

Abstract
Contrary to the prevailing professional opinion of the past few decades, recent experimental and clinical data support the fact that protein replacement therapy by allogeneic blood and marrow transplantation is not limited to freely diffusible molecules such as enzymes, but also large structural proteins such as collagens. A prime example is the cross-correction of type VII collagen deficiency in generalised severe recessive dystrophic epidermolysis bullosa, in which blood and marrow transplantation can attenuate the mucocutaneous manifestations of the disease and improve patients’ quality of life. Although allogeneic blood and marrow transplantation can improve the integrity of the skin and mucous membranes, today’s accomplishments are only the first steps on the long pathway to cure. Future strategies will be built on the lessons learned from these first transplant studies.
Introduction
Dystrophic epidermolysis bullosa is a group of heritable mechanobullous skin diseases typified by skin fragility, blister formation, and scarring. The most severe forms of the disease are characterised by mutilating scarring, blisters covering large proportions of the body surface, and, later in the disease course, mitten deformities, joint contractures, oesophageal strictures, corneal erosions, chronic cutaneous infections, and aggressive squamous cell carcinoma.1–3 Children and adults with recessive dystrophic epidermolysis bullosa face a life of pain and a high risk of developing squamous cell carcinoma, which can occur as early as 13 years of age. Of the patients who survive to reach 40 years of age, 50% have this cancer. Nearly all patients with dystrophic epidermolysis bullosa and squamous cell carcinoma die from metastatic disease. Patients with severe dystrophic epidermolysis bullosa have profound physical disabilities, and daily activities (eg, going to the toilet, feeding, bathing, and walking) are major challenges. Children with the disease need round-the-clock daily care, and the quality of life of patients tends to decline with age.
Pathophysiology
Type VII collagen (C7) is synthesised by both human keratinocytes and fibroblasts. The protein is secreted within the basement membrane zone that lies between the epidermis and dermis of the skin. C7 is the major component of anchoring fibrils, which are necessary for normal epidermal-dermal adherence. Genetic defects in the C7 gene, COL7A1, lead to recessive dystrophic epidermolysis bullosa. Ultrastructurally abnormal morphology and the absence or scarcity of anchoring fibrils result in extreme skin fragility caused by cleavage and detachment of the sublamina densa from the underlying dermis (figure 1). Dystrophic epidermolysis bullosa can be inherited as an autosomal dominant or autosomal recessive disease, with blisters and scarring present at birth or shortly afterwards. The blisters in the neonate often cover the whole body, including the oral and oesophageal mucosa, and persist throughout life.
Figure 1. Structure of the dermal-epidermal junction and phenotype of generalised severe recessive dystrophic epidermolysis bullosa.

(A) Ultrastructural features of a healthy human skin basement membrane zone with anchoring fibrils (arrows) extending from the basement membrane to the papillary dermis. (B) Immune fluorescence-aided visualisation of healthy human type VII collagen (in red) at the dermal-epidermal junction. Nuclei are blue (DAPI stain). (C) Wounds in recessive dystrophic epidermolysis bullosa. (D) Disfiguring scarring in recessive dystrophic epidermolysis bullosa.
Wound healing and chronic skin infections
After injury, healthy skin regenerates with remarkable efficiency. All layers of the skin and its cellular compartments are engaged in this robust and carefully regulated process. Recent accumulating experimental and clinical experience, partly from cellular transplantation studies in recessive dystrophic epidermolysis bullosa, provides evidence that extracutaneous cells and factors are also operational in skin repair, thus increasing our understanding of the underlying molecular and cellular mechanisms responsible for the maintenance of skin integrity.
