

Abstract
Clinical oncology is hampered by a lack of tools to accurately assess a patient’s response to pathway-targeted therapies. Serum and tumor cell surface proteins whose abundance, or change in abundance in response to therapy, differentiates patients responding to a therapy from patients not-responding to a therapy could be usefully incorporated into tools for monitoring response. Here we posit and then verify that proteomic discovery in in vitro tissue culture models can identify proteins with concordant in vivo behavior and further, can be a valuable approach for identifying tumor-derived serum proteins. In this study we use Stable Isotope Labeling of Amino acids in Culture (SILAC) with proteomic technologies to quantitatively analyze the gefitinib-related protein changes in a model system for sensitivity to EGFR targeted tyrosine kinase inhibitors. We identified 3,707 intracellular proteins, 1,276 cell surface proteins, and 879 shed proteins. More than 75% of the proteins identified had quantitative information and a subset consisting of [400] proteins showed a statistically significant change in abundance following gefitinib treatment. We validated the change in expression profile in vitro and screened our panel of response markers in an in vivo isogenic resistant model and demonstrated that these were markers of gefitinib response and not simply markers of phospho-EGFR downregulation. In doing so, we also were able to identify which proteins might be useful as markers for monitoring response and which proteins might be useful as markers for a priori prediction of response.
Keywords: NSCLC, EGFR, Gefitinib, Proteomics, SILAC
Introduction
The epidermal growth factor receptor (EGFR) signaling pathway, which has been implicated in a range of cancers, including breast, lung, and colon carcinomas (1, 2), has been a particular focus of pathway-targeted therapeutics(3). The multilayered EGFR pathway, composed of ligands, receptors, and signaling molecules, impacts fundamental cellular processes such as differentiation, growth, motility, and division of epithelial cells (4, 5). Significant effort has been made to identify the constituents of the EGFR pathway, and demarcate the relationships among those constituents, their post-translational modifications (6), and more broadly to elucidate mechanisms for how the pathway interacts with diverse effector molecules which lead to changes in cellular behavior (7).
In addition to increasing our understanding of the EGFR axis, there may be significant clinical utility to identifying novel pathway members whose abundance is indicative of pathway dependence and activation status. This is especially important because lung cancer patients with activating mutations in EGFR demonstrate dramatic sensitivity to EGFR targeted tyrosine kinase inhibitors (TKI) (8, 9), such as gefitinib (ZD1839, Iressa, AstraZeneca) and erlotinib (OSI-774, Tarceva, Genentech) (10, 11). These compounds inhibit the downstream activity of the EGFR axis by competitively inhibiting ATP binding in the catalytic core of the kinase domain of EGFR. Though effective in some patients, those initially demonstrating clinical response to EGFR targeted TKI’s ultimately develop resistance(12). Diverse transcriptomic and phosphoproteomic studies, have also been undertaken to explore the impact of EGFR TKI inhibition on sensitive and resistant cells(6, 13-15). These approaches have greatly enhanced our understanding of EGFR biology and uncovered diverse mechanisms of resistance(16-19). Despite these studies, little is known about the relationship between EGFR inhibition and the proteome itself. In addition, there is still a significant unmet biologic and clinical need for proteins predictive of response or indicative of therapy effectiveness to EGFR TKI’s.
To bridge this gap, we first assessed the broad protein network effects of inhibiting EGFR kinase with gefitinib in an epidermoid cancer cell line (A431) that over-expresses EGFR. This cell line is a model system for sensitivity to EGFR targeted therapies (e.g., gefitinib) (20-22) and has been studied extensively. To quantify the gefitinib induced changes in protein abundance, we used Stable Isotope Labeling of Amino Acids in Culture (SILAC) with mass spectrometry (MS). Following our initial broadscale profiling, a panel of proteins was selected for extensive follow-up analysis including interrogation with various EGFR-axis specific and non-specific therapeutic agents. In order to assess the portability of these protein markers beyond A431 tissue we subsequently assessed the dose-dependent protein level change of our panel, and its generality both in vivo and to non small-cell lung cancer (NSCLC) tissue. These experiments suggested that our results are not restricted to in vitro studies of the A431 cell line, and may have potential utility for biologic and clinical studies of EGFR TKI inhibition.
Materials and Methods
Reagents and cell lines
All chemicals were purchased from Sigma (St. Louis, MO) unless stated otherwise. Antibodies directed to ELAVL-1, GLTSCR2, KLF5, QARS, were purchased from Abnova (Taipei City, Taiwan). Antibodies directed to HSPG2, BAG4, S100A9, SERPINE1, TNFAIP2, Lipocalin-2, and VAMP3 were purchased from Novus Biologicals (Littleton, CO). Antibody directed to Claudin-1 was purchased from Zymed/Invitrogen (Carlsbad, CA). Antibodies directed to ALB, Apo-L, C3, CBFB, EpCAM, PRDX6, and Transferrin were purchased from Abcam (Cambridge, MA). Antibodies directed to Testican 2 (SPOCK2), and TROP-2 were purchased from R&D Systems (Minneapolis, MN). Antibodies directed to EGFR, p-EGFR (Y-1068), and SNX5 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody directed to PDCD4 was purchased from Rockland Immunochemicals (Gilbertsville, PA). The antibody directed to actin was purchased from Sigma. A431, HCC827, H1650, H23, and H1975 cells lines were obtained from the standardized tissue bank, ATCC (Manassas, VA), which authenticates tissues by genotype and used within six months. Cells were verified to be free of mycoplasma (USC core facility). The MTS assay for cell viability at 48 hours (Figure S3) was conducted as directed by the manufacturer (Promega, Madison, WI).
