

Abstract
Asymmetric epoxidation of various styrenes using carbocyclic oxazolidinone-containing ketone 3 has been investigated. High enantioselectivity (89–93% enantiomeric excess) has been attained for this challenging class of alkenes. Mechanistic studies show that substituents on the ketone catalyst can have electronic influences on secondary orbital interactions, which affects the competition between spiro and planar transition states and, ultimately, enantioselectivity. The results described herein not only reveal the potential of chiral dioxirane catalyzed asymmetric epoxidation as a viable entry into this important class of olefins but also further enhance the understanding of the mechanistic aspects of chiral ketone-catalyzed asymmetric epoxidation.
Epoxides are important intermediates for the synthesis of complex molecules. Asymmetric epoxidation of prochiral alkenes presents a powerful strategy for the synthesis of enantiomerically enriched epoxides. Great progress has been made in the areas of asymmetric epoxidation of allylic alcohols (1–3), chiral metal complex-catalyzed epoxidation of unfunctionalized alkenes (particularly with conjugated cis- and trisubstituted alkenes) (4–7), and asymmetric epoxidation of electron-deficient alkenes under nucleophilic conditions (8–10). Styrene oxides are extremely useful and can be prepared by a number of methods such as asymmetric reduction of α-halo acetophenones (refs. 11 and 12 and references therein), asymmetric dihydroxylation of styrenes (13, 14), and kinetic resolution of racemic epoxides (15). The asymmetric epoxidation of styrenes also has received a considerable amount of interest. Various chiral catalysts and reagents have been investigated for the epoxidation of styrenes, including chiral porphyrin complexes (16–34), chiral salen complexes (35–42), chiral oxaziridines and oxaziridinium salts (43–46), and enzymes (47–51). Metal catalysts such as chiral porphyrin and salen complexes have been studied extensively for the epoxidation of these alkenes, and the enantioselectivities have reached the 80% range in a number of cases (21, 23, 29, 31, 37, 38, 40, 41), with 96% enantiomeric excess (ee) being obtained in one case (3,5-dinitrostyrene) (23).
Among other oxidants, dioxiranes either isolated or generated in situ are powerful epoxidation agents (Scheme 1; refs. 52–54). Chiral dioxiranes were shown recently to be effective for asymmetric epoxidation of trans-, trisubstituted (55–91), and certain cis-alkenes (92–95). Asymmetric epoxidation of styrenes with chiral dioxiranes has been studied also (61, 66, 68, 81, 88, 90, 93–95); however, the enantioselectivity has not exceeded 85% (93–95). Generally speaking, the highly enantioselective epoxidation of styrenes still remains a formidable challenge.
Scheme 1.

During our studies, we found that varying the catalyst structure could have a dramatic effect on enantioselectivity for the dioxirane-mediated epoxidation of styrene. Fructose-derived ketone 1 (Scheme 2), a very effective catalyst for trans- and trisubstituted alkenes, gave only 24% ee for styrene (89–91). On the other hand, 81% ee was obtained for styrene when ketone 2 was used (93). In our efforts to further study the conformational and electronic effects of ketone catalysts on epoxidation, we found that ketone 3 (prepared from quinic acid in 13 steps with 1.1% overall yield), a carbocyclic analog of 2, provides very high enantioselectivity for the epoxidation of styrenes. Herein we report our studies on this subject.
Scheme 2.

Materials and Methods
General. All chemicals were purchased and used without additional purification unless otherwise noted. Experimental details of the synthesis of ketone 3 as well as the GC spectra for all the styrenes are reported in Supporting Methods and Materials, which is published as supporting information on the PNAS web site.
Representative Asymmetric Epoxidation Procedure (Table 1, Entry 1). To a solution of styrene (0.0104 g, 0.10 mmol) and ketone 3 (0.0060 g, 0.02 mmol) in dimethoxyethane/dimethoxymethane [5:1 (vol/vol)] (1.6 ml) were added buffer (0.2 M K2CO3-AcOH in 4 × 10–4 M aqueous EDTA, pH 8.0) (1.0 ml) and Bu4NHSO4 (0.0075 g, 0.02 mmol) with stirring. After the mixture was cooled to approximately –10°C (bath temperature) via an NaCl-ice bath, a solution of oxone (0.212 M in 4 × 10–4 M aqueous EDTA) (1.6 ml) and a solution of K2CO3 (0.479 M in 4 × 10–4 M aqueous EDTA) (1.6 ml) were added dropwise separately over a period of 8 h via syringe pump. The reaction mixture was quenched by addition of pentane and extracted with pentane. The combined organic layers were dried (Na2SO4), filtered, concentrated, and purified by flash chromatography [the silica gel was buffered by 1% Et3N in pentane; pentane/ether (1:0 to 10:1) was used as the eluent] to give styrene oxide as a colorless oil (0.0076 g, 63% yield, 90% ee).
