituto Superiore di Sanità (grant number 526D/34, 2006); I.F. received salary support from ‘Amici del Centro Dino Ferrari’; V.S. acknowledges support from the ‘Amici del Centro Dino Ferrari’. A.R., C.G., B.C., N.T. and V.S. were supported by the Ministero della Salute Italiano (ALS-FTD, RF-2009-1473856); G.Si. was supported by Monte dei Paschi di Siena Foundation (grant 39167) and the Ministero della Salute Italiano (grant number RF-INP-2007-627227); A.C. was supported in part by Federazione Italiana Giuoco Calcio and Fondazione Vialli e Mauro per la Sclerosi Laterale Amiotrofica onlus, Ministero della Salute Italiano (grant number RF-PIE-2007-635695); AF was supported by Progetto Regione Emilia Romagna-Università ‘RARER’. The Italian replica control samples were obtained from the ‘Parkinson Institute Biobank’ (http://www.parkinsonbiobank.com) of the Telethon Genetic Biobank Network (http://www.biobanknetwork.org) supported by TELETHON Italy (grant number GTB07001) and by ‘Fondazione Grigioni per il Morbo di Parkinson’. ALSAGEN Consortium is supported by funds from the National Institutes for Health Research Biomedical Research Centre for Mental Health at the South London and Maudsley National Health Service Foundation Trust and Institute of Psychiatry, King's College London, the University of Leuven (grant number GOA/11/014), the Interuniversity Attraction Poles (IUAP) program P7/16 of the Belgian Federal Science Policy Office and the 7th Framework Programme of the European Union (ADAMS project, grant numbers HEALTH-F4-2009-242257 and FP7/2007-2013, grant agreements 259867 and 278611). P.V.D. holds a clinical investigatorship from FWO-Vlaanderen; W.R. is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders. The UK National DNA Bank for Motor Neuron Disease Research was funded by the Motor Neurone Disease Association (grant 3/3), the Wellcome Trust (grant number 070122/A/02/Z) and the National Institute for Health Research Dementias and Neurodegenerative Diseases Research Network. A.A.-C. and J.F.P. receive salary support from the National Institute for Health Research Dementia Biomedical Research Unit at South London and Maudsley National Health Service Foundation Trust and King's College London. The RADIANT depression and bipolar case control studies (DeCC, BACC) were genotyped under Medical Research Council (grant number G0701420). A.A.-C. thanks the Motor Neurone Disease Association of Great Britain and Northern Ireland, the ALS Association, the ALS Therapy Alliance and the Medical Research Council (grant number G0600974) for support. R.H.B. acknowledges support from the National Institute of Neurological Disorders and Stroke (grant number 5RO1-NS050557-05) and the National Institute of Neurological Disorders and Stroke American Recovery and Reinvestment Act Award (grant number RC2-NS070-342), the Angel Fund, the ALS Association, P2ALS, Project ALS, the Pierre L. de Bourgknecht ALS Research Foundation and the ALS Therapy Alliance. J.E.L. acknowledges grant support from National Institute of Health/ National Institute of Neurological Disorders and Stroke (grant number 1R01NS073873). The Dutch, Belgian and Swedish GWAS data generation was supported by the Prinses Beatrix Fonds, VSB fonds, H. Kersten and M. Kersten (Kersten Foundation), The Netherlands ALS Foundation, and J.R. van Dijk and the Adessium Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
Genetic and statistical analysis was performed using a Linux cluster supported by the National Institute for Health Research Biomedical Research Centre for Mental Health at the South London and Maudsley National Health Service Foundation Trust and Institute of Psychiatry King's College London. G.So. acknowledges EuroBiobank for ALS DNA samples; K.M. acknowledges Motor Neurone Disease Association UK, Queen Elizabeth Hospital Birmingham Charity. M.S. acknowledges support through EU via the Euro-MOTOR consortium and German BMBF Motoneuron disease network.