Culture, isotopic labeling, and treatment of cells with therapeutic agents
The A431 human epithelial carcinoma cells were grown in DMEM media (Invitrogen) containing 1% of dialyzed fetal bovine serum (FBS, Invitrogen) with 13C - lysine (Invitrogen) substituted for lysine for seven passages (1:2) according the previously published SILAC protocol (23). We used a concentration of one percent serum (instead of the more typical 10% or higher) for our shed protein studies because it increases our ability to reliably quantify cell-derived signals amongst the background bovine serum proteins by an order of magnitude compared to 10% serum without compromising phenotype. Specifically, 1% serum does not compromise cell growth, viability, or response to therapy. To confirm this we preformed cell viability assays on A431 cells and did not observe differences in cell growth rate or gefitinib IC50 upon dosing between 1-10% serum. Cells remained attached to plates and there was no evidence of rounding-up. Incorporation of 13C Lys isotope exceeded 97% of the total protein lysine content. Samples from the same batch of cells were used for analysis of cell surface proteins and for analysis of conditioned media and whole cell lysate proteins. Cells were grown in the presence of 100 nM of gefitinib (Protein Kinases, Inc., Germany) for 16 h. The shed proteins were obtained directly from the cell conditioned media after 16 h of treatment. Cells and debris were removed by centrifugation at 5,000 × g and filtration through a 0.22-μm filter. Total extracts were obtained by sonication of ~2×107 cells in 1 ml of PBS containing the detergent octyl-glucoside (OG, 1% w/v) and protease inhibitors (complete protease inhibitor cocktail, Roche Diagnostics, Penzberg, Germany) followed by centrifugation at 20,000 × g.
Protein identification by LC-MS/MS
Protein digestion and identification by LC-MS/MS was performed as described previously (24). Briefly, each one of the reversed-phase fractions were individually digested in solution with trypsin (400 ng/fraction) and grouped into 15 to 21 pools for each cell line and each compartment (i.e., cell surface, conditioned media, and soluble whole cell lysate) based on chromatographic features. Pools were individually analyzed by LC-MS/MS in a LTQ-FTICR or LTQ-ORBITRAP mass spectrometer (Thermo-Finnigan, Waltham, MA) coupled to a nanoflow chromatography system (Eksigent, Dublin, CA) using a 25-cm column (Picofrit 75 μm ID, New Objectives, Woburn, MA) packed in-house with MagicC18 resin (Michrome Bioresources, Auburn, CA) over a 90 minute linear gradient. Acquired data was automatically processed using default parameters, except where noted, by the Computational Proteomics Analysis System V8.2 – CPAS (25). The tandem mass spectra were searched against version 3.13 of the human IPI database (60,428 protein entries) with five sequences for human and bovine trypsin added. The search was performed with X!Tandem (2005.12.01). The mass tolerance for precursor ions was set during the search to 1 AMU with a mass tolerance for fragment ions set to 0.5 Daltons. However, matches with less than 5ppm mass accuracy were considered to false positives and discarded. A fixed modification of 6.020129 mass units was added to lysine residues for database searching to account for incorporation of the heavy lysine isotope. All identifications with a PeptideProphet (26) probability greater than 0.9 were submitted to ProteinProphet (27) and the subsequent protein identifications were filtered at a 1% error rate with tryptic fragments (1 missed cleavage) with allowance for fixed modification on C = 57.021 and variable modifications on C = -17.027, E = -18.011, K = 6.020, M= 15.995, and Q = -17.027. Detailed proteomic methods in supplemental text.
In vivo xenografts
The A431 resistant model was derived in vivo from tumors with demonstrated resistance upon gefitinib dosing over a 9 month period by serial passage of A431 subcutaneous xenografts in presence of 50 mg/kg of gefitinib (AstraZeneca, London, UK) for nine months. Eight to 10 week-old nude athymic BALB/c female mice were obtained from Charles River Breeding Laboratories (Wilmington, MA) and were maintained in pressurized ventilated cages at the Cedars-Sinai Medical Center vivarium. All animal experiments were performed as per the institutional guidelines and were approved by the Institutional Animal Care and Use Committee at CSMC. Gefitinib was administered orally daily to twenty animals. Control animals received vehicle alone (20 animals). The tumor volumes were measured twice a week with a digital vernier caliper and were calculated as: π/6 × (larger diameter) × (smaller diameter)2. The data are represented as a plot of mean tumor volumes versus time measured in days from 5 animals.
Verification
In vitro and in vivo verification of proteomics data was accomplished by western blot. For in vitro experiments, cells were incubated in heavy SILAC media (1% FBS) for 16 h with 0, 100, 500, and 1,000 nM gefitinib, erlotinib (LC Labs, Woburn, MA), AKT inhibitor (A6730, Sigma), or MAPK inhibitor (PD98059, Calbiochem) or 5, 25, 100 nM taxol (LC Labs). The cells were washed three times with PBS and lysed in RAF buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EDTA; 1 mM PMSF; 1 mg/ml each aprotinin, leupeptin, pepstatin; 1 mM Na3VO4, 1 mM NaF) supplemented with 1% SDS. Lysates were sonicated for 10 minutes, heated at 95°C for 10 minutes, and centrifuged for 15 minutes at 20,000 × g. The supernatant was cleared through a 0.22-micron filter and protein concentration was determined (BCA, BioRad, Hercules, CA). Lysates were subject to SDS-PAGE and subsequent immunoblot. For in vivo experiments, mouse tumor pieces from 5 mice were pooled and suspended in RAF buffer and homogenized with a high-speed blender. The homogenate was centrifuged for 5 min at 200 × g. The supernatant was sonicated on ice for 2 minutes and centrifuged for 1 hr at 12,000 × g. The supernatant (soluble fraction) was cleared through a 0.22-micron filter and processed as above. The pellet (insoluble fraction) was resuspended in RAF buffer with 2% SDS and DTT, heated at 95°C and sonicated prior to processing. Serum from 5 mice was pooled and depleted using two MARS-3 columns (Agilent) connected in tandem with HPLC. The unbound fraction was concentrated to a final concentration of 2 mg/ml and processed as above.