Wound care in people with epidermolysis bullosa poses unique challenges. Highly personalised treatment plans are necessary because of variations in clinical severity, pathogenicity of the bacterial and fungal organisms present, degree of skin involvement, and the patient’s age. Furthermore, as patients get older, they have a higher risk of colonisation with antibiotic-resistant bacteria, such as meticillin-resistant Staphylococcus aureus and pan-resistant Pseudomonas aeruginosa. The implications for wound healing depend on the bacterial load and virulence. Whereas contamination refers to smaller bacterial loads with little effect on wound healing, critical colonisation occurs when bacterial proliferation causes local damage and wound healing is prevented.4 In this situation, chronic, non-healing wounds occur, despite intact epithelial repair mechanisms. In some cases, these wounds are due to the development of bacterial biofilms—a state in which bacteria and secreted extracellular substances adhere to the wound bed and become almost impenetrable to antibiotic treatment. Such wounds are debilitating and potentially life-threatening. Treatment strategies for the removal of biofilms include topical and systemic antibiotics, silver dressings, and debridement.5
Blood and marrow transplantation
Potential of blood and marrow stem cells
The biology of stem-cell transplantation has been fortunate in its rich history of constant innovation and adaptation, and in its ability to transfer experimental findings directly into patients with life-threatening diseases. Haemopoietic stem-cell transplantation, the prototypical stem-cell therapy, has been a life-saving approach for people with more than 70 different haematological diseases for the past 45 years.6 Furthermore, the discovery that a few blood or bone marrow stem cells taken from a donor can give rise to a fully functional lympho-haemopoietic system in the recipient enabled therapy for some equally fatal non-malignant disorders, such as genetic enzymopathies.7 This treatment effect is due to the ability of donor cells to secrete an enzyme (such as lysosomal hydrolase in mucopolysaccharidoses) and the ability of the recipient cells to take it up even when they are themselves deficient. Every year, more than 50 000 people now receive haemopoietic stem-cell trans plantation for various, otherwise fatal, malignant and non-malignant disorders.8
Successful cell and tissue therapies have been mostly confined to the repair of the same tissue, such as when bone marrow or umbilical cord blood as a source of haemopoietic stem cells is transplanted into people with lympho-haemopoietic disorders, or similarly when skin grafts are used to replace skin damaged or lost by burns, trauma, or surgery. However, two notable exceptions are that bone marrow can be used to treat the bone disorder osteogenesis imperfecta9 and the skin disease recessive dystrophic epidermolysis bullosa.10
The fascinating cellular journey from blood to skin by virtue of the transplantation of bone marrow is both mechanistically significant and clinically relevant. The pathway of treatment of children with generalised severe recessive dystrophic epidermolysis bullosa, which began with near-complete clinical helplessness, has led to an amelioration of their blistering and pain, and improvement to their quality of life.
Repurposing of blood and bone marrow
The problem
The pain that people with recessive dystrophic epidermolysis bullosa endure is often overwhelming, and the consequences of the disease are tragic. Motivated by the suffering of the patients and despair of their families, the progress achieved in skin biology applied to pre clinical approaches has been equally extraordinary.
Several new treatments that target wounds in the disease have been developed, including local cell therapy with skin fibroblasts11,12 and mesenchymal stem or stromal cells,13 gene therapy with gammaretroviral14 or lentiviral vectors,15–17 and C7 protein replacement therapy.18–20 These innovations emanated from decades of preclinical molecular and cellular studies by highly motivated teams worldwide,3,12,21–27 and the commitment of both national funding organisations and patient support groups.
Despite the unforgettable appearance of a child with recessive dystrophic epidermolysis bullosa, the pathological process of generalised severe disease is best understood in its entirety, with several external and internal organ systems being affected (cutaneous and gastro intestinal) and whole body responses (such as compromised nutrition, anaemia, pruritus, chronic inflammation, and local and systemic infections) amplifying the injury of the underlying C7 deficiency. There fore, as an alternative to local gene, cell, and protein therapies, we sought systemic and possibly permanent cross-correction of C7 in the extracellular matrix with durable transplantation of self-renewing blood and marrow stem cells.
Proof of concept
To investigate this idea, we transplanted congenic wild-type bone marrow cells (among a range of other stem-cell types) into neonatal mice with a targeted disruption of the Col7a1 gene28 that results in widespread blistering and early death because of the absence of C7 expression. We hypothesised that a stem-cell population existed in bone marrow that homed to injured skin close to the dermal-epidermal junction and produced cellular progeny that secreted wild-type C7 protein where it was needed. Although various non-haemopoietic and haemopoietic cell populations from bone marrow did not successfully correct the disease, it turned out that an infusion of highly purified bone marrow progenitors (CD150+ CD48− cells)29 migrated to injured skin and secreted C7. Anchoring fibrils were partly restored and blisters on paws healed,30 whereas untreated affected pups died within 14 days.
Thus, donor cells capable of both secreting C7 and homing to the injured mucocutaneous membranes led to part correction of the disease phenotype. This functional correction of C7 was done in a mouse model of human recessive dystrophic epidermolysis bullosa; and this achievement, along with data from Chino and colleagues31 that also showed similar effects with prenatal CD90-depleted bone marrow (in the absence of any other curative approach for severe disease), led us to examine the safety and efficacy of allogeneic blood and marrow transplantation as a treatment for children with the most severe forms of the illness.
First-in-human clinical trial
So far, only the results for the first seven patients have been reported in the scientific literature. Here, we provide a general summary of our experience so far.
This first clinical trial10 of systemic cellular therapy for a genodermatosis showed that donor cells home to injured skin and do so in unexpectedly high numbers, expression of C7 is increased and sustained for many years after blood and marrow transplantation, and anchoring fibrils gradually appear and increase in number. Clinically, nearly all patients have experienced some improvement in maintenance of overall skin integrity (figure 2).
Figure 2. Increased C7 expression and clinical benefit after blood and marrow transplantation for severe recessive dystrophic epidermolysis bullosa.