Conflict of Interest statement: None declared.
APPENDIX
Slagen Consortium members
Cereda Cristina (Laboratory of Experimental Neurobiology, IRCCS ‘C. Mondino’ National Institute of Neurology Foundation, Pavia, Italy), Comi Giacomo (Neurologic Unit, IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), D'Alfonso Sandra (Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, ‘A. Avogadro’ University, Novara, Italy), Fogh Isabella (Department of Neuroscience, Institute of Psychiatry, King's College London, London, UK; and Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milano, Italy), Gellera Cinzia (Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milano, Italy), Ratti Antonia (Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano; and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), Silani Vincenzo (Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano; and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), Sorarù Gianni (Department of Neurosciences, University of Padova, Padova, Italy).
Slagen collaborators
Bongioanni Paolo (Neurorehabilitation Unit, Azienda Ospedaliero-Universitaria Pisana, Pisa, Italy), Brescia Morra Vincenzo (Department of Neurological Sciences, University Federico II, Napoli, Italy), Calini Daniela (Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milan, Italy), Castellotti Barbara (Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milano, Italy), Ceroni Mauro (Laboratory of Experimental Neurobiology, IRCCS ‘C. Mondino’ National Institute of Neurology Foundation, Pavia, Italy), Conforti Francesca Luisa (Institute of Neurological Sciences, National Research Council, Mangone, Italy), Corbo Massimo (NeuroMuscular Omnicentre-NEMO, Fondazione Serena Onlus, Niguarda Ca' Granda Hospital, Milano, Italy), Corrado Lucia (Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, ‘A. Avogadro’ University, Novara, Italy), Corti Stefania (Neurologic Unit, IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico; and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), Del Bo Roberto (Neurologic Unit, IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico; and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), Ferlini Alessandra (Department of Medical Sciences, University of Ferrara, Ferrara, Italy), Filla Alessandro (Department of Neurological Sciences, University Federico II, Napoli, Italy), Filosto Massimo (Section for Neuromuscular Diseases and Neuropathies, Clinical Neurology, University Hospital ‘Spedali Civili’, Brescia, Italy), Gagliardi Stella (Laboratory of Experimental Neurobiology, IRCCS ‘C. Mondino’ National Institute of Neurology Foundation, Pavia, Italy), Inghilleri Maurizio (Department of Neurology and Psychiatry, University of Rome ‘Sapienza’, Roma, Italy), Marsili Angela (Department of Neurological Sciences, University Federico II, Napoli, Italy), Mazzini Letizia (Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, ‘A. Avogadro’ University; and ALS Center Department of Neurology, ‘Maggiore della Carità’ University Hospital, Novara, Italy), Pegoraro Elena (Department of Neurosciences, University of Padova, Padova, Italy), Penco Silvana (Department of Laboratory Medicine, Medical Genetics Unit, Niguarda Ca' Granda Hospital, Milano, Italy), Pensato Viviana (Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milano, Italy), Puorro Giorgia (Department of Neurological Sciences, University Federico II, Napoli, Italy), Querin Giorgia (Department of Neurosciences, University of Padova, Padova, Italy), Saccà Francesco (Department of Neurological Sciences, University Federico II, Napoli, Italy), Siciliano Gabriele (Division of Neurology, Department of Clinical and Experimental Medicine, University of Pisa, Nuovo Ospedale S.Chiara-Cisanello, Pisa, Italy), Sorbi Sandro (Department of Neurological and Psychiatric Sciences, University of Firenze, Firenze, Italy), Taroni Franco (Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico ‘Carlo Besta’, Milano, Italy), Ticozzi Nicola (Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano; and Department of Pathophysiology and Tranplantation, ‘Dino Ferrari’ Center, Università degli Studi di Milano, Milano, Italy), Tiloca Cinzia (Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milan, Italy).