Proteomics Data will be deposited at http://proteomics.fhcrc.org/CPL/home.html and will be accessible to the public.
Results
Deep quantitative proteomics of three sub-proteomes of A431 cells
In order to quantitatively enumerate the changes associated with the inhibition of EGFR activity, we compared untreated A431 cells with A431 cells treated with 100 nM gefitinib for either 2 or 16 hours prior to multi-compartment proteomic analysis. Three separate proteome fractionation techniques were used to interrogate cellular changes: a) shotgun LC/MS/MS to assess the intracellular proteome [referred to as the “whole cell lysate”]; b) biotin-capture-based cell-surface profiling; and c) Solid-Phase Extraction of Glycoprotein (SPEG) profiling (28) for the enrichment and subsequent study of shed proteins. By using three separate techniques we enabled a diversity of analysis; characterization of intracellular proteins is critical to understanding drug mechanism whereas the cell-surface and shed proteins are potentially relevant to molecular diagnostics, tumor imaging, and targeted therapies. Analysis was performed in a reciprocal fashion (e.g., in one experiment we treated isotopically ‘heavy’ cells and in a second experiment we treated isotopically ‘light’ cells). The dose of 100 nM gefitinib inhibits EGFR phosphorylation (IC50 40 – 80 nM) while minimizing suppression of other kinases (for example, the IC50 for HER2 is 1,200 – 3,700 nM) (29).
As a reference for non-TKI induced proteome changes, we admixed untreated A431(L) with untreated A431(H) and observed a standard deviation of 0.25 in the H/L fold change (data not shown). This enabled us to establish a threshold for significant quantitative fold change (1.4). We identified a total of 4,935 unique proteins (total peptides = 329,435; unique peptides = 82,269) with notable overlap between sub-proteomes (Fig. 1B). A complete list of observed proteins and their coverage is given in Table S1 and quantitation data are given in Table S2. The distribution of proteins with a fold change of ±1.4 was significantly higher in the 16-hour experiment and this time point was used for subsequent analyses (Fig. 1C).
Figure 1. Sub-proteome analysis of A431 cells with and without gefitinib treatment.
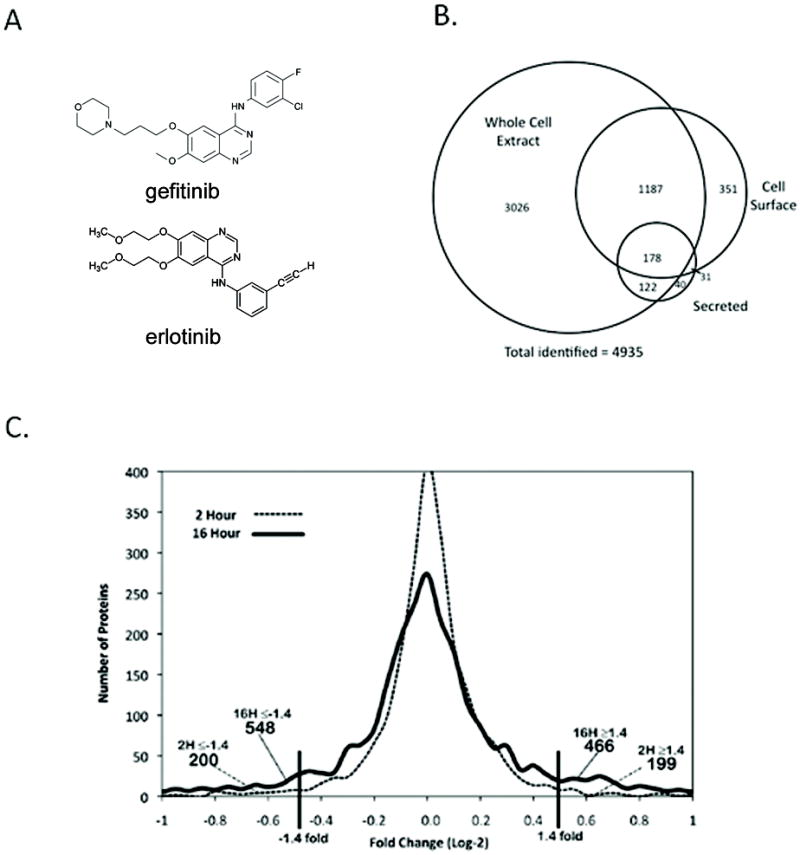
A431 cells were maintained in SILAC media and treated with 100 nM gefitinib for either 2 or 16 hours and analyzed by LC-MS/MS. Cell-surface proteins were isolated by biotin labeling and subsequently captured by avidin chromatography. Solid-phase extraction of glycoproteins was used to enrich in shed proteins. A) Chemical structure of gefitinib (top) and erlotinib (bottom). B) Total protein identifications based on compartment with a PeptideProphet score ≥ 0.9 and ProteinProphet score of ≥ 0.90. C) Histogram of (log2) fold change versus unique protein identifications for the 2 hr and 16 hr treatments shows quantitative changes in A431 proteomes were greater after 16 hours.