Table 1. Asymmetric epoxidation of styrenes catalyzed by ketone 3.
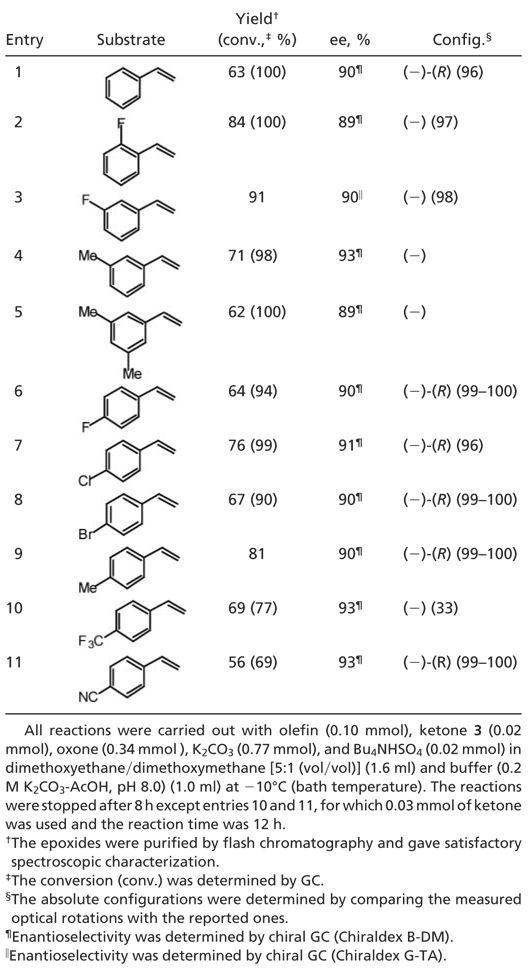 |
Results and Discussion
Styrene was used as a test substrate for the initial asymmetric epoxidation study. When the reaction was carried out with 20 mol % of ketone 3 at –10°C, (R)-styrene oxide was obtained with 100% conversion and 90% ee (Table 1, entry 1). The high ee obtained for styrene encouraged us to extend the epoxidation to other styrenes. The ees obtained are slightly dependent on the substituents on the phenyl groups of the alkenes (Table 1, entries 2–11). High ees (89–93%) were obtained for various styrenes containing both electron-donating and electron-withdrawing groups.
Our earlier studies suggest that the stereodifferentiation for the epoxidation of cis-olefins with ketone 2 likely involves electronic interactions. It seems that there is an attractive interaction between the Rπ group and the oxazolidinone moiety of the ketone catalyst in the transition state (Scheme 3; refs. 92–95). As a result, spiro transition state A overcomes the competing spiro B, providing high ees.
Scheme 3.

For styrenes, in addition to spiro D, planar transition state E also competes with the favored transition state spiro C (Scheme 4; refs. 93–95). The higher ees obtained for styrenes with ketone 3 compared with 2 suggest that the replacement of the pyranose oxygen with a carbon has a noticeable impact on the competition between these transition states. The increased ees obtained with 3 could result from further favoring spiro F or disfavoring spiro G and/or planar H (Scheme 5).
Scheme 4.

Scheme 5.

To probe the competition between spiro F and spiro G further, the epoxidation of cis-β-methylstyrene by using both ketones 2 and 3 was performed, because planar K is unlikely to be competitive because of the unfavorable interaction between the methyl group of the alkene and the spiro oxazolidinone of the ketone (Scheme 6). As shown in Table 2, ketone 3 did not give a better ee than ketone 2, which suggests that the replacement of the pyranose oxygen with a carbon has little effect on the competition between spiro I and spiro J. It could be inferred from this result that the higher ee obtained for styrene with ketone 3 is probably not caused by the favoring of spiro F over spiro G (Scheme 5).
Scheme 6.