Collaborators
La Bella Vincenzo (ALS Clinical Research Center, Department of Experimental BioMedicine and Clinical Neurosciences, Bio.Ne.C, University of Palermo, Palermo, Italy), Logroscino Giancarlo (Department of Neurosciences and Sense Organs, University of Bari, Policlinico, Bari, Italy), Monsurrò Maria Rosaria (Second Division of Neurology, Second University of Naples, Naples, Italy), Quattrone Aldo (Institute of Neurology, University Magna Graecia, Catanzaro, Italy), Simone Isabella Laura (Department of Neurosciences and Sense Organs, University of Bari, Policlinico, Bari, Italy).
Alsgen consortium collaborators
Ahmeti Kreshnik B. (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Ajroud-Driss Senda (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Al-Chalabi Ammar (MRC Centre for Neurodegeneration Research, Institute of Psychiatry, King's College London, London, UK), Andersen Peter M. (Institute of Pharmacology and Clinical Neuroscience, Section for Neurology, Umeå University, Umeå, Sweden), Armstrong Jennifer (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Birve Anne (Institute of Pharmacology and Clinical Neuroscience, Section for Neurology, Umeå University, Umeå, Sweden), Blauw Hylke M. (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), Brown Robert H. (Department of Neurology, University of Massachusetts Medical School, Worcester, MA, USA.), Bruijn Lucie (The ALS Association), Chen Wenjie (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Chiò Adriano (ALS Center, Department of Neuroscience, University of Turin, Torino, Italy), Comeau Mary C. (Wake Forest University Health Sciences, Medical Center Blvrd, Winston-Salem, NC, USA), Cronin Simon (Department of Neurology, Beaumont Hospital, Dublin, Ireland), Diekstra Frank P. (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), Soraya Gkazi Athina (King's Health Partners Centre for Neurodegeneration Research and Department of Clinical Neurosciences, Institute of Psychiatry Kings College London, London, UK), Glass Jonathan D. (Department of Neurology, Center for Neurodegenerative Disease, Emory University School of Medicine, Atlanta, GA, USA), Grab Josh D. (Wake Forest University Health Sciences, Medical Center Blvrd, Winston-Salem, NC, USA), Groen Ewout J. (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), Haines Jonathan L. (Vanderbilt University Medical Center, Nashville, TN, USA), Hardiman Orla (Dept Neurology, Beaumont Hospital and Trinity College Dublin, Dublin, Ireland), Heller Scott (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Huang Jie (Wellcome Trust Sanger Institute, Hinxton, UK), Hung Wu-Yen (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), ITALSGEN Consortium, Jaworski James M. (University of Miami, Miller School of Medicine, Biomedical Research Building, Miami, FL, USA), Jones Ashley (MRC Centre for Neurodegeneration Research, Institute of Psychiatry, King's College London, London, UK), Khan Humaira, Landers John E. (Department of Neurology, University of Massachusetts Medical School, Worcester, MA, USA.), Langefeld Carl D. (Wake Forest University Health Sciences, Medical Center Blvrd, Winston-Salem, NC, USA), Leigh Nigel P. (Brighton and Sussex medical School, Trafford Centre for Medical Research, Falmer, Brighton,UK), Marion Miranda C. (Wake Forest University Health Sciences, Medical Center Blvrd, Winston-Salem, NC, USA), McLaughlin Russell L. (Dept Population Genetics, Smurfit Institute, Trinity College Dublin, Dublin, Ireland), Meininger Vincent (Centre Référent Maladies Rares, APHP, UPMC, Hôpital de la Salpêtrière, Paris, France), Melki Judith (Institut National de la Santé et de la Recherche Médicale UMR-788, University of Paris 11, Biomedical Institute of Bicetre, Le Kremlin-Bicêtre, France), Miller Jack W. (King's Health Partners Centre for Neurodegeneration Research and Department of Clinical Neurosciences, Institute of Psychiatry, Kings College