

Abstract
Catalytic asymmetric ring-opening metathesis (AROM) provides an efficient method for the synthesis of a variety of optically enriched small organic molecules that cannot be easily prepared by alternative methods. The development of Mo-catalyzed AROM transformations that occur in tandem with ring-closing metathesis are described. The utility of the Mo-catalyzed AROM/ring-closing metathesis is demonstrated through an enantioselective approach to the synthesis of (+)-africanol.
One program of research in these laboratories during the past several years has focused on the design and development of chiral catalysts for efficient enantioselective olefin metathesis (1). As part of this initiative, we have synthesized, characterized, and examined the catalytic activity of high-oxidation-state Mo-based chiral complexes such as 1–4 (Scheme 1) (1). These investigations have led to the development of a range of methods for catalytic asymmetric ring-closing metathesis (2–6) and asymmetric ring-opening metathesis (AROM) (7–10). Through such protocols, optically enriched organic molecules can be prepared that cannot be easily accessed by alternative approaches.
Scheme 1.

With the availability of various chiral catalysts and the enantioselective transformations that can be promoted by such complexes, we have recently begun to explore the utility of catalytic asymmetric metathesis in target-oriented synthesis. Through application of catalytic asymmetric metathesis to multistep synthesis, we aim to demonstrate the unique features of asymmetric olefin metathesis, as well as identify and address any related shortcomings. We report herein key findings in connection with Mo-catalyzed AROM/RCM (11). Furthermore, we disclose an enantioselective synthesis of sesquiterpenoid (+)-africanol (12–17), where a Mo-catalyzed AROM/RCM is used to establish the stereochemical identity of an early intermediate (Fig. 1). In addition to the crucial catalytic asymmetric metathesis step, the total synthesis addresses the utility of other metal-catalyzed transformations as a means to differentiate alkenes formed through metal-catalyzed enantioselective desymmetrizations.
Fig. 1.
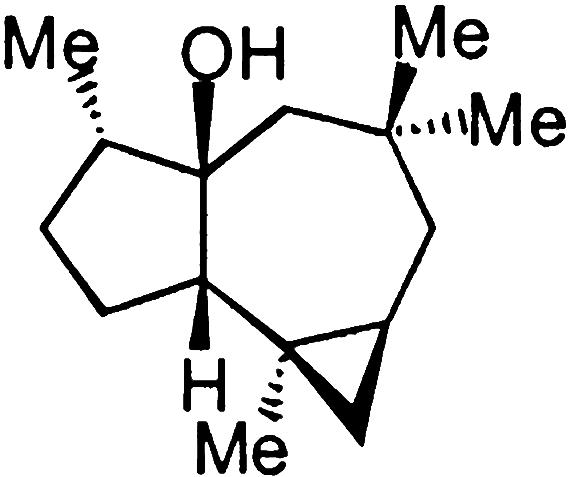
(+)-africanol.
Catalytic ROM can be induced to occur in tandem with RCM to yield synthetically versatile bicyclic structures (18). Representative examples of catalytic AROM/RCM, which may also be considered rearrangement processes, are illustrated in Schemes 2 and 3. It should be mentioned that it is not always clear whether cleavage of the more strained cyclic olefin occurs in an intramolecular fashion, where RCM (by means of alkylidene ii) gives rise to rupture of the ring, or ring-opening occurs first (19) (by means of iii formed by reaction of i with the substrate) followed by RCM. The different levels of asymmetric induction observed in the formation of 6 versus 8 (69% and 92% ee) may be partly the result of such mechanistic intricacies. The terminal olefin in 5 may be converted to ii (R = H) more readily than would be the case with the more substituted alkene of substrate 7, which might prefer to proceed through the catalytic cycle as iii (R = Me). The requirement for the use of diallylether as an additive (20) in the reaction of 7 (<2% conversion without the additive) is likely due to in situ generation of Mo-methylidene i, which is expected to be a more effective initiator (less sterically hindered) compared with the neophylidene precatalyst (see Scheme 1). It merits mention that in this class of transformations reaction efficiency can be significantly affected depending on the stereochemical identity of the substrate, as exemplified by rapid polymerization of 9 (Scheme 3) (7).
Scheme 2.

Scheme 3.