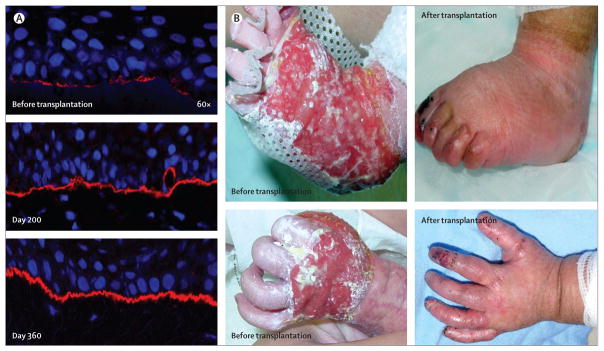
(A) Immune fluorescence-aided visualisation of human type VII collagen (in red) at the dermal epidermal junction before blood and marrow transplantation (top panel), 200 days after blood and marrow transplantation (middle panel), and 360 days after blood and marrow transplantation (bottom panel). Nuclei are blue (DAPI stain). (B) Clinical comparison of extremity wounds before (left panels) and after (right panels) blood and marrow transplantation. Reprinted with permission from reference 10.
To provide unequivocal proof for this outcome is challenging. Serial whole-body photography, weighing of dressings used over time, and referencing of parental and patient logs are not objective proof. Without question, fluctuations in condition occur on a daily and weekly basis. We developed a process to assess skin fragility by exposing small areas of skin to negative pressure and establishing the length of time to blister formation (video). Additional biochemical and ultrastructural assessments are being incorporated, new devices to assess quality of life from the perspective of the patient and caregivers are being used, and new photographic methods to assess total body wound involvement are being implemented.
Iterative progress
Protocol changes have been made to reduce side-effects and improve the speed of recovery. For example, the intensity of the conditioning regimen has been reduced from a combination of busulfan, fludarabine, and cyclophosphamide to combination therapy with fludarabine and low doses of cyclophosphamide and radiation (figure 3). Of the 20 patients with recessive dystrophic epidermolysis bullosa who received blood and marrow transplantation, five died from disease progression or complications of transplantation, and all those treated with a non-myeloablative regimen are alive. Although the new conditioning regimen is clearly associated with substantially less toxicity, there is an increased chance that mixed chimerism (ie, presence of both host and donor cells) will occur in the blood and marrow compartment, which will occasionally necessitate an intervention, such as the infusion of donor lymphocytes, to achieve nearly complete chimerism. The effect of the mesenchymal stromal cell infusions (figure 3) on skin integrity is less clear. Although transient engraftment of mesenchymal stromal cells has been detected in the skin and blood, the incidence of acute graft-versus-host disease has been extraordinarily low so far. Whether something different about the skin in recessive dystrophic epidermolysis bullosa reduces the risk of graft-versus-host disease or whether the presence of mesenchymal stromal cells at the time of blood and marrow transplantation prevents graft-versus-host disease is not yet known.
Figure 3. Evolution of the conditioning regimen for the treatment of severe recessive dystrophic epidermolysis bullosa.

Day 0=day of stem cell infusion. HSCT=haemopoietic stem-cell transplantation. GVHD=graft-versus-host disease.
MMF=mycophenolate mofetil. MSC=mesenchymal stromal cell. BU=busulfan. Flu=fludarabine.
CY=cyclophosphamide. TBI=total body irradiation.
Thus, of the 20 transplant recipients, many, but not all, have significant benefit from allogeneic blood and marrow transplantation—chronic wounds, once always open and infected, change to acute ones that heal and remain free of blisters; skin is cleared of colonisation with bacteria and fungi; anaemia is resolved or ameliorated; periodical oesophageal dilatations are needed less frequently; and nutrition and growth improve. From these outcomes, we can now think that the specialty can move away from traditional palliative care with its focus on infection and pain control, complex bandaging, and nutritional support. Instead, parents and caretakers could seek more effective treatment for severe forms of epidermolysis bullosa when the risks of allogeneic blood and marrow transplantation are justifiable.32
Nonetheless, in none of the patients has all the skin and mucosal lesions healed, none has had all dressings removed, and none has achieved an entirely normal number and structure of anchoring fibrils. Importantly, this situation is similar to that with other genetic disorders treatable with blood and marrow transplantation, such as mucopolysaccharidosis type I (Hurler syndrome), in which the fatal natural history of the disease is averted but not all disease manifestations are eliminated. However, results achieved so far provide a strong incentive to improve the treatment, to make it more effective and less risky.
Clinical findings and lessons learned
Although data are consistent with a model in which donor lympho-haemopoietic cells engraft in a permissive skin niche and provide a continuous source of wild-type cells to mediate skin healing, we need to better understand the biological mechanisms that underlie the potential of the blood and marrow graft to stabilise external and internal mucocutaneous epithelia.