London, London, UK), Mora Gabriele (Department of Neurorehabilitation, Salvatore Maugeri Foundation, IRCCS, Scientific Institute of Milan, Milan, Italy), Pericak-Vance Margaret A. (University of Miami, Miller School of Medicine, Biomedical Research Building (BRB) #313, Miami, FL, USA), Rampersaud Evadnie (University of Miami, Miller School of Medicine, Biomedical Research Building (BRB) #313, Miami, FL, USA), Robberecht Wim (Neurology Department, KU Leuven, Belgium; and Vesalius Research Center, VIB, Leuven, Belgium), Shaw Christopher E. (PO43, Department of Clinical Neurosciences, Institute of Psychiatry, Kings College London, London, UK), Siddique Nailah (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Siddique Teepu (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Smith Bradley N. (King's Health Partners Centre for Neurodegeneration Research; PO43, Department of Clinical Neurosciences, Institute of Psychiatry Kings College London, London, UK), Sufit Robert (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Topp Simon (King's Health Partners Centre for Neurodegeneration Research; PO43, Department of Clinical Neurosciences, Institute of Psychiatry, Kings College London, London, UK), Traynor Bryan J. (Laboratory of Neurogenetics, National Institutes of Health, Bethesda, MD, USA), Vance Caroline (King's Health Partners Centre for Neurodegeneration Research, London, UK), van Damme Philip (Neurology Department, KU Leuven, Belgium; and Vesalius Research Center, VIB, Leuven, Belgium), van den Berg Leonard H. (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), van Es Michael (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), van Vught Paul (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), Veldink Jan H. (Rudolf Magnus Institute of Neuroscience, Department of Neurology, University Medical Centre Utrecht, Utrecht, The Netherlands), Yang Yi (Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA), Zheng J.G.(Northwestern University School of Medicine, Davee Department of Neurology, Division of Neuromuscular Medicine, Chicago, IL, USA).
REFERENCES
- 1.Mitchell J.D., Borasio G.D. Amyotrophic lateral sclerosis. Lancet. 2007;369:2031–2041. doi: 10.1016/S0140-6736(07)60944-1. [DOI] [PubMed] [Google Scholar]
- 2.Beghi E., Logroscino G., Chiò A., Hardiman O., Mitchell D., Swingler R., Traynor B.J. EURALS Consortium. The epidemiology of ALS and the role of population-based registries. Biochim. Biophys. Acta. 2006;1762:1150–1157. doi: 10.1016/j.bbadis.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Armon C. An evidence based medicine approach to the evaluation of the role of exogenous risk factors in sporadic amyotrophic lateral sclerosis. Neuroepidemiology. 2003;22:217–228. doi: 10.1159/000070562. [DOI] [PubMed] [Google Scholar]
- 4.Manjaly Z.R., Scott K.M., Abhinav K., Wijesekera L., Ganesalingam J., Goldstein L.H., Janssen A., Dougherty A., Willey E., Stanton B.R., et al. The sex ratio in amyotrophic lateral sclerosis, A population based study. Amyotroph. Lateral. Scler. 2010;11:439–442. doi: 10.3109/17482961003610853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Es M.A., van Vught P.W., Blauw H.M., Franke L., Saris C.G., Van den Bosch L., de Jong S.W., de Jong V., Baas F., van't Slot R., et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2007;40:29–31. doi: 10.1038/ng.2007.52. [DOI] [PubMed] [Google Scholar]
- 6.Cronin S., Berger S., Ding J., Schymick J.C., Washecka N., Hernandez D.G., Greenway M.J., Bradley D.G., Traynor B.J., Hardiman O. A genome-wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 2008;17:768–774. doi: 10.1093/hmg/ddm361. [DOI] [PubMed] [Google Scholar]
- 7.Schymick J.C., Scholz S.W., Fung H.C., Britton A., Arepalli S., Gibbs J.R., Lombardo F., Matarin M., Kasperaviciute D., Hernandez D.G., et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls, first stage analysis and public release of data. Lancet Neurol. 2007;6:322–328. doi: 10.1016/S1474-4422(07)70037-6. [DOI] [PubMed] [Google Scholar]