Table 2. Epoxidation of olefins with ketones 2 and 3.
| Ketone 2
|
Ketone 3
|
||||
|---|---|---|---|---|---|
| Entry | Olefin | conv., %† | ee, %† | conv., %† | ee, %† |
| 1 |
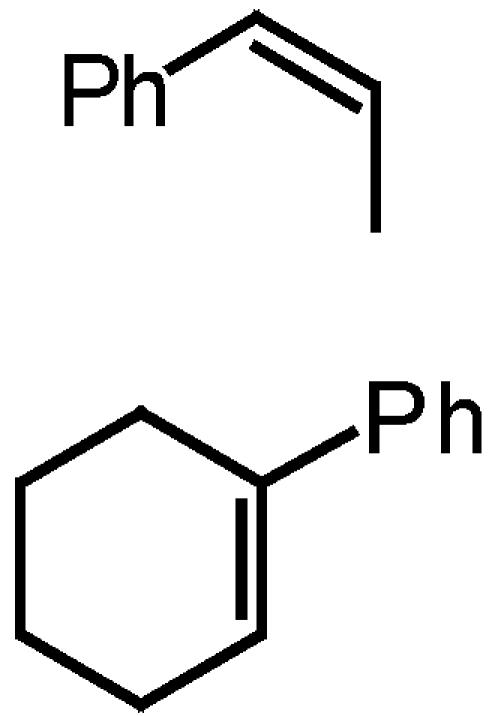
|
100 | 91 (1R,2S) | 100 | 89 (1R,2S) |
| 2 | 100 | 39 (S,S) | 96 | 40 (R,R) | |
All reactions were carried out with olefin (0.10 mmol), ketone 3 (0.02 mmol), oxone (0.34 mmol), K2CO3 (0.77 mmol), and Bu4NHSO4 (0.02 mmol) in dimethoxyethane/dimethoxymethane [5:1 (vol/vol)] (1.6 ml) and buffer (0.2 M K2CO3-AcOH, pH 8.0) (1.0 ml) at -10°C (bath temperature).
Enantioselectivity was determined by chiral GC (Chiraldex B-DM). The conversion (conv.) and ee were determined from the crude extracts.
To probe the competition between spiro F and planar H further, the epoxidation of 1-phenylcyclohexene was investigated with ketones 2 and 3. As shown in our earlier studies (94, 95), planar M is favored over spiro L for ketone 2 because of the attractive interaction between the phenyl group of the olefin and the oxazolidinone moiety of the ketone catalyst, and 39% ee of the (S,S) isomer was obtained (Table 2 and Scheme 7). In stark contrast, when ketone 3 was used, 40% ee of the (R,R) isomer of the epoxide was obtained (Table 2 and Scheme 7), indicating spiro N being favored over planar O. This result indicates that the replacement of the pyranose oxygen with a carbon leads to the favoring of spiro transition states over planar transition states, consequently suggesting that the higher ee obtained for styrene with ketone 3 is largely caused by the favoring of spiro F and G over planar H (Scheme 5).
Scheme 7.

The effect of the pyranose oxygen on the competition between spiro and planar transition states could be conformational and/or electronic in nature. Conformationally, replacing the oxygen of the ring in ketone 2 with a carbon would allow more flexibility in the system by eliminating the anomeric effect. However, an overlay of the x-ray structures of ketones 2 (94) and 3 shows that the two lie almost directly over each other (Figs. 1, 2, 3), suggesting that these two ketones have similar conformations at least in the solid state. Therefore, the difference in ees observed with styrene for ketones 2 and 3 is unlikely due to the conformational difference between these two ketones.
Fig. 1.

The stereoview of ketone 2.
Fig. 2.

The stereoview of ketone 3.
Fig. 3.

Crystal structure overlay of ketones 2 and 3.
A more likely explanation is that the pyranose oxygen influences the transition states via an electronic effect. Spiro and planar are the two extreme transition state geometries proposed for dioxirane-mediated epoxidations (Fig. 4; refs. 52–56, 62, 68, 69, 89, 90, and 101–107). The primary frontier orbital interaction for both spiro and planar approaches is between the highest occupied molecular orbital of the alkene (π of C C) and the lowest unoccupied molecular orbital of the dioxirane (σ* of O—O), which accounts for the electrophilic nature of the dioxirane (52–54). Both experimental and theoretical studies show that, in general, spiro transition states are favored over planar transition states (52–56, 62, 68, 69, 89, 90, 101–107). The favoring of a spiro transition state over a planar transition state is attributed to the stabilizing secondary orbital interaction of the nonbonding orbital (lone pair) of the oxygen of the dioxirane with the π* orbital of the alkene in the spiro transition state (stereoelectronic origin) (Fig. 4). This stabilizing orbital interaction cannot be achieved geometrically in the planar transition state (Fig. 4; refs. 103 and 104). Because the spiro and planar modes usually give two opposite enantiomers, factors influencing these two transition states would consequently affect the enantioselectivity of the epoxidation.