Verification of proteins indicative of response to gefitinib treatment
Of the 180 proteins with greater than 1.4 fold changes in abundance upon gefitinib-treatment (Table S2), 19 were initially selected for immunoassay-based verification based on magnitude of change in abundance, availability of commercially available affinity reagents of sufficient quality, and our confidence that the proteins were correctly identified and quantified (criteria are described in detail in the Methods section). Protein selection was unbiased with regard to biological function. We specifically focused on proteins whose abundance was up-regulated in response to treatment, as these are likely to be useful markers of response. We performed immunoblots on treated and untreated A431 lysates and were able to verify the treatment-response for 19 proteins. Densitometry was used to determine the quantitative fold change in protein concentration from three independent experiments (Fig. 2). Dose-titration is a common technique to verify the dose-dependent impact of a perturbation. We recognize, however, that higher concentrations of gefitinib may lead to off-target effects. However, it is most likely that such off target effects would mute the dose-dependent effect rather than producing or amplifying it. ELAVL-1 was the only protein whose response to gefitinib treatment at 100 nM was not amplified at higher doses (500 and 1,000 nM). All other proteins had a dose-dependent increase when extracts of cells treated with 100, 500, and 1,000 nM gefitinib as determined by western blot. VAMP3, KLF5, and GLTSCR2 demonstrated a marginal increase upon gefitinib dosing at 100 nM and as a result were excluded from further study as quantifying small fold changes can be potentially misleading. Overall, 16/19 (84%) immune-blot studies supported our proteomic predictions. This percentage may increase as new commercially available antibodies become available and as more sophisticated analytic techniques are employed.
Figure 2. In vitro verification of SILAC ratios.
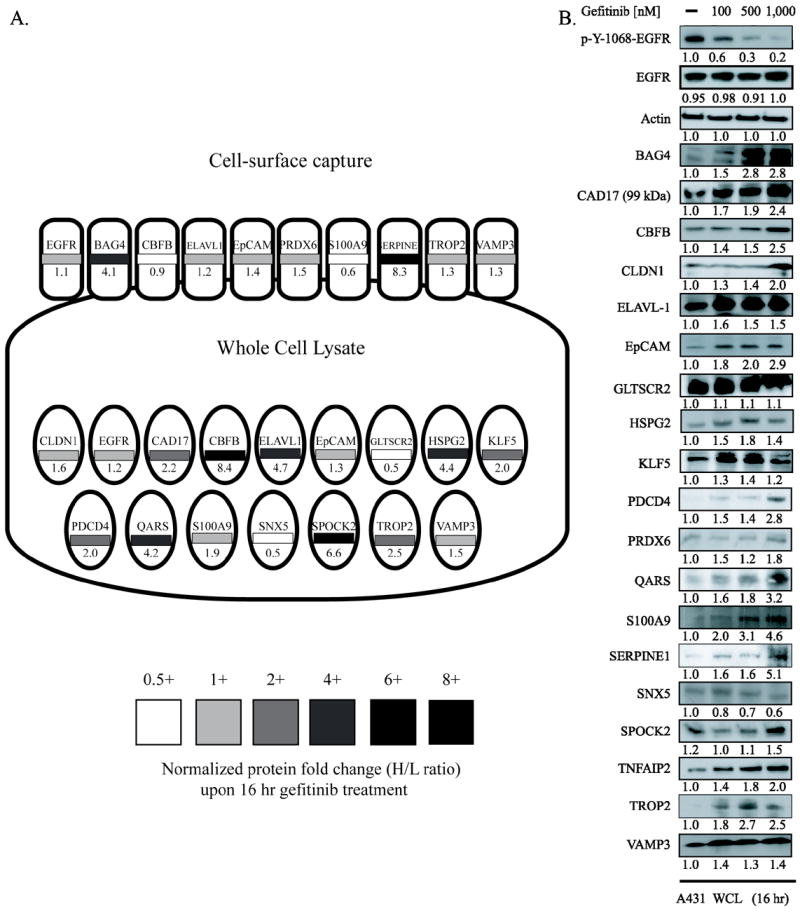
A) Fold change of candidate biomarkers is given as a function of compartment of identification with 16 hour gefitinib treatment. Proteins identified by cell-surface capture are depicted by rounded rectangles while proteins identified in the whole cell lysate are ellipses. Normalized fold change for each protein is given numerically and by the grayscale heat-map. (B) A431 cells were treated with vehicle control, 100nM, 500nM, and 1,000nM gefitinib for 16 hours. Densitometry analysis for three independent biological replicate experiments are listed below each immunoblot.
Pathway analysis of protein panel demonstrates specificity to EGFR pathway
In order to ascertain if the protein level changes associated with gefitinib dosing were a result of downregulation of EGFR tyrosine phosphorylation we examined the enrichment of known phosphoproteins amongst the 180 upregulated proteins. We observed that 56% of these proteins were previously characterized as phosphoproteins (Fig. S1B). This suggests that our proteomic experiment successfully impacted proteins associated with phosphorylation events. We used gene-set enrichment to identify protein networks associated with the perturbed proteins resulting from gefitinib treatment and observed network changes associated with non-canonical EGFR networks (Fig. S1C). As the 16 verified proteins were not members of any obvious, known bio-module (though most were within 1-hop of canonical EGFR pathway members), we sought to confirm that the impact of treatment on the proteins in our panel was specific to EGFR inhibition. A431 cells were treated with either erlotinib (100 – 1,000 nM) or with the off-axis control chemotherapeutic paclitaxel (10 – 100 nM) and the changes in levels of the 16 validated proteins and controls were investigated using immunoblots (Table I, Fig. S2A). Treatment with erlotinib produced protein abundance changes that mirrored the effect of gefitinib, whereas none of the proteins tested demonstrated a change in abundance in response to paclitaxel treatment. Since the IC50 of paclitaxel in A431 cells was previously shown to be ~ 10 nM (30), the lack of protein abundance changes with paclitaxel treatment at concentrations above the drug’s IC50 suggests that the proteins in our panel are not impacted by off-axis chemotherapeutics. The responses of the proteins in the panel to gefitinib and erlotinib and the lack of responses to paclitaxel suggest that levels of these proteins are perturbed as a function of the catalytic activity of the EGFR, but their known roles do not immediately suggest a mechanism for the observed behavior.