- 8.van Es M.A., Van Vught P.W., Blauw H.M., Franke L., Saris C.G., Andersen P.M., Van Den Bosch L., de Jong S.W., van’ t Slot R., Birve A., et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis, a genome-wide association study. Lancet Neurol. 2007;6:869–877. doi: 10.1016/S1474-4422(07)70222-3. [DOI] [PubMed] [Google Scholar]
- 9.Chiò A., Schymick J.C., Restagno G., Scholz S.W., Lombardo F., Lai S.L., Mora G., Fung H.C., Britton A., Arepalli S., et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009;18:1524–1532. doi: 10.1093/hmg/ddp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunckley T., Huentelman M.J., Craig D.W., Pearson J.V., Szelinger S., Joshipura K., Halperin R.F., Stamper C., Jensen K.R., Letizia D., et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 8. 2007:775–788. doi: 10.1056/NEJMoa070174. [DOI] [PubMed] [Google Scholar]
- 11.van Es M.A., Veldink J.H., Saris C.G., Blauw H.M., van Vught P.W., Birve A., Lemmens R., Schelhaas H.J., Groen E.J., Huisman M.H., et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 12.Fogh I., D'Alfonso S., Gellera C., Ratti A., Cereda C., Penco S., Corrado L., Sorarù G., Castellotti B., Tiloca C., et al. No association of DPP6 with amyotrophic lateral sclerosis in an Italian population. Neurobiol. Aging. 2009;32:966–967. doi: 10.1016/j.neurobiolaging.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Li X.G., Zhang J.H., Xie M.Q., Liu M.S., Li B.H., Zhao Y.H., Ren H.T., Cui L.Y. Association between DPP6 polymorphism and the risk of sporadic amyotrophic lateral sclerosis in Chinese patients. Chin. Med. J. (Engl) 2009;24:2989–2992. [PubMed] [Google Scholar]
- 14.Fernández-Santiago R., Sharma M., Berg D., Illig T., Anneser J., Meyer T., Ludolph A., Gasser T., et al. No evidence of association of FLJ10986 and ITPR2 with ALS in a large German cohort. Neurobiol. Aging. 2011;32:551–554. doi: 10.1016/j.neurobiolaging.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 15.Shatunov A., Mok K., Newhouse S., Weale M.E., Smith B., Vance C., Johnson L., Veldink J.H., van Es M.A., van den Berg L.H., et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries, a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmeti K.B., Ajroud-Driss S., Al-Chalabi A., Andersen P.M., Armstrong J., Birve A., Blauw H.M., Brown R.H., Bruijn L., et al. ALSGEN Consortium. Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol. Aging. 2012;34:357.e7–357.e19. doi: 10.1016/j.neurobiolaging.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., Kleinberger G., Janssens J., Bettens K., Van Cauwenberghe C., Pereson S., et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 20.Ratti A., Corrado L., Castellotti B., Del Bo R., Fogh I., Cereda C., Tiloca C., D'Ascenzo C., Bagarotti A., Pensato V., et al. C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol. Aging. 2012;33:2528.e7–2528.e14. doi: 10.1016/j.neurobiolaging.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Al-Chalabi A., Fang F., Hanby M.F., Leigh P.N., Shaw C.E., Ye W., Rijsdijk F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry. 2010;81:1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falconer D.S. Introduction to quantitative genetics. Longman Scientific & Technical, Wiley; 1986. Burnt Mill, Harlow, Essex, England New York. [Google Scholar]
- 23.Zaitlen N., Kraft P., Patterson N., Pasaniuc B., Bhatia G., Pollack S., Price A.L. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits. PLoS Genet. 2013;5:e1003520. doi: 10.1371/journal.pgen.1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 26.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 27.Willer C.J., Li Y., Abecasis G.R. METAL, fast and efficient meta-analysis of genome wide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pirinen M., Donnelly P., Spencer C.C. Including known covariates can reduce power to detect genetic effects in case-control studies. Nat. Genet. 2012;44:848–851. doi: 10.1038/ng.2346. [DOI] [PubMed] [Google Scholar]