C) and the lowest unoccupied molecular orbital of the dioxirane (σ* of O—O), which accounts for the electrophilic nature of the dioxirane (52–54). Both experimental and theoretical studies show that, in general, spiro transition states are favored over planar transition states (52–56, 62, 68, 69, 89, 90, 101–107). The favoring of a spiro transition state over a planar transition state is attributed to the stabilizing secondary orbital interaction of the nonbonding orbital (lone pair) of the oxygen of the dioxirane with the π* orbital of the alkene in the spiro transition state (stereoelectronic origin) (Fig. 4). This stabilizing orbital interaction cannot be achieved geometrically in the planar transition state (Fig. 4; refs. 103 and 104). Because the spiro and planar modes usually give two opposite enantiomers, factors influencing these two transition states would consequently affect the enantioselectivity of the epoxidation.
Fig. 4.

The spiro and planar transition states for the dioxirane epoxidation of olefins.
Based on the aforementioned model, it is anticipated that the spiro transition state could be further favored over the planar transition state by enhancing the secondary orbital interaction by either lowering the energy of the π* orbital of the alkene or raising the energy of the nonbonding orbital of the oxygen of the dioxirane. The former case has been well illustrated, and higher ees are usually obtained when the reacting alkenes are conjugated with groups such as phenyl, alkene, alkyne, etc. (55–95, 108, 109). However, the latter case is less well addressed, at least experimentally. The switch of the epoxide configuration observed for the two structurally similar ketones for 1-phenylcyclohexene (Scheme 7) could be due to the fact that the replacement of the pyranose oxygen in ketone 2 with a carbon in ketone 3 increases the interaction of the nonbonding orbital of the dioxirane with the π* orbital of the alkene by raising the energy of the nonbonding orbital of the dioxirane, consequently favoring spiro N over planar O. In the case of styrene, this change from an oxygen to a carbon leads to the favoring of desired spiro F and undesired spiro G over undesired planar H (Scheme 5), thus reducing the minor enantiomer generated via planar H pathway and enhancing the enantioselectivity of the reaction overall. Additional improvement of the enantioselectivity for styrene seems to require a catalyst that can further suppress undesired spiro G along with undesired planar H.
The electronic effect of a substituent on the reactivity and stability of the ketone catalyst has been well investigated (52–95). The different enantioselectivities obtained with structurally similar ketones 2 and 3 show that the substituent could also electronically alter the competition between spiro and planar transition states, thus affecting the enantioselectivity of the epoxidation. Although an electron-withdrawing substituent may frequently increase the reactivity and stability of a ketone catalyst, such a substituent at the same time could also have an unfavorable effect on the enantioselectivity of the reaction by lowering the energy of the nonbonding orbital of the dioxirane, thus disfavoring the major spiro transition state. To design an effective catalyst, a delicate balance between reactivity and enantioselectivity needs to be achieved. The comparative studies between ketones 2 and 3 described herein provide valuable insight for future catalyst design.
Conclusions
The asymmetric epoxidation of styrenes using a carbocyclic oxazolidinone-containing ketone (3) as catalyst has been investigated. High enantioselectivity (89–93% ee) has been attained for this challenging type of alkene. The results described herein reveal the potential of chiral dioxirane-mediated asymmetric epoxidation as a viable entry into this important class of olefins with high enantioselectivity in addition to cis-, trans-, and trisubstituted alkenes. Ketone 3 provides a promising lead for additional optimization of the ketone structure to enhance the enantioselectivity for terminal olefins. Mechanistic studies of the origin of high enantioselectivity of ketone 3 have uncovered the electronic effects of ketone substituents on the secondary orbital interaction, which directly affects the competition between spiro and planar transition states and, ultimately, enantioselectivity. The information obtained from these studies provides valuable insight for the design of new ketone catalysts.
Supplementary Material
Acknowledgments
We thank Ms. Susie M. Miller and Professor Oren P. Anderson of the X-Ray Crystallographic Laboratory at Colorado State University for the determination of the x-ray structures of ketones 2 and 3. We also are grateful for generous financial support from the National Institutes of Health, General Medical Sciences (Grant GM59705-05), the Camille and Henry Dreyfus Foundation, and the Monfort Foundation (Colorado State University).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: ee, enantiomeric excess.
References
- 1.Johnson, R. A. & Sharpless, K. B. (1993) in Catalytic Asymmetric Synthesis, ed. Ojima, I. (VCH, New York), pp. 103–158.