Table I. Cross Validation of Protein Panel across NSCLC Tissue and Treatments.
Dose-dependent change in protein levels were measured by western blot (data shown in Figures S2 and S3) and are marked with (+) for increase, (-) for decrease, (N.C) for no change, and (~) for not detected. The cell lines have various sensitivities for gefitinib: A431, IC50 = 40 - 80 nM; HCC827, 100 nM; H1650, 1 μM; H23, 8 μM; and H1975, 12 μM. Protein level changes of the panel of proteins after treatment with selective inhibitors of MAPK (PD 98059), AKT (LY294002), erlotinib (on-axis control), and paclitaxel (off-axis control) are shown.
| Tissue | A431 | HCC827 | H1650 | H23 | H1975 | A431 | A431 | A431 | A431 |
|---|---|---|---|---|---|---|---|---|---|
| gefitinib IC50 [nM] | 0.5 | 0.1 | 1.0 | 8.0 | 12.0 | ||||
| Treatment | gefitinib | gefitinib | gefitinib | gefitinib | gefitinib | AKT (A6730) | MAPK (PD98059) | erlotinib | paclitaxel |
| BAG4 | + | N.C. | N.C. | + | N.C. | + | N.C. | + | N.C. |
| CAD17 | + | + | + | + | N.C. | N.C. | + | + | N.C. |
| CBFB | + | + | + | N.C. | N.C. | + | N.C. | + | N.C. |
| Claudin 1 | + | N.C. | + | N.C. | N.C. | + | + | + | N.C. |
| ELAVL-1 | + | + | + | N.C. | N.C. | N.C. | N.C. | + | N.C. |
| EpCAM | + | + | + | N.C. | N.C. | N.C. | N.C. | + | N.C. |
| HSPG2 | + | N.C. | + | N.C. | N.C. | N.C. | N.C. | + | N.C. |
| PDCD4 | + | + | + | N.C. | N.C. | N.C. | + | + | N.C. |
| PRDX6 | + | + | + | N.C. | N.C. | N.C. | + | + | N.C. |
| QARS | + | + | + | N.C. | ~ | + | N.C. | + | N.C. |
| S100A9 | + | + | N.C. | N.C. | N.C. | N.C. | + | + | N.C. |
| SNX5 | - | - | N.C. | N.C. | N.C. | N.C. | N.C. | - | N.C. |
| SERPINE1 | + | + | + | N.C. | N.C. | N.C. | + | + | N.C. |
| TNFAIP2 | + | N.C. | + | + | N.C. | + | N.C. | + | N.C. |
| TESTICAN2 | + | + | N.C. | ~ | N.C. | N.C. | + | + | N.C. |
| TROP-2 | + | + | + | N.C. | N.C. | N.C. | N.C. | + | N.C. |
To demarcate the relative positions of each of our panel proteins within the broader EGFR signaling axis, we assessed how their abundance was impacted by inhibition of MAPK and AKT pathways. A431 cells were treated with either an AKT pathway specific tyrosine kinase inhibitor (LY294002) or a MAPK-specific TKI (PD98059). The majority of the panel proteins showed a change in abundance with one of these treatments (Table I, Fig. S2A). Specifically BAG4, CBFB, QARS, and TNFAIP2 showed a change in abundance in response to AKT inhibition and CAD17, PDCD4, S100A9, SERPINE1, and Testican-2 showed a change in abundance in response to MAPK inhibition. ELAVL-1, EpCAM, HSPG2, TROP2, and SNX5 were unchanged by treatment of cells with either TKI. Claudin-1 was the only protein that had changes in expression in the presence of both TKIs. In aggregate, the TKI inhibition data suggest that when EGFR signaling is perturbed, protein level changes are effected at various biological nodes. The proteins that were not perturbed upon treatment with either a MAPK or an AKT TKI are either upstream of these pathways or are part of another pathway. A schematic of the EGFR signaling pathway with inlays of the 16 proteins evaluated is depicted in Figure. S2B.
Potential a priori and post-therapy predictors of therapeutic response in lung cancer cell lines
As noted above, EGFR targeted therapies are extensively used in the treatment of non-small cell lung cancer. Consequently, to demonstrate that the gefitinib induced protein level change is not specific to the A431 model, we sought to investigate the generality of our protein panel across a set of NSCLC cell lines with varying sensitivity to gefitinib (Table I, Fig. S3). Gefitinib inhibits the phosphorylation of EGFR in HCC827, H1650, and H23 cell lines and is ineffective in H1975 cells as a result of the T790M mutation in EGFR kinase (31). There was clear concordance of the treatment-induced changes of protein abundance in the gefitinib-sensitive (HCC827, H1650) lines than in the gefitinib-resistant (H23, H1975) lines. Significant increase in the abundances of EpCAM, HSPG2, PDCD4, ELAVL-1, and TNFAIP2 were observed in the gefitinib-sensitive lines upon treatment. We also measured basal protein abundance across eight NSCLC lines (HCC827, H2935, H3255, H1666, H1650, H1975, H2235, H23) and found a relation between protein level and IC50 for many proteins in our panel suggests that, in addition to monitoring of therapeutic response, the levels of these proteins may correlate with gefitinib response a priori (Fig. S3).