As the data in Scheme 3 illustrate (10 → 11), Mo-catalyzed tandem ROM/RCM can be effected to afford heterocyclic structures that bear a tertiary ether site (21). Considering the paucity of effective methods for the preparation of nonracemic tertiary alcohols and ethers (22), the present protocol offers a convenient and enantioselective approach to a difficult problem in organic synthesis. As also depicted in Scheme 3 (12 → 13), meso trienes can be converted to structurally complex polycycles in 80% yield and excellent levels of enantioselectivity (96% to >98% ee) (23).
Efficient AROM/cross metathesis (AROM/CM) of norbornyl substrates with terminal olefins can be promoted by Mo-based chiral catalysts (7). Within this context, we have examined various AROM/RCM reactions carried out in the presence of styrene (two equivalents) to establish whether, under such conditions, AROM/CM effectively competes with AROM/RCM pathways. As shown in Scheme 4, reaction of diene 5, bearing a terminal olefin, proceeds to afford 6 in nearly the same yield and enantioselectivity as observed in the absence of the olefin additive (see Scheme 2 for comparison); none of the corresponding AROM/CM product (e.g., 14) is observed. In contrast, diene 7 affords only triene 14, the product of a catalytic AROM/CM, in >98% ee and 85% isolated yield (see Scheme 2 for reaction outcome in the absence of styrene). Similar to diene 7, substrate 15, bearing a trisubstituted olefin, undergoes AROM/CM exclusively to afford optically pure 16 in 77% isolated yield. In reactions of 7 and 15, compounds arising from AROM/RCM (compare 6) or meso products from an additional CM of 14 or 16 with styrene are not detected (net AROM/CM/CM). Similarly, products corresponding to CM of 6 with styrene are not observed.
Scheme 4.

The difference in the outcome of reactions depicted in Scheme 4 may be attributed to a number of mechanistic factors. It is plausible that in the case of 5, the rapidly formed (7) benzylidene iv (Scheme 4) reacts with the substrate to regenerate styrene and affords terminal alkylidene v, which undergoes facile RCM to deliver 6. With substrates 7 and 15, which bear the more substituted and less reactive olefinic side chains, the catalytic cycle might commence with an AROM through reaction of benzylidene iv with the substrate to afford vi. The resulting Mo-alkylidene may then react more readily with another molecule of styrene to afford 14 or 16 rather than participate in an RCM with the neighboring di- or trisubstituted alkene. Whether the five-membered chelate structure vi shown in Scheme 4, which is expected to cause lowering of reactivity of the Mo-alkylidene, plays a role in reducing the facility of ring closure in reactions involving the more substituted olefinic chains (i.e., 7 and 15) is unclear at the present time (internal chelation in v involves a less favorable four-membered structure).
With an effective catalytic AROM/RCM available, we began to explore the utility of this method through its use in an enantioselective synthesis of africanol. The retrosynthesis, illustrated in Scheme 5, involves access to a through structural modification of b, which in turn may be synthesized enantioselectively through Mo-catalyzed AROM/RCM of diene c. This retrosynthetic analysis highlights a key characteristic of enantioselective olefin metathesis: Asymmetric desymmetrizations carried out through olefin metathesis afford products that bear the same (e.g., 5 → 6, Scheme 4) or a larger number (e.g., 15 → 16, Scheme 4) of olefin sites, compared with the corresponding substrate. This attribute is in contrast to the majority of other enantioselective desymmetrizations (24), where, typically, one enantiotopic site is converted to another distinctly different functional group. Accordingly, use of catalytic metathesis as a stereochemistry-determining step in a total synthesis must be accompanied with effective strategies that allow for differentiation of the resulting olefinic sites. In the case of intermediate b (Scheme 5), differentiation of the terminal alkene from the cyclic disubstituted olefin was not expected to be particularly problematic, but in certain cases (e.g., 14 or 16 in Scheme 4), differential functionalization of various olefins may require highly regio- and site-selective methods.
Scheme 5.

We devised two separate routes for the conversion of b to the target molecule. One possibility (Option 1 in Scheme 5), would involve rearrangement of d to e through catalytic ROM/RCM. After rupture, the disubstituted cyclic alkene in d would likely be readily regenerated. However, we argued that, if closure to the more stable trisubstituted olefin were to occur, even in minor quantities, the lower reactivity of the trisubstituted olefin of e would force the equilibrium to its favor, rendering it the predominant product. This proposal was based on previous observations in our laboratories regarding the reversibility of olefin metathesis and successful application of similar principles in the enantioselective total synthesis of fluvirucin B1 (25) and nebivolol (26). Alternatively, AROM/RCM product b could be converted to aldehyde f; a subsequent sequential decarbonylation/hydroformylation would deliver g, which could then be converted to a. Through the route represented by Option 2, we would be able to examine the possibility of regioselective hydroformylation of the disubstituted olefin in f. It should be noted that enantioselective synthesis of africanol through Option 1 (Scheme 5) would require reaction of ent-b, whereas Option 2 may proceed through b.
Based on the above planning, we prepared unsaturated ketone 17, which was alkylated in >95% diastereoselectivity with alkyllithium 18 to afford diene 19 after silylation of the resulting tertiary carbinol in 81% overall isolated yield. To identify the optimal chiral catalyst for desymmetrization of 19, various chiral Mo complexes were screened; these studies led us to establish that in the presence of 3 mol percent 2a (Scheme 1), 20 is formed in 87% ee and 97% isolated yield (Scheme 6). Two additional points in connection with the catalytic AROM/RCM are noteworthy. (i) Mo-catalyzed enantioselective synthesis of 20 was carried out in the presence of minimal solvent (8 M in pentane). Sufficient pentane was added only to dissolve the chiral catalyst; homodimeric products were not detected under such condition. (ii) Catalytic AROM/RCM of the diene related to 19, but lacking the geminal dimethyl group (compare 22, Scheme 6), resulted in exclusive formation of the derived homodimer, even though the catalytic process was effected at 0.1 M concentration in benzene. Dienes bearing less sterically bulky protecting groups such as 21 (Scheme 6) underwent AROM/RCM efficiently but with significantly lower enantioselectivity (35% ee). Based on previous studies (21) regarding the positive effect of tetrahydrofuran as an additive in Mo-catalyzed olefin metathesis reactions, as shown in Scheme 6, we determined that in the presence of one equivalent of tetrahydrofuran, enantioselectivity of formation of 22 can be improved but did not prove to be competitive with the asymmetric rearrangement of 19 (75% ee versus 87% ee).
Scheme 6.

With optically enriched 20 in hand, we examined the possibility of the catalytic metathesis-based rearrangement illustrated as Option 1 in Scheme 5. Toward this end, 23 was obtained regioselectively (>98%) in 71% isolated yield through Wacker oxidation (27) of the terminal alkene (Scheme 7). Although conversion of 23 to diene 24 can in principle be accomplished by an olefination reaction, in practice, we found that the two-step alkylation/elimination is a significantly more efficient and reliable route, delivering the desired 24 in 72% overall yield. Extensive experimentation with a wide variety of Mo- and Ru-based metathesis catalysts under a myriad of reaction conditions, however, failed to convert 24 to 25 (starting material recovered in most cases). Lack of reactivity may be due several factors: (i) resistance of the cyclic alkene toward catalytic ROM; (ii) ROM may occur with the undesired regioselectivity, positioning the metal-alkylidene at the α-carbon (subsequent RCM of the alkylidene with the 1,1-disubstituted alkene would generate a highly strained compound); and (iii) the ring-opened product might revert back to 24 rapidly such that the resulting metal-alkylidene does not have the opportunity to react with the neighboring 1,1-disubstituted olefin to afford 25. Further information can be found in Supporting Text, which is published as supporting information on the PNAS web site.
Scheme 7.

We next turned our attention to establishing the feasibility of Option 2 (Scheme 5). Accordingly, aldehyde 26 was synthesized from optically enriched 20 in 86% overall yield through a two-step procedure (Scheme 8). Catalytic decarbonylation (28–30) of 26 in the presence of 10 mol percent Rh(dppe)2Cl at elevated temperature proceeded slowly (96 h) but effectively to afford 27 in 96% isolated yield. At this point, we focused on catalytic regioselective hydroformylation of the disubstituted olefin in 27. Initial conditions, such as that labeled condition A (31, 32) in Scheme 8, led to highly efficient C—C bond formation but with predominant generation of the undesired regioisomer 28. After extensive experimentation, we were able to determine that in the presence of 2 mol percent Rh(acac) (CO)2 and 2 mol percebt P(o-t-BuC6H4)3 under 800 psi of CO/H2 (1:1) gas (toluene, 70°C, 6 h), an equal mixture of 28 and the desired isomer 29 are formed in 96% overall yield (33). Reduction of the aldehyde with NaBH4 afforded the corresponding primary alcohol; the undesired regioisomer formed during the previous step (catalytic hydroformylation) was readily removed at this point (48% isolated overall yield of the desired regioisomer after two steps). Subsequent elimination of the primary alcohol according to the method of Grieco et al. (34) proceeded to afford exocyclic olefin 30. Efficient isomerization of the exocyclic alkene to the desired trisubstituted olefin isomer through treatment of 30 with eight equivalents of KH in 1,3-diaminopropane (35–39) at 22°C and subsequent removal of the silylether delivered the desired tertiary carbinol 31 in 48% overall yield. The structural identity of the synthetic sample was ascertained through comparison of the spectral data with those reported (17). Bicyclic alcohol 31 has been readily converted to africanol (17).
Scheme 8.

In brief, we have developed a highly efficient and enantioselective method for the synthesis of various functionalized cyclic and polycyclic structures through Mo-catalyzed asymmetric metathesis. The utility of the Mo-catalyzed AROM/RCM metathesis to target-oriented synthesis is demonstrated through its application to the synthesis of (+)-africanol. Perhaps the most noteworthy feature of the synthesis is the facility with which the tertiary alcohol stereogenic center is established enantioselectively through the combination of a highly diastereoselective alkylation of ketone 17 and an enantioselective rearrangement of the derived silyl ether to bicyclic structure 20 (Scheme 6).
Efforts in total synthesis are valuable because they allow us to identify critical deficiencies in the arsenal of available reaction methods. The present effort underlines the need for more efficient and selective methods that allow for regio- and stereoselective alkylations of unactivated olefins (40).
Supplementary Material
Acknowledgments
We thank Professor James White for providing us with spectral data, and Shayne Guliano for helpful experimental assistance. G.S.W. thanks Bristol Myers-Squibb for a Graduate Fellowship Award in organic synthesis (2001–2002). This work was supported by National Institutes of Health Grant GM-59426.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: RCM, ring-closing metathesis; ROM, ring-opening metathesis; AROM, asymmetric ROM; CM, cross metathesis.
References
- 1.Schrock, R. R. & Hoveyda, A. H. (2003) Angew. Chem. Int. Ed. 42, 4592–4633. [DOI] [PubMed] [Google Scholar]
- 2.Alexander, J. B., La, D. S., Cefalo, D. R., Hoveyda, A. H. & Schrock, R. R. (1998) J. Am. Chem. Soc. 120, 4041–4042. [Google Scholar]
- 3.Zhu, S. S., Cefalo, D. R., La, D. S., Jamieson, J. Y., Davis, W. M., Hoveyda, A. H. & Schrock, R. R. (1999) J. Am. Chem. Soc. 121, 8251–8259. [Google Scholar]
- 4.Kiely, A. F., Jernelius, J. A., Schrock, R. R. & Hoveyda, A. H. (2002) J. Am. Chem. Soc. 124, 2868–2869. [DOI] [PubMed] [Google Scholar]
- 5.Dolman, S. J., Sattely, E. S., Hoveyda, A. H. & Schrock, R. R. (2002) J. Am. Chem. Soc. 124, 6991–6997. [DOI] [PubMed] [Google Scholar]
- 6.Dolman, S. J., Schrock, R. R. & Hoveyda, A. H. (2003) Org. Lett. 5, 4899–4902. [DOI] [PubMed] [Google Scholar]
- 7.La, D. S., Sattely, E. S., Ford, J. G., Schrock, R. R. & Hoveyda, A. H. (2001) J. Am. Chem. Soc. 123, 7767–7778. [DOI] [PubMed] [Google Scholar]
- 8.Van Veldhuizen, J. J., Garber, S. B., Kingsbury, J. S. & Hoveyda, A. H. (2002) J. Am. Chem. Soc. 124, 4954–4955. [DOI] [PubMed] [Google Scholar]
- 9.Van Veldhuizen, J. J., Gillingham, D. G., Garber, S. B., Kataoka, O. & Hoveyda, A. H. (2003) J. Am. Chem. Soc. 125, 12502–12508. [DOI] [PubMed] [Google Scholar]
- 10.Seiders, T. J., Ward, D. W. & Grubbs, R. H. (2001) Org. Lett. 3, 3225–3228. [DOI] [PubMed] [Google Scholar]
- 11.Weatherhead, G. S., Ford, J. G., Alexanian, E. J., Schrock, R. R. & Hoveyda, A. H. (2000) J. Am. Chem. Soc. 122, 1828–1829. [Google Scholar]
- 12.Tursch, B., Braekman, J. C., Daloze, D., Fritz, P., Kelecom, A., Karlsson & Losman, R. (1974) Tetrahedron Lett. 747–750.
- 13.Sugimura, T., Futagawa, T. & Tai, A. (1990) Chem. Lett. 12, 2295–2298. [Google Scholar]
- 14.Hayasaka, K., Ohtsuka, T., Shirahama, H. & Matsumoto, T. (1985) Tetrahedron Lett. 873–876.
- 15.Shirahama, H. H., Hayano, K., Kanemoto, Y., Misumi, S., Ohtsuka, T., Hashiba, N., Furusaki, A., Murata, S., Noyori, R. & Matsumoto, T. (1980) Tetrahedron Lett. 4835–4838.
- 16.Paquette, L. A. & Ham, W. H. (1987) J. Am. Chem. Soc., 109, 3025–3036. [Google Scholar]
- 17.Fan, W. & White, J. B. (1993) J. Org. Chem. 58, 3557–3562. [Google Scholar]
- 18.Schrader, T. O. & Snapper, M. L. (2003) in Handbook of Metathesis, ed. Grubbs, R. H. (Wiley, New York), pp. 205–237.
- 19.Cefalo, D. R., Kiely, A. F., Wuchrer, M., Jamieson, J. Y., Schrock, R. R. & Hoveyda, A. H. (2001) J. Am. Chem. Soc. 123, 3139–3140. [Google Scholar]
- 20.Harrity, J. P. A., La, D. S., Cefalo, D. R., Visser, M. S. & Hoveyda, A. H. (1998) J. Am. Chem. Soc. 120, 2343–2351. [Google Scholar]
- 21.Teng, X., Cefalo, D. R., Schrock, R. R. & Hoveyda, A. H. (2002) J. Am. Chem. Soc. 124, 10779–10784. [DOI] [PubMed] [Google Scholar]
- 22.DiMauro, D. F. & Kozlowski, M. C. (2002) J. Am. Chem. Soc. 124, 12668–12669. [DOI] [PubMed] [Google Scholar]
- 23.Tsang, W. C. P., Jernelius, J. A., Cortez, G. A., Weatherhead, G. S., Schrock, R. R. & Hoveyda, A. H. (2003) J. Am. Chem. Soc. 125, 2591–2596. [DOI] [PubMed] [Google Scholar]
- 24.Willis, M. C. (1999) J. Chem. Soc. Perkin Trans. 1, 1765–1784. [Google Scholar]
- 25.Xu, Z., Johannes, C. W., Houri, A. F., La, D. S., Cogan, D. A., Hofilena, G. E. & Hoveyda, A. H. (1997) J. Am. Chem. Soc. 119, 10302–10316. [Google Scholar]
- 26.Johannes, C. W., Visser, M. S., Weatherhead, G. S. & Hoveyda, A. H. (1998) J. Am. Chem. Soc. 120, 8340–8347. [Google Scholar]
- 27.Smith, A. B., Cho, Y. S. & Friestad, G. K. (1998) Tetrahedron Lett. 8765–8768.
- 28.Dougherty, D. H. & Pignolet, L. H. (1978) J. Am. Chem. Soc. 100, 7083–7085. [Google Scholar]
- 29.Meyer, M. D. & Kruse, L. I. (1984) J. Org. Chem. 49, 3195–3199. [Google Scholar]
- 30.Boeckman, R. K., Zhang, J. & Reeder, M. R. (2002) Org. Lett. 4, 3891–3894. [DOI] [PubMed] [Google Scholar]
- 31.Jardine, F. H. (1982) Polyhedron 1, 569–605. [Google Scholar]
- 32.Breit, B. & Seiche, W. (2001) Synthesis 1, 1–36. [Google Scholar]
- 33.van Leeuwen, P. W. M. N. & Roobeek, C. F. (1981) Tetrahedron 37, 1973–1983. [Google Scholar]
- 34.Grieco, P. A., Gilman, S. & Nishizawa, M. (1976) J. Org. Chem. 41, 1485.–1486. [Google Scholar]
- 35.Abrams, S. R. (1984) Can. J. Chem. 62, 1333–1334. [Google Scholar]
- 36.Brown, C. A. & Jadhav, P. K. (1993) Org. Synth. Coll. Ed. 8, 553–556. [Google Scholar]
- 37.Macaulay, S. R. (1980) J. Org. Chem. 45, 734–735. [Google Scholar]
- 38.Brown, C. A. & Yamashita, A. (1975) J. Am. Chem. Soc. 97, 891–892. [Google Scholar]
- 39.Midland, M. M., Halterman, R. L., Brown, C. A. & Yamaichi, A. (1981) Tetrahedron Lett. 4171–4172.
- 40.Hoveyda, A. H. & Heron, N. M. (1999) in Comprehensive Asymmetric Catalysis, eds. Jacobsen, E. N., Pfaltz, A. & Yamamoto, H. (Springer, Berlin), pp. 431–454.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.