- 2.Katsuki, T. & Martin, V. S. (1996) Org. React. 48, 1–299. [Google Scholar]
- 3.Johnson, R. A. & Sharpless, K. B. (2000) in Catalytic Asymmetric Synthesis, ed. Ojima, I. (VCH, New York), pp. 231–280.
- 4.Jacobsen, E. N. (1993) in Catalytic Asymmetric Synthesis, ed. Ojima, I. (VCH, New York), pp. 159–202.
- 5.Collman, J. P., Zhang, X., Lee, V. J., Uffelman, E. S. & Brauman, J. I. (1993) Science 261, 1404–1411. [DOI] [PubMed] [Google Scholar]
- 6.Mukaiyama, T. (1996) Aldrichim. Acta 29, 59–76. [Google Scholar]
- 7.Katsuki, T. (2000) in Catalytic Asymmetric Synthesis, ed. Ojima, I. (VCH, New York), pp. 287–325.
- 8.Porter, M. J. & Skidmore, J. (2000) Chem. Commun., 1215–1225.
- 9.Lauret, C. & Roberts, S. M. (2002) Aldrichim. Acta 35, 47–51. [Google Scholar]
- 10.Nemoto, T., Ohshima, T. & Shibasaki, M. (2002) J. Synth. Org. Chem. Jpn. 60, 94–105. [Google Scholar]
- 11.Corey, E. J., Shibata, S. & Bakshi, R. K. (1988) J. Org. Chem. 53, 2861–2863. [Google Scholar]
- 12.Hamada, T., Torii, T., Izawa, K., Noyori, R. & Ikariya, T. (2002) Org. Lett. 4, 4373–4376. [DOI] [PubMed] [Google Scholar]
- 13.Kolb, H. C. & Sharpless, K. B. (1992) Tetrahedron 48, 10515–10530. [Google Scholar]
- 14.Weissman, S. A., Rossen, K. & Reider, P. J. (2001) Org. Lett. 3, 2513–2515. [DOI] [PubMed] [Google Scholar]
- 15.Tokunaga, M., Larrow, J. F., Kakiuchi, F. & Jacobsen, E. N. (1997) Science 277, 936–938. [DOI] [PubMed] [Google Scholar]
- 16.Groves, J. T. & Myers, R. S. (1983) J. Am. Chem. Soc. 105, 5791–5796. [Google Scholar]
- 17.Mansuy, D., Battioni, P., Renaud, J.-P. & Guerin, P. (1985) J. Chem. Soc. Chem. Commun., 155–156.
- 18.O'Malley, S. & Kodadek, T. (1989) J. Am. Chem. Soc. 111, 9116–9117. [Google Scholar]
- 19.Groves, J. T. & Viski, P. (1990) J. Org. Chem. 55, 3628–3634. [Google Scholar]
- 20.Halterman, R. L. & Jan, S.-T. (1991) J. Org. Chem. 56, 5253–5254. [Google Scholar]
- 21.Naruta, Y., Tani, F., Ishihara, N. & Maruyama, K. (1991) J. Am. Chem. Soc. 113, 6865–6872. [Google Scholar]
- 22.Konishi, K., Oda, K.-I., Nishida, K., Aida, T. & Inoue, S. (1992) J. Am. Chem. Soc. 114, 1313–1317. [Google Scholar]
- 23.Naruta, Y., Ishihara, N., Tani, F. & Maruyama, K. (1993) Bull. Chem. Soc. Jpn. 66, 158–166. [Google Scholar]
- 24.Collman, J. P., Lee, V. J., Zhang, X., Ibers, J. A. & Brauman, J. I. (1993) J. Am. Chem. Soc. 115, 3834–3835. [Google Scholar]
- 25.Collman, J. P., Lee, V. J., Kellen-Yuen, C. J., Zhang, X., Ibers, J. A. & Brauman, J. I. (1995) J. Am. Chem. Soc. 117, 692–703. [Google Scholar]
- 26.Berkessel, A. & Frauenkron, M. (1997) J. Chem. Soc. Perkin Trans. 1, 2265–2266.
- 27.Gross, Z. & Ini, S. (1997) J. Org. Chem. 62, 5514–5523. [Google Scholar]
- 28.Lai, T.-S., Zhang, R., Cheung, K.-K., Kwong, H.-L. & Che, C.-M. (1998) Chem. Commun., 1583–1584.
- 29.Collman, J. P., Wang, Z., Straumanis, A., Quelquejeu, M. & Rose, E. (1999) J. Am. Chem. Soc. 121, 460–461. [Google Scholar]
- 30.Zhang, R., Yu, W.-Y., Lai, T.-S. & Che, C.-M. (1999) Chem. Commun., 409–410.