In vitro response profile also observed in vivo
We next evaluated the expression and gefitinib induced perturbation of the proteins in our panel in an A431 xenograft model. Mice were treated with 50 mg/kg gefitinib or vehicle control after tumors became palpable. In mice treated with gefitinib, tumors were significantly smaller than those in untreated mice and grew at a much slower rate (Fig. 3A). Tumors and sera were collected at the experimental endpoint and abundance of our panel proteins was determined. Tumor lysates were fractionated to separate cell lysates into intracellular and membrane fractions. We measured the dose-dependent changes in abundance for the 16 proteins and compared these data to levels in untreated animals. The changes observed in vivo for 12 of the 16 proteins were in concordance with results from in vitro experiments (Fig. 3B). The dose-dependent changes in abundance were not identical in the cell-surface and the intracellular sub-proteomes. For proteins like PRDX6, which was chosen for validation based on its fold change in the whole-cell profiling experiments, the change in abundance in the insoluble (membrane) fraction was significantly greater than that in the soluble fraction.
Figure 3. A431 tumors are responsive to gefitinib in vivo.

A) Tumor growth curve for A431 xenografts in animals treated with 50 mg/kg gefitinib or left untreated and tumor growth curve for gefitinib-resistant tumors (A431-ZDR) in animals treated with 50 mg/kg gefitinib or left untreated. B) Tumor lysates were generated from A431 xenografts and analyzed by immunoblot for the 16 proteins in the panel. Lysates were fractionated to enrich for soluble and insoluble proteins.
Protein panel differentiates sensitive and resistant A431 tumors in vivo
To test the hypothesis that some of these proteins may distinguish the response phenotype of tumors to EGFR kinase inhibitors in vivo, we compare the basal levels and response to therapy of the markers in an in vivo derived xenograft model resistant to gefitinib (A431-ZDR). This line maintains wild-type EGFR. In addition, K-ras alleles and PTEN levels are identical to those in the parent A431 tumors. No differences in tumor growth rates were observed when animals treated with gefitinib were compared with untreated controls (Fig. 3A) even though EGFR phosphorylation was down-regulated in tumors from treated animals (Fig. 3B). In all xenograft experiments, the serum concentration of gefitinib was 150 +/- 50 nM. CAD17, CBFB, ELAVL-1, EpCAM, HSPG-2, QARS, S100A9, SNX5, SERPINE1, Testican 2, and TROP-2 expression changes were different in the A431-ZDR tumors than in the A431 tumors (Fig. 3B). This supports the hypothesis that certain panel proteins are correlative to response to gefitinib.
Protein panel differentiates sensitive and resistant A431 tumors in analysis of serum from tumor-bearing mice
Sera from mice implanted with either A431 or A431-ZDR tumors were collected and immunoblots were performed. When these mice were treated with gefitinib, dose-dependent changes were observed in EpCAM, TROP2, and PRDX6 that differed in mice implanted with gefitinib sensitive and resistant tumor cells. In addition, we were able to differentiate sera from sensitive and resistant mice as the serum concentrations of EpCAM and PRDX6 were significantly higher in the A431-ZDR serum than in A431 serum (Fig. 4).
Figure 4. Analysis of panel proteins from serum of animals bearing gefitinib-sensitive and gefitinib-resistant tumors.
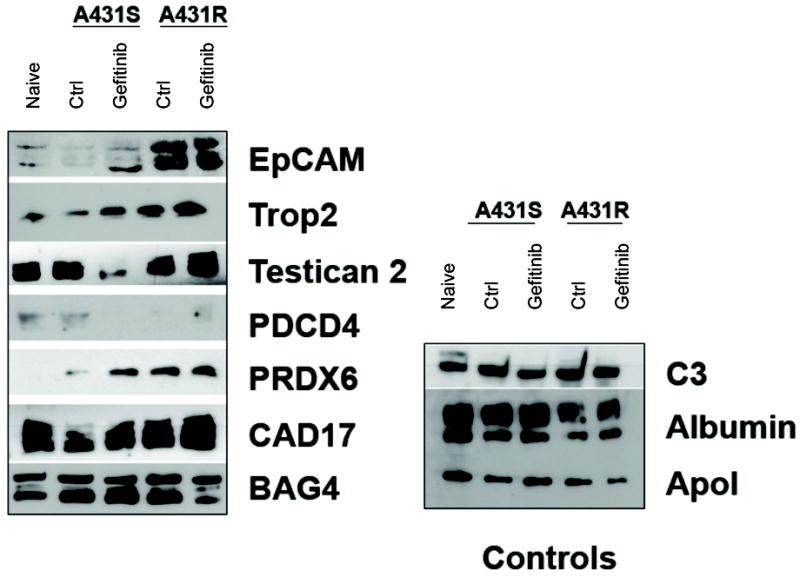
Sera were collected from naïve mice and mice implanted with A431 cells and A431-ZDR tumors and analyzed by immunoblot for the 16 proteins in our panel. Seven of the 16 proteins were detectable in sera.
Discussion
In this study, we employed quantitative proteomics to identify a panel of proteins whose change in abundance upon treatment correlates with sensitivity to EGFR tyrosine kinase inhibition across a number of cell lines and in an in vivo (iso-genic sensitive/resistant) xenograft model. We also demonstrated differences between baseline abundance and levels of certain proteins after gefitinib treatment in gefitinib-sensitive versus resistant NSCLC cell lines (Figure S4 & Table I). Several proteins showed changes dependent on gefitinib treatment in an in vivo tumor model (Fig. 3). The results presented highlight a proteomics paradigm where discovery in vitro successfully translates to concordant behavior in vivo. In addition, our model-based, but broad-scale approach allowed for significant and diverse investigation of the specificity and generality of discovered proteins. An alternate panel discovery strategy may have been to investigate the known members of the EGFR signaling axis. However, as noted, none of the discovered candidate proteins were previously characterized as connected to the EGFR signaling axis. Furthermore, though forty-eight proteins previously associated with EGFR signaling (32) were identified and quantified in our analysis (Table S2), they showed no significant changes in abundance in response to EGFR targeted therapies. The average fold increase in abundance of these proteins was 0.98 in the intracellular fraction and 1.04 in the cell-surface sub-proteome. Since the predominant response to gefitinib is known to be decreased phosphorylation of EGFR, it is not surprising that expression levels of the 48 pathway members did not change significantly in response to gefitinib treatment. Ontology enrichment and gene set analysis on the proteins with more than a ±1.4 fold change in abundance showed no significant enrichment in the EGFR ontology tree or in any other gene set (GO, IPA (33)) (Fig. S1).