Several findings have offered guidance. First, in mice, only highly purified haemopoietic stem cells rescued animals with recessive dystrophic epidermolysis bullosa from lethality. In human beings, both haemopoietic and non-haemopoietic donor cells engrafted in human skin after blood and marrow transplant, as evidenced by detection of male donor cells with and without expression of pan-haemopoietic CD45 antigen in the skin of a female transplant recipient (figures 4 and 5). Whether these non-haemopoietic donor-derived cells in the skin are actually derived from the haemopoietic stem cells or from another cell population in the marrow remains unknown. Nonetheless, the skin seems to be able to support other cell populations at high frequency. Second, engraftment is typically higher in the vicinity of active wounds, which suggests that cells home to areas of tissue damage or injury, where local injured cells guide systemic reparative cells to the wounds. Moreover, chimerism in recipient skin is sustained for long periods (at least 5 years so far) and is related to the degree of mucocutaneous improvement.
Figure 4. Engraftment of both haemopoietic and non-haemopoietic cells in recessive dystrophic epidermolysis bullosa skin after bone marrow transplantation.

Y chromosome (red dot and arrow) and X chromosome (green dot and arrow) of male donor cells in skin of female recipient after sex-mismatched bone marrow transplantation. Some donor cells express pan-haemopoietic antigen CD45 (blue rectangles); other donor cells (non-haemopoietic cells in both epidermis and dermis) do not (white ovals). As would be expected in a recipient with an engrafted lymphohaemopoietic system, occasional donor haemopoietic cells were detected inside a dermal blood vessel (endothelium stained green with anti-CD31 antibody). A linear band (fuchsia) at the dermal-epidermal junction (dotted line) shows type VII collagen. Reprinted with permission from reference 10.
Figure 5. Anchoring fibrils in recessive dystrophic epidermolysis bullosa skin after bone marrow transplantation.
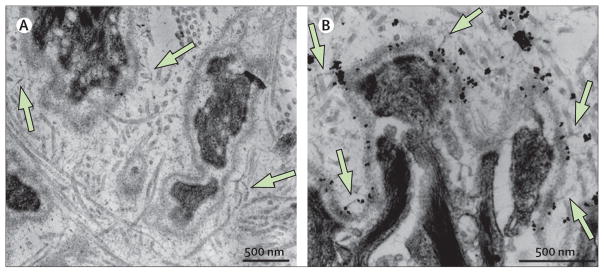
Anchoring fibrils (homopolymers of type VII collagen) more than 3 years after bone marrow transplantation on ultrastructural examination by transmission electronmicroscopy (A) and anti-type VII collagen antibody-aided immune electronmicroscopy (B). The antibody is mAb185 (from Lynn Sakai, Microimaging Center, Shriner’s Hospital for Children, Portland, OR, USA) and is conjugated to gold particles (black dots).
With this evidence, we can propose a model for consideration. Donor blood and bone marrow (sources that contain both haemopoietic and non-haemopoietic stem cells) home towards the gradient of stress signals or proinflammatory factors secreted systemically from active wounds. For example, high-mobility group protein B1, identified in the elegant studies of Tamai and colleagues33 and Petrof and coworkers,34 has been shown to cross the vascular wall dermis and locate a permissive environment of skin niches. Once integrated in these so-called docking stations, donor cells secrete C7 chains that homopolymerise in the extracellular matrix35 and, as they reach the barrier of the basement membrane zone, form anchoring fibrils, resulting in increased skin stability (figures 4–6).
Figure 6. Cross-correction for recessive dystrophic epidermolysis bullosa after blood and marrow transplantation.

Models of (A) healthy skin and (B) skin in recessive dystrophic epidermolysis bullosa. Presence of patches of mutant type VII collagen at the dermal-epidermal junction does not prevent easy separation of the epidermis from the dermis upon minimal trauma. (C) Model of skin repair in recessive dystrophic epidermolysis bullosa after allogeneic blood and marrow transplantation. Presence of donor cells (yellow) in the dermis and epidermis, indicating the presence of haemopoietic (neutrophils and lymphocytes) and non-haemopoietic cells (stromal cells and possibly keratinocytes) of donor origin that have migrated from the blood or bone marrow graft, homed to the injured skin, and secreted wild-type (normal) type VII collagen above and below the dermal-epidermal junction, leading to enhanced integrity of the skin and resistance to trauma.
Allogeneic blood and marrow transplant is the first form of gene therapy (ie, delivery of wild-type genes—in this case via haemopoietic stem cells from healthy allogeneic donors) in patients with many genetic diseases, such as severe combined immune deficiency, mucopolysaccharidosis type I, and now epidermolysis bullosa. However, equally relevant to the gene therapy of skin is the rare event of somatic mosaicism. In some patients with recessive dystrophic epidermolysis bullosa, the pathogenic COL7A1 mutation self-corrects by unequal crossover or secondary mutation.36–39 This self-correction of a single host skin cell results in patches of skin where the pathomechanistic consequences of COL7A1 mutation are reversed: normal or near-normal C7 expression, anchoring fibrils, and skin integrity are achieved. Together, these findings suggest that gene correction of cells resident in the skin can ameliorate the manifestations of the disease. This theory has provided substantial impetus for gene therapy that uses COL7A1 augmentation with viral vectors,40 corrective trans-splicing of mutant COL7A1 RNA,41 or gene editing in which a specific COL7A1 mutation is restored to the wild-type sequence in its natural genomic location.42 Correction of autologous stem-cell populations could reduce, or perhaps eliminate, the existing need for high-dose chemotherapy and immunosuppression for donor-host tolerance induction in the setting of allogeneic blood and marrow transplantation.