- 31.Gross, Z. & Ini, S. (1999) Org. Lett. 1, 2077–2080. [Google Scholar]
- 32.Reginato, G., Bari, L. D., Salvadori, P. & Guilard, R. (2000) Eur. J. Org. Chem., 1165–1171.
- 33.Zhang, R., Yu, W.-Y., Wong, K.-Y. & Che, C.-M. (2001) J. Org. Chem. 66, 8145–8153. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, R., Yu, W.-Y., Sun, H.-Z., Liu, W.-S. & Che, C.-M. (2002) Chem. Eur. J. 8, 2495–2507. [DOI] [PubMed] [Google Scholar]
- 35.Zhang, W., Loebach, J. L., Wilson, S. R. & Jacobsen, E. N. (1990) J. Am. Chem. Soc. 112, 2801–2803. [Google Scholar]
- 36.O'Connor, K. J., Wey, S.-J. & Burrows, C. J. (1992) Tetrahedron Lett. 33, 1001–1004. [Google Scholar]
- 37.Palucki, M., Pospisil, P. J., Zhang, W. & Jacobsen, E. N. (1994) J. Am. Chem. Soc. 116, 9333–9334. [Google Scholar]
- 38.Palucki, M., McCormick, G.-J. & Jacobsen, E. N. (1995) Tetrahedron Lett. 36, 5457–5460. [Google Scholar]
- 39.Hashihayata, T., Ito, Y. & Katsuki, T. (1997) Tetrahedron 53, 9541–9552. [Google Scholar]
- 40.Takeda, T., Irie, R., Shinoda, Y. & Katsuki, T. (1999) Synlett, 1157–1159.
- 41.Kim, G.-J. & Shin, J.-H. (1999) Tetrahedron Lett. 40, 6827–6830. [Google Scholar]
- 42.Nakata, K., Takeda, T., Mihara, J., Hamada, T., Irie, R. & Katsuki, T. (2001) Chem. Eur. J. 7, 3776–3782. [DOI] [PubMed] [Google Scholar]
- 43.Davis, F. A., Harakal, M. E. & Awad, S. B. (1983) J. Am. Chem. Soc. 105, 3123–3126. [Google Scholar]
- 44.Davis, F. A. & Chattopadhyay, S. (1986) Tetrahedron Lett. 27, 5079–5082. [Google Scholar]
- 45.Aggarwal, V. K. & Wang, M. F. (1996) Chem. Commun., 191–192.
- 46.Bohe, L., Lusinchi, M. & Lusinchi, X. (1999) Tetrahedron 55, 141–154. [Google Scholar]
- 47.Takahashi, O., Umezawa, J., Furuhashi, K. & Takagi, M. (1989) Tetrahedron Lett. 30, 1583–1584. [Google Scholar]
- 48.Allain, E. J., Hager, L. P., Deng, L. & Jacobsen, E. N. (1993) J. Am. Chem. Soc. 115, 4415–4416. [Google Scholar]
- 49.Zaks, A. & Dodds, D. R. (1995) J. Am. Chem. Soc. 117, 10419–10424. [Google Scholar]
- 50.Lakner, F. J., Cain, K. P. & Hager, L. P. (1997) J. Am. Chem. Soc. 119, 443–444. [Google Scholar]
- 51.Hollmann, F., Lin, P.-C., Witholt, B. & Schmid, A. (2003) J. Am. Chem. Soc. 125, 8209–8217. [DOI] [PubMed] [Google Scholar]
- 52.Murray, R. W. (1989) Chem. Rev. (Washington, D.C.) 89, 1187–1201. [Google Scholar]
- 53.Adam, W., Curci, R. & Edwards, J. O. (1989) Acc. Chem. Res. 22, 205–211. [Google Scholar]
- 54.Curci, R., Dinoi, A. & Rubino, M. F. (1995) Pure Appl. Chem. 67, 811–822. [Google Scholar]
- 55.Denmark, S. E. & Wu, Z. (1999) Synlett, 847–859.
- 56.Frohn, M. & Shi, Y. (2000) Synthesis (Mass), 1979–2000.
- 57.Curci, R., Fiorentino, M. & Serio, M. R. (1984) J. Chem. Soc. Chem. Commun., 155–156.