The biological functions of the proteins identified in this study are diverse and (with the exception of SNX5, which has been implicated in membrane trafficking and degradation of the EGFR (34)) have not been previously implicated in EGFR function. Two of the proteins we identified, EpCAM and TROP2, have been implicated in gefitinib resistance (35, 36). In A431 cells and gefitinib-sensitive NSCLC cell lines, the expression of EpCAM and TROP2 was up-regulated upon gefitinib treatment (Figs. 2-4) and the serum concentration of EpCAM was higher in mice implanted with gefitinib-resistant xenografts than gefitinib-sensitive cells. The basal levels of EpCAM in NSCLC cell lines also correlated with gefitinib resistance (Fig. S4). PDCD4 is the only protein we identified that was previously shown to function downstream of the EGFR signaling pathway. PDCD4 up-regulation is associated with increased c-jun transcriptional activity (37). We found that PDCD4 expression was modulated by RAS/MAPK activity and gefitinib treatment in gefitinib-sensitive NSCLC cell lines (Table I). The other proteins in our panel have been implicated in various biological functions ranging from protein biosynthesis to regulation of fibrinolysis, demonstrating the biological reach of the EGFR signaling pathway. The reasons for the perturbation of levels of these proteins observed in A431 and NSCLC cell lines are unclear but an understanding of the involvement of these proteins in gefitinib resistance is worth pursuing
We also observed that EGFR changes glycosylation state, but not total abundance, in response to gefitinib treatment(Fig. S1). By overlaying total protein fold change (+1.18 compared to untreated cells) with the N-linked glycosylation data, we identified a region within domain III of EGFR extracellular domain that demonstrated a dose-dependent decrease in glycosylation at N356 (Table S3). This is relevant to EGFR biology because N-linked glycosylation of EGFR impacts ligand binding(38), alters receptor self-association and activity(39), and modulates the effectiveness of EGFR-TKIs(40). This observation highlights the importance of glycobiology within the framework of EGFR signaling.
A central component for our model-based discovery-based proteomic experiments is the translation of in vitro, model-based, perturbations to complex, in vivo, model systems. There are many steps in this process, from the initial discovery, through diverse verification steps to the development of assays for clinical application. In this study, we hypothesized and demonstrated that an in vitro model system for EGFR signaling may be utilized to predict gefitinib induced changes in NSCLC cell lines and in vivo xenografts. We additionally suggested that an important component of such studies are diverse, mechanism-based verification steps that go beyond verifying the accuracy of mass-spectrometry results, but instead delve into the biological origins and consequences of observed quantitative events. Such studies are highly unique and allow an investigation into both the generality and specificity of putative marker studies. In a study by Myers, et al, the combined inhibition/excitation profile of A431 cells were used to predict response in colorectal cancer tissue(41). Both studies demonstrate that in vitro model-based systems may be used to identify protein level changes that are recapitulated in complex systems. Our study further hypothesized that blood biomarkers are typically comprised of either shed or cell surface proteins(42) and hence utilized a compartment based strategy to enhance the likelihood for the translation of any putative biomarker to a clinical tool. Furthermore, we found it particularly interesting that most of these markers were not immediate family members of EGFR, but instead a few steps away. As a result, we compared the gefitinib inhibition profile with that of two downstream nodes of EGFR signaling (MAPK and AKT). By investigating the protein profile of candidates across a large number of lung cancer cell lines, we demonstrated the generality of the marker panel. By contrasting the impact of inhibition with EGFR (gefitinib) to MAPK(PD98059), AKT(LY294002), to that of a non-EGFR cytotoxic agent (paclitaxel) we were able to determine specificity of response. It remains not only crucial to identify putative biomarkers but equally important to perform experiments that hasten clinical translation of such proteins by clarifying the specificity and generality of putative markers coordinated changes in abundance.
Here we have presented a study in which a panel of proteins was obtained via quantitative proteomic comparison of cells that overexpresses the EGFR before and after treatment with an inhibitor of EGFR kinase. The proteins up-regulated upon gefitinib treatment appear to be connected to the EGFR-kinase activity and changes in expression are indicative of cellular network responsiveness to the targeted therapeutic agent. Expression changes were significantly concordant between in vitro and in vivo systems, suggesting that in vitro models may be generally used as a discovery environment. Notably, we have observed five of the sixteen proteins in the sera of lung cancer patients (private communication, Hanash Lab) and at least two representative peptides from each of the sixteen proteins identified in this study have been identified by various research groups and deposited in Human Plasma PeptideAtlas(43) and/or PRIDE(44). The biological relevance of tumor-derived proteins may be easier to assess and a greater variety of quantitative proteomic techniques are currently applicable in vitro than in vivo. The results indicated herein may ultimately have tremendous practical impact, as there is a significant clinical need for serum-based tools that can stratify and characterize tumor behavior.
Supplementary Material
Acknowledgments
We thank Sanjiv Sam Gambhir from Stanford University for the suggestions and comments in this project. We thank Damien Wood for his expertise in HPLC and LC-MS/MS. We thank Spielberg Family for funding.
Grant Support:
CCNE-TR 5U54CA119367, CCNE-T 1U54CA151459, PSOC-MCSTART 5U54CA143907
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, et al. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–70. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- 3.Arteaga CL, Baselga J. Tyrosine kinase inhibitors: why does the current process of clinical development not apply to them? Cancer Cell. 2004;5:525–31. doi: 10.1016/j.ccr.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 4.Ullrich A, Riedel H, Yarden Y, Coussens L, Gray A, Dull T, et al. Protein kinases in cellular signal transduction: tyrosine kinase growth factor receptors and protein kinase C. Cold Spring Harb Symp Quant Biol. 1986;51(Pt 2):713–24. doi: 10.1101/sqb.1986.051.01.084. [DOI] [PubMed] [Google Scholar]
- 5.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 6.Morandell S, Stasyk T, Skvortsov S, Ascher S, Huber LA. Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics. 2008;8:4383–401. doi: 10.1002/pmic.200800204. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–4. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 9.Riely GJ. The use of first-generation tyrosine kinase inhibitors in patients with NSCLC and somatic EGFR mutations. Lung Cancer. 2008;60(Suppl 2):S19–22. doi: 10.1016/S0169-5002(08)70101-6. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. Oncologist. 2003;8:303–6. doi: 10.1634/theoncologist.8-4-303. [DOI] [PubMed] [Google Scholar]
- 11.Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer. 2004;11:689–708. doi: 10.1677/erc.1.00600. [DOI] [PubMed] [Google Scholar]
- 12.Rubin BP, Duensing A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab Invest. 2006;86:981–6. doi: 10.1038/labinvest.3700466. [DOI] [PubMed] [Google Scholar]
- 13.Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II, Yang SH, et al. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009;63:219–26. doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25:843–54. doi: 10.1007/s10585-008-9200-4. [DOI] [PubMed] [Google Scholar]
- 15.Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–7. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 17.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 19.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 20.Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–25. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 21.Bryant JA, Finn RS, Slamon DJ, Cloughesy TF, Charles AC. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol Ther. 2004;3:1243–9. doi: 10.4161/cbt.3.12.1233. [DOI] [PubMed] [Google Scholar]
- 22.Oyama M, Kozuka-Hata H, Tasaki S, Semba K, Hattori S, Sugano S, et al. Temporal perturbation of tyrosine phosphoproteome dynamics reveals the system-wide regulatory networks. Mol Cell Proteomics. 2009;8:226–31. doi: 10.1074/mcp.M800186-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1:2650–60. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 24.Faca V, Pitteri SJ, Newcomb L, Glukhova V, Phanstiel D, Krasnoselsky A, et al. Contribution of protein fractionation to depth of analysis of the serum and plasma proteomes. J Proteome Res. 2007;6:3558–65. doi: 10.1021/pr070233q. [DOI] [PubMed] [Google Scholar]
- 25.Rauch A, Bellew M, Eng J, Fitzgibbon M, Holzman T, Hussey P, et al. Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J Proteome Res. 2006;5:112–21. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 26.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 27.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y, Aebersold R, Zhang H. Isolation of N-linked glycopeptides from plasma. Anal Chem. 2007;79:5826–37. doi: 10.1021/ac0623181. [DOI] [PubMed] [Google Scholar]
- 29.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–54. [PubMed] [Google Scholar]
- 30.Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, et al. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12:4695–701. doi: 10.1158/1078-0432.CCR-06-0346. [DOI] [PubMed] [Google Scholar]
- 31.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 33.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–7. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Liu ZQ, Chen CX, Magill S, Jiang Y, Liu YJ. Inhibitory regulation of EGF receptor degradation by sorting nexin 5. Biochem Biophys Res Commun. 2006;342:537–46. doi: 10.1016/j.bbrc.2006.01.179. [DOI] [PubMed] [Google Scholar]
- 35.Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA, Jr, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–91. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 36.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 37.Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, et al. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006;26:1297–306. doi: 10.1128/MCB.26.4.1297-1306.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cummings RD, Soderquist AM, Carpenter G. The oligosaccharide moieties of the epidermal growth factor receptor in A-431 cells. Presence of complex-type N-linked chains that contain terminal N-acetylgalactosamine residues. J Biol Chem. 1985;260:11944–52. [PubMed] [Google Scholar]
- 39.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–83. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 40.Ling YH, Li T, Perez-Soler R, Haigentz M., Jr Activation of ER stress and inhibition of EGFR N-glycosylation by tunicamycin enhances susceptibility of human non-small cell lung cancer cells to erlotinib. Cancer Chemother Pharmacol. 2009;64:539–48. doi: 10.1007/s00280-008-0902-8. [DOI] [PubMed] [Google Scholar]
- 41.Myers MV, Manning HC, Coffey RJ, Liebler DC. Protein expression signatures for inhibition of epidermal growth factor receptor mediated signaling. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang Q, Kani K, Faca VM, Zhang W, Zhang Q, Jain A, et al. Impact of protein stability, cellular localization, and abundance on proteomic detection of tumor-derived proteins in plasma. PLoS One. 2011;6:e23090. doi: 10.1371/journal.pone.0023090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deutsch EW, Eng JK, Zhang H, King NL, Nesvizhskii AI, Lin B, et al. Human Plasma PeptideAtlas. Proteomics. 2005;5:3497–500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 44.Barsnes H, Vizcaino JA, Eidhammer I, Martens L. PRIDE Converter: making proteomics data-sharing easy. Nat Biotechnol. 2009;27:598–9. doi: 10.1038/nbt0709-598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.