- 29.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA, a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A., et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X.M., Luo L. dSarm-ing axon degeneration. Science. 2012;337:418–419. doi: 10.1126/science.1226150. [DOI] [PubMed] [Google Scholar]
- 32.Fischer L.R., Culver D.G., Davis A.A., Tennant P., Wang M., Coleman M., Asress S., Adalbert R., Alexander G.M., Glass J.D., et al. WldS gene modestly prolongs survival in the SOD1G93A. Neurobiol. Dis. 2005;19:293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Coleman M.P., Freeman M.R. Wallerian degeneration, wld(s) and nmnat. Annu. Rev. Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller M.F., Saad M., Bras J., Bettella F., Nicolaou N., Simón-Sánchez J., Mittag F., Büchel F., Sharma M., Gibbs J.R., et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson's disease. Hum. Mol. Genet. 2012;21:4996–5009. doi: 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S.H., Harold D., Nyholt D.R. Goddard M.E., Zondervan K.T., Williams J., Montgomery G.W., Wray N.R., Visscher P.M., editors. ANZGene Consortium, International Endogene Consortium, Genetic and Environmental Risk for Alzheimer's disease Consortium. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer's disease, multiple sclerosis and endometriosis. Hum. Mol. Genet. 2013;22:832–841. doi: 10.1093/hmg/dds491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simón-Sánchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G., et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M., Combarros O., Zelenika D., Bullido M.J., Tavernier B., et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 38.Deng M., Wei L., Zuo X., Tian Y., Xie F., Hu P., Zhu C., Yu F., Meng Y., Wang H., et al. Genome-wide association analyses in Han Chinese identify two new susceptibility loci for amyotrophic lateral sclerosis. Nat. Genet. 2013;45:697–700. doi: 10.1038/ng.2627. [DOI] [PubMed] [Google Scholar]
- 39.Iida A., Takahashi A., Deng M., Zhang Y., Wang J., Atsuta N., Tanaka F., Kamei T., Sano M., Oshima S., et al. Replication analysis of SNPs on 9p21.2 and 19p13.3 with amyotrophic lateral sclerosis in East Asians. Neurobiol. Aging. 2011;32:713–754. doi: 10.1016/j.neurobiolaging.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Ogaki K., Li Y., Atsuta N., Tomiyama H., Funayama M., Watanabe H., Nakamura R., Yoshino H., Yato S., Tamura A., et al. Analysis of C9orf72 repeat expansion in 563 Japanese patients with amyotrophic lateral sclerosis. Neurobiol. Aging. 2012;33:2527.e11–2527.e16. doi: 10.1016/j.neurobiolaging.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Miller R.G., Munsat T.L., Swash M., Brooks B.R. Consensus guidelines for the design and implementation of clinical trials in ALS. World Federation of Neurology committee on Research. J. Neurol. Sci. 1999;169:2–12. doi: 10.1016/s0022-510x(99)00209-9. [DOI] [PubMed] [Google Scholar]
- 42.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK, a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 45.Higgins J.P., Thompson S.G., Deeks J.J., Altman D.G. Measuring inconsistency in meta-analyses. Br. Med. J. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ioannidis J.P., Patsopoulos N.A., Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS One. 2007;2:e841. doi: 10.1371/journal.pone.0000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abel O., Powell J.F., Andersen P.M., Al-Chalabi A. ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012;9:1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were accessed by formal request to the consortia members. The complete summary meta-analysis data (SNP, genomic position, odds ratio and P-value) are freely available at the ALSOD website (http://alsod.iop.kcl.ac.uk/) in a searchable format (47).