- 58.Curci, R., D'Accolti, L., Fiorentino, M. & Rosa, A. (1995) Tetrahedron Lett. 36, 5831–5834. [Google Scholar]
- 59.Denmark, S. E., Forbes, D. C., Hays, D. S., DePue, J. S. & Wilde, R. G. (1995) J. Org. Chem. 60, 1391–1407. [Google Scholar]
- 60.Brown, D. S., Marples, B. A., Smith, P. & Walton, L. (1995) Tetrahedron 51, 3587–3606. [Google Scholar]
- 61.Yang, D., Yip, Y.-C., Tang, M.-W., Wong, M.-K., Zheng, J.-H. & Cheung, K.-K. (1996) J. Am. Chem. Soc. 118, 491–492. [Google Scholar]
- 62.Yang, D., Wang, X.-C., Wong, M.-K., Yip, Y.-C. & Tang, M.-W. (1996) J. Am. Chem. Soc. 118, 11311–11312. [Google Scholar]
- 63.Song, C. E., Kim, Y. H., Lee, K. C., Lee, S. G. & Jin, B. W. (1997) Tetrahedron Asymmetry 8, 2921–2926. [Google Scholar]
- 64.Adam, W. & Zhao, C.-G. (1997) Tetrahedron Asymmetry 8, 3995–3998. [Google Scholar]
- 65.Denmark, S. E., Wu, Z., Crudden, C. M. & Matsuhashi, H. (1997) J. Org. Chem. 62, 8288–8289. [DOI] [PubMed] [Google Scholar]
- 66.Wang, Z.-X. & Shi, Y. (1997) J. Org. Chem. 62, 8622–8623. [Google Scholar]
- 67.Adam, W., Fell, R. T., Saha-Moller, C. R. & Zhao, C.-G. (1998) Tetrahedron Asymmetry 9, 397–401. [Google Scholar]
- 68.Armstrong, A. & Hayter, B. R. (1998) Chem. Commun., 621–622.
- 69.Yang, D., Wong, M.-K., Yip, Y.-C., Wang, X.-C., Tang, M.-W., Zheng, J.-H. & Cheung, K.-K. (1998) J. Am. Chem. Soc. 120, 5943–5952. [Google Scholar]
- 70.Yang, D., Yip, Y.-C., Chen, J. & Cheung, K.-K. (1998) J. Am. Chem. Soc. 120, 7659–7660. [Google Scholar]
- 71.Adam, W., Saha-Moller, C. R. & Zhao, C.-G. (1999) Tetrahedron Asymmetry 10, 2749–2755. [Google Scholar]
- 72.Carnell, A. J., Johnstone, R. A. W., Parsy, C. C. & Sanderson, W. R. (1999) Tetrahedron Lett. 40, 8029–8032. [Google Scholar]
- 73.Armstrong, A. & Hayter, B. R. (1999) Tetrahedron 55, 11119–11126. [Google Scholar]
- 74.Armstrong, A., Hayter, B. R., Moss, W. O., Reeves, J. R. & Wailes, J. S. (2000) Tetrahedron Asymmetry 11, 2057–2061. [Google Scholar]
- 75.Solladie-Cavallo, A. & Bouerat, L. (2000) Org. Lett. 2, 3531–3534. [DOI] [PubMed] [Google Scholar]
- 76.Bortolini, O., Fogagnolo, M., Fantin, G., Maietti, S. & Medici, A. (2001) Tetrahedron Asymmetry 12, 1113–1115. [Google Scholar]
- 77.Seki, M., Furutani, T., Imashiro, R., Kuroda, T., Yamanaka, T., Harada, N., Arakawa, H., Kusama, M. & Hashiyama, T. (2001) Tetrahedron Lett. 42, 8201–8205. [Google Scholar]
- 78.Armstrong, A., Moss, W. O. & Reeves, J. R. (2001) Tetrahedron Asymmetry 12, 2779–2781. [Google Scholar]
- 79.Matsumoto, K. & Tomioka, K. (2002) Tetrahedron Lett. 43, 631–633. [Google Scholar]
- 80.Stearman, C. J. & Behar, V. (2002) Tetrahedron Lett. 43, 1943–1946. [Google Scholar]
- 81.Denmark, S. E. & Matsuhashi, H. (2002) J. Org. Chem. 67, 3479–3486. [DOI] [PubMed] [Google Scholar]
- 82.Shing, T. K. M. & Leung, G. Y. C. (2002) Tetrahedron 58, 7545–7552. [Google Scholar]
- 83.Bortolini, O., Fantin, G., Fogagnolo, M., Forlani, R., Maietti, S. & Pedrini, P. (2002) J. Org. Chem. 67, 5802–5806. [DOI] [PubMed] [Google Scholar]
- 84.Armstrong, A., Ahmed, G., Dominguez-Fernandez, B., Hayter, B. R. & Wailes, J. S. (2002) J. Org. Chem. 67, 8610–8617. [DOI] [PubMed] [Google Scholar]
- 85.Klein, S. & Roberts, S. M. (2002) J. Chem. Soc. Perkin Trans. 1, 2686–2691.
- 86.Shing, T. K. M., Leung, Y. C. & Yeung, K. W. (2003) Tetrahedron 59, 2159–2168. [Google Scholar]
- 87.Sartori, G., Armstrong, A., Maggi, R., Mazzacani, A., Sartorio, R., Bigi, F. & Dominguez-Fernandez, B. (2003) J. Org. Chem. 68, 3232–3237. [DOI] [PubMed] [Google Scholar]
- 88.Chan, W.-K., Yu, W.-Y., Che, C.-M. & Wong, M.-K. (2003) J. Org. Chem. 68, 6576–6582. [DOI] [PubMed] [Google Scholar]
- 89.Tu, Y., Wang, Z.-X. & Shi, Y. (1996) J. Am. Chem. Soc. 118, 9806–9807. [Google Scholar]
- 90.Wang, Z.-X., Tu, Y., Frohn, M., Zhang J.-R. & Shi, Y. (1997) J. Am. Chem. Soc. 119, 11224–11235. [Google Scholar]
- 91.Shu, L. & Shi, Y. (2001) Tetrahedron 57, 5213–5218. [Google Scholar]
- 92.Tian, H., She, X., Shu, L., Yu, H. & Shi, Y. (2000) J. Am. Chem. Soc. 122, 11551–11552. [Google Scholar]
- 93.Tian, H., She, X., Xu, J. & Shi, Y. (2001) Org. Lett. 3, 1929–1931. [DOI] [PubMed] [Google Scholar]
- 94.Tian, H., She, X., Yu, H., Shu, L. & Shi, Y. (2002) J. Org. Chem. 67, 2435–2446. [DOI] [PubMed] [Google Scholar]
- 95.Shu, L., Wang, P., Gan, Y. & Shi, Y. (2003) Org. Lett. 5, 293–296. [DOI] [PubMed] [Google Scholar]
- 96.Archelas, A. & Furstoss, R. (1999) J. Org. Chem. 64, 6112–6114. [DOI] [PubMed] [Google Scholar]
- 97.Doussot, J., Guy, A., Siaugue, J.-M., Ferroud, C. & Guieres, A. F. (1999) Chirality 11, 541–545. [DOI] [PubMed] [Google Scholar]
- 98.Pews, R. G. (1967) J. Am. Chem. Soc. 89, 5605–5608. [Google Scholar]
- 99.Pedragosa-Moreau, S., Morisseau, C., Zylber, J., Archelas, A., Baratti, J. & Furstoss, R. (1996) J. Org. Chem. 61, 7402–7407. [DOI] [PubMed] [Google Scholar]
- 100.Moussou, P., Archelas, A., Baratti, J. & Furstoss, R. (1998) J. Org. Chem. 63, 3532–3537. [Google Scholar]
- 101.Baumstark, A. L. & McCloskey, C. J. (1987) Tetrahedron Lett. 28, 3311–3314. [Google Scholar]
- 102.Baumstark, A. L. & Vasquez, P. C. (1988) J. Org. Chem. 53, 3437–3439. [Google Scholar]
- 103.Bach, R. D., Andres, J. L., Owensby, A. L., Schlegel, H. B. & McDouall, J. J. W. (1992) J. Am. Chem. Soc. 114, 7207–7217. [Google Scholar]
- 104.Houk, K. N., Liu, J., DeMello, N. C. & Condroski, K. R. (1997) J. Am. Chem. Soc. 119, 10147–10152. [Google Scholar]
- 105.Jenson, C., Liu, J., Houk, K. N. & Jorgensen, W. L. (1997) J. Am. Chem. Soc. 119, 12982–12983. [Google Scholar]
- 106.Armstrong, A., Washington, I. & Houk, K. N. (2000) J. Am. Chem. Soc. 122, 6297–6298. [Google Scholar]
- 107.Deubel, D. V. (2001) J. Org. Chem. 66, 3790–3796. [DOI] [PubMed] [Google Scholar]
- 108.Frohn, M., Dalkiewicz, M., Tu, Y., Wang, Z.-X. & Shi, Y. (1998) J. Org. Chem. 63, 2948–2953. [Google Scholar]
- 109.Wang, Z.-X., Cao, G.-A. & Shi, Y. (1999) J. Org. Chem. 64, 7646–7650. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.