Future directions for treatment of recessive dystrophic epidermolysis bullosa
Although substantial improvements have already been made for patients with epidermolysis bullosa undergoing allogeneic blood and marrow transplant, such as the use of a non-myeloablative conditioning regimen (figure 3), further advances are necessary. Some promising strategies are listed here.
Gene correction of autologous cells
On the basis of the pioneering work of Mavilio and colleagues in a related form of epidermolysis bullosa—junctional epidermolysis bullosa, which is caused by deficiency of type XVII collagen43—at least two groups are pursuing treatment of individual wounds with autologous cells corrected by viral-based ex vivo therapy. These trials have benefited from transformative knowledge gained from improved vector designs that aimed to decrease the pro-neoplastic side-effects associated with an earlier generation of retroviral vectors in immune deficiencies (eg, X-linked severe combined immune deficiency, Wiscott-Aldrich syndrome, and chronic granulomatous disease).44,45 The problem of an autonomous so-called selfish transgene46,47 delivered by a viral vector, however, can be circumvented, in theory, by gene editing.40
Such gene repair is initiated by binding and cleaving of DNA close to the site of the targeted gene mutation (with a hybrid synthetic molecule such as zinc-finger nuclease, homing endonuclease, or transcription activator-like effector nuclease [TALEN])48,49 and completed by homologous recombination between exogenously provided wild-type COL7A1 sequence and mutant COL7A1 DNA. We have recently used gene editing to permanently change a pathogenic mutation in COL7A1 to the wild-type sequence, which is, to our knowledge, the first demonstration of TALEN-mediated gene correction of any disease-causing mutation within the natural context of the human genome.42 This work was undertaken in dermal fibroblasts, which were then amplified to numbers sufficient for local therapy (as has been done with allogeneic fibroblasts). Of note, fibroblasts are probably superior to keratinocytes for local wound therapy in recessive dystrophic epidermolysis bullosa. Both cell types secrete C7 protein, but fibroblasts are more robust and can be cultured extensively. However, to extend the effect to systemic therapy, engraftable gene-edited stem cells would be needed. Haemopoietic stem cells might, of course, not be the ideal cell type for systemic therapy, but we have yet to identify another population of stem cells (presumably epidermal or mesenchymal) that are capable of long-term engraftment in injured skin after intravenous administration. As an alternative to the challenging transgenesis of haemopoietic stem cells, several groups have pursued an indirect pathway by reprogramming dermal fibroblasts into pluripotent cells (which function much like embryonic stem cells), termed induced pluripotent stem (iPS) cells.50,51
iPS cells are engineered stem cells that have the potential to develop into almost any cell type in the body. For that reason, they have become a popular device for the investigation of tissue formation in health and disease, early stages of development, and so-called disease in a dish (to substitute, in part, for clinical testing of drug inter vention strategies with an in-vitro model), all of which are relevant to the biology and treatment of epidermolysis bullosa.50,52 As a first example of induced pluripotency in a disorder of the extracellular matrix, we have shown that iPS cells can be generated from skin fibroblasts of patients with recessive dystrophic epidermolysis bullosa, differentiated into artificial skin equivalents and haemopoietic progenitors, and transduced with wild-type COL7A1 to produce C7 protein.50 Essential iPS cell issues, such as transgene integration-free reprogramming, genomic fidelity, and transplantability, are being investigated in an effort to develop genomically stable, person alised iPS cell-derived cellular grafts51 that are free of contamination by undifferentiated cells and are less susceptible to immune rejection after transplantation than are iPS cells made with existing methods.53–55
Crucially, the data from iPS cell biology can now facilitate the previously impossible advances in cell therapy for specific clinical needs, such as expansion of disease-free cells from autologous gene-corrected cells or cells from skin patches with somatic mosaicism. In theory, personalised iPS cells can provide an inexhaustible supply of progeny cells with dermal and bone marrow phenotypes,56–58 applicable—independently or together—to both the local and systemic treatment of recessive dystrophic epidermolysis bullosa.
The concept of combining drugs to achieve the same goal by different mechanisms has been supported by many examples from oncology and infectious disease medicine. More recently, we hypothesised that the combination of enzyme replacement therapy with haemopoietic stem-cell transplantion for severe forms of mucopolysaccharidosis type I is superior to either modality alone.59 A similar combination of protein and cellular therapy is conceivable for recessive dystrophic epidermolysis bullosa. An even greater benefit might be derived from systemic therapy with blood and marrow transplantation combined with local treatment of the remaining wounds that are resistant because of major previous tissue damage and scarring. In principle, this benefit can be accomplished with autologous gene-corrected fibroblasts (local therapy) and blood and marrow transplantation (systemic lifelong therapy), or—even more imminently—with allogeneic fibroblasts, keratinocytes, or mesenchymal stromal cells from the donor used for local injection in an engrafted recipient of blood and marrow transplantation. In the setting of either matched sibling or unrelated donor, the recipient would be expected to perceive the locally injected cells as self. Thus, the persistence of the cells in and around wounds will probably extend beyond the time observed after injection of allogeneic fibroblasts in patients without previous blood and marrow transplantation,11,12 and result in superior clinical benefit (figure 7).
Figure 7. Therapeutic opportunities for recessive dystrophic epidermolysis bullosa.

Several strategies can be used locally or systemically, and alone or in combination. Existing treatments include injection of third-party fibroblasts adjacent to wounds, direct injection of recombinant type VII collagen, and infusion of mesenchymal stromal cells or HLA-matched haemopoietic stem cells. New treatments in development include the use of genetically corrected keratinocytes and haemopoietic stem cells, and cellular reprogramming.
From bedside to bench: new hypotheses
The goal of innovative clinical trials in recessive dystrophic epidermolysis bullosa is to alleviate the suffering caused by this progressively debilitating disease. As is the case for several rare disorders, changes in the practice of medicine for recessive dystrophic epidermolysis bullosa can also have major implications for much more common afflictions, such as thermal or chemical burns. To bring about further improvements, we need to carefully evaluate each patient’s response in the quest to understand the mechanisms underlying the disease and its response to treatment.
One of the main challenges is to understand the effect of donor cells on upregulation of mutant versus wild-type C7. New C7 was expressed in the vicinity of donor cells at the skin basal membrane zone in mice with recessive dystrophic epidermolysis bullosa that were genetically unable to express any endogenous C7;28 however, in several human recipients with residual expression of mutant C7 before blood and marrow transplantation we have recorded increased total C7 and clinical improvement afterwards, even when detectable donor cells are absent in the skin.10,60 Microchimerism could be operational in that donor cells are merely below the detection limit of the chimerism assays (PCR for a variable number of tandem repeats). Another possibility is that expression of COL7A1 gene is somehow enhanced by the chemotherapy used in the conditioning regimen before allogeneic cell infusion or immunosuppression, but this effect would be expected to be transient. By contrast, in some cases, a sustained benefit has been recorded over several years.
Intriguing data from McGrath’s group suggest that local inflammation induces donor cells to secrete factors (such as heparin-binding epidermal growth factor-like growth factor) that can increase C7 protein concentrations in recipients’ skin cells in the murine model of epidermolysis bullosa.61 Allogeneic cells might also be able to boost the endogenous regenerative capacity of damaged skin through non-C7-based mechanisms.62 The preferential deposit of C7 protein in the skin and wounds in recessive dystrophic epidermolysis bullosa after systemic application19 supports not only the role of C7 in wound closure by an increase in keratinocyte mobility and re-epithelialisation, but also the existence of a robust and specific homing mechanism that mediates skin repair in the disease.
These two mechanisms (secretion of fully functional C7 from donor cells and partly functional C7 from primed recipient cells) might indeed operate in concert.25,63–65 However, importantly, the sum of direct and indirect release of C7 is not simple and might not be always benign.12,66 Some mutations known to cause dominant dystrophic epidermolysis bullosa also occur in recessive disease. Therefore, overexpression of recipient’s mutant C7 might act in a dominant negative way when homotrimerised with donor (wild-type) C7. Alternatively, mutant wild-type anchoring fibril hybrids can be at least partly functional (consistent with our finding of rudimentary anchoring fibrils after blood and marrow transplant) or function as scaffolds for assembly of the triple helices of C7 (we owe this latter idea to John A McGrath, King’s College, London, UK).
Additionally, donor cells might mediate increased expression of other skin proteins relevant to skin adhesion, such as laminin 322 or fibronectin. This process could result in improved stability of the skin and a decrease in disfiguring skin fibrosis, even when the concentration of C7 is low and by itself inadequate to prevent blistering. Finally, the combined expression of these compensatory proteins could be key to the creation of a so-called educational gate for entry of donor cells to permissive niches in wounded skin.67
Another challenge is that injury is the key to homing to skin. Skin is a dynamic, constantly changing organ with high turnover of haemopoietic cells, which are involved in skin immunity, integrity maintenance, and healing of injury.68 The groups of Socie69 and Storek70 have shown that transplanted bone marrow cells can be detected in skin and mucosa (up to 5·5% donor cells, including donor-derived keratinocytes in patients with skin graft-versus-host disease). Moreover, the work of Krause’s team71 suggested that bone marrow contains epidermal progenitors. The cell fate of such cells from systemic circulation depends on their proliferation potential, permissive ness of the skin niche, and local cell-cell metabolic wiring. Taken together with several examples of context similarities in the niche regulation of the skin, thymus, and bone marrow,72–79 these findings support a tentative model of homing, engraftment, and functional integration of blood and bone marrow donor cells in the injured skin of patients with recessive dystrophic epidermolysis bullosa who have received a transplant (figure 6).
Conclusions
Data from innovative clinical trials are never as clear as we would like them to be. In some patients, the efficacy has been incomplete, and in all—even those with a very favourable increase in quality of life—long-term follow-up with quantitative scoring instruments for severity, pain, itching, disability, and cost of care will be needed to ascertain the durability of the initial clinical benefit.80 Fortunately, several blood and marrow transplant recipients from the initial clinical trial have sustained dramatically improved skin integrity and quality of life up to 5 years after transplantation.
Although the idea of allogeneic blood and marrow transplantation for recessive dystrophic epidermolysis bullosa sparked controversy and excitement in equal measure, an increasing amount of data support a conclusion that systemic therapy with bone marrow and umbilical cord blood cells for a genodermatosis is possible, and that it can ameliorate the acute life-threatening features of generalised severe disease. Advances in bioengineered skin, cellular reprogramming, graft engineering (eg, identification of cells capable of both homing to injured skin and secreting C7),33 and the convergence of cellular and gene therapy are poised to go further in changing the natural history of this disease.
Perhaps it is also instructive to think about how this work began. The use of stem cells and their potential power to replace diseased and damaged tissues was originally developed by haematologists. Even now, despite all the work with stem cells for many diseases, the only proven curative stem-cell therapy has been haemopoietic stem-cell transplantation for the treatment of high-risk leukaemia, lymphoma, and various non-malignant diseases of the lymphohaemopoietic system, such as aplastic anaemia and severe combined immune deficiency. Moreover, blood and marrow transplant physicians are familiar with offering very high-risk treatments, especially for diseases that themselves are associated with a high risk of death.
Louis Pasteur once said that in the fields of observation chance favours only the prepared mind.81 We began by speculating which stem cells were needed for the repair of this disease. The original hypothesis that multipotent stem cells or epidermal stem cells would correct the disease proved to be incorrect. When bone-marrow-derived stem cells were shown to be able to ameliorate recessive dystrophic epidermolysis bullosa, this finding led to new hypotheses that were not bound to any single preconceived idea. Did circulating leucocytes secrete sufficient C7 to promote the integrity of the dermal-epidermal junction in the disorder? Did a non-haemopoietic stem cell in the bone marrow home to wounds and engraft in the skin?
Few overnight successes occur in medicine, but the first clinical trials have shown a new treatment with substantial benefit. Lessons have been learned and the treatment and research have progressed with improved support and funding. Patients and families have renewed hope that one day the whole person, not just a patch of skin, could be cured.
Supplementary Material
Search strategy and selection criteria.
We searched PubMed for articles with the phrases “recessive dystrophic epidermolysis bullosa”, “stem cell”, “stem cell therapy”, “hematopoietic stem cell transplantation”, and “skin regeneration”. Most of the selected articles were published since 2010. We included reviews to accommodate formatting limitations, and older publications with a major effect on the subject.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01 AR063070 and R01AR059947), US Department of Defense (W81XWH-12-1-0609), Epidermolysis Bullosa Research Fund, Jackson Gabriel Silver Foundation, DEBRA, University of Minnesota Academic Health Center, Pioneering Unique Cures for Kids Foundation, Liao Family Fund, Sarah Moreland Fund, and the Children’s Cancer Research Fund (Minneapolis, MN, USA). We could not have completed this work without the generous support of many people, but especially John A McGrath (St John’s Institute of Dermatology, King’s College, London, UK) who reviewed the results, challenged our assumptions, and provided a measure of objectivity to our interpretation of the data. We also thank Alain Hovnanian (Inserm U781, Imagine Institut, Necker Hospital for Sick Children, Paris, France) and Katsuto Tamai (Department of Dermatology, Osaka University, Osaka, Japan) who with McGrath reviewed every patient record to verify the appropriateness for enrolment onto the trial; Colleen Delaney, who continues to chair the Data Safety and Monitoring Board for this trial; Douglas R Keene (Microimaging Center at the Shriner’s Hospital for Children, Portland, OR, USA) for doing nearly all the electron microscopic studies; Megan Riddle (University of Minnesota, MN, USA) for immunofluorescence imaging; and David Woodley and Mei Chen (Keck School of Medicine, University of Southern California, Los Angeles, CA, USA) for assessments of anti-C7 antibodies.
Footnotes
Contributors
JT and JW both contributed to the design and implementation of the study, writing of the report, and development of the figures. Additionally, both authors have reviewed the final draft of the report and figures before its submission.
Conflicts of interest
We declare that we have no conflicts of interest.
References
- 1.Bruckner-Tuderman L. Dystrophic epidermolysis bullosa: pathogenesis and clinical features. Dermatol Clin. 2010;28:107–14. doi: 10.1016/j.det.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 2.Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Uitto J, McGrath JA, Rodeck U, Bruckner-Tuderman L, Robinson EC. Progress in epidermolysis bullosa research: toward treatment and cure. J Invest Dermatol. 2010;130:1778–84. doi: 10.1038/jid.2010.90. [DOI] [PubMed] [Google Scholar]
- 4.Mellerio JE. Infection and colonization in epidermolysis bullosa. Dermatol Clin. 2010;28:267–69. doi: 10.1016/j.det.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Pope E, Lara-Corrales I, Mellerio JE, Martinez AE, Sibbald C, Sibbald RG. Epidermolysis bullosa and chronic wounds: a model for wound bed preparation of fragile skin. Adv Skin Wound Care. 2013;26:177–88. doi: 10.1097/01.ASW.0000428864.72412.b7. [DOI] [PubMed] [Google Scholar]
- 6.Thomas ED, Lochte HL, Jr, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957;257:491–96. doi: 10.1056/NEJM195709122571102. [DOI] [PubMed] [Google Scholar]
- 7.Hobbs JR, Hugh-Jones K, Barrett AJ, et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;318:709–12. doi: 10.1016/s0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 8.Appelbaum FR. Hematopoietic-cell transplantation at 50. N Engl J Med. 2007;357:1472–75. doi: 10.1056/NEJMp078166. [DOI] [PubMed] [Google Scholar]
- 9.Otsuru S, Gordon PL, Shimono K, et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood. 2012;120:1933–41. doi: 10.1182/blood-2011-12-400085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–39. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179–89. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]
- 12.Kern JS, Loeckermann S, Fritsch A, et al. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol Ther. 2009;17:1605–15. doi: 10.1038/mt.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conget P, Rodriguez F, Kramer S, et al. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12:429–31. doi: 10.3109/14653241003587637. [DOI] [PubMed] [Google Scholar]
- 14.Ortiz-Urda S, Lin Q, Green CL, Keene DR, Marinkovich MP, Khavari PA. Injection of genetically engineered fibroblasts corrects regenerated human epidermolysis bullosa skin tissue. J Clin Invest. 2003;111:251–55. doi: 10.1172/JCI17193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Titeux M, Pendaries V, Hovnanian A. Gene therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:361–66. doi: 10.1016/j.det.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Titeux M, Pendaries V, Zanta-Boussif MA, et al. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther. 2010;18:1509–18. doi: 10.1038/mt.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woodley DT, Keene DR, Atha T, et al. Intradermal injection of lentiviral vectors corrects regenerated human dystrophic epidermolysis bullosa skin tissue in vivo. Mol Ther. 2004;10:318–26. doi: 10.1016/j.ymthe.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 18.Remington J, Wang X, Hou Y, et al. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 2009;17:26–33. doi: 10.1038/mt.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodley DT, Remington J, Huang Y, et al. Intravenously injected human fibroblasts home to skin wounds, deliver type VII collagen, and promote wound healing. Mol Ther. 2007;15:628–35. doi: 10.1038/sj.mt.6300041. [DOI] [PubMed] [Google Scholar]
- 20.Yasuda M, Claypool DJ, Guevara E, Roop DR, Chen J. Genetic manipulation of keratinocyte stem cells with lentiviral vectors. Methods Mol Biol. 2013;989:143–51. doi: 10.1007/978-1-62703-330-5_12. [DOI] [PubMed] [Google Scholar]
- 21.Chen M, Kasahara N, Keene DR, et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat Genet. 2002;32:670–75. doi: 10.1038/ng1041. [DOI] [PubMed] [Google Scholar]
- 22.De Luca M, Pellegrini G, Mavilio F. Gene therapy of inherited skin adhesion disorders: a critical overview. Br J Dermatol. 2009;161:19–24. doi: 10.1111/j.1365-2133.2009.09243.x. [DOI] [PubMed] [Google Scholar]
- 23.Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann NY Acad Sci. 2010;1194:213–22. doi: 10.1111/j.1749-6632.2010.05463.x. [DOI] [PubMed] [Google Scholar]
- 24.Petrova A, Ilic D, McGrath JA. Stem cell therapies for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2010;163:1149–56. doi: 10.1111/j.1365-2133.2010.09981.x. [DOI] [PubMed] [Google Scholar]
- 25.Abdul-Wahab A, Petrof G, McGrath JA. Bone marrow transplantation in epidermolysis bullosa. Immunotherapy. 2012;4:1859–67. doi: 10.2217/imt.12.120. [DOI] [PubMed] [Google Scholar]
- 26.Uitto J, Christiano AM, McLean WH, McGrath JA. Novel molecular therapies for heritable skin disorders. J Invest Dermatol. 2012;132:820–28. doi: 10.1038/jid.2011.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan WF, Murrell DF. Fibroblast-based cell therapy strategy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28:367–70. doi: 10.1016/j.det.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Heinonen S, Mannikko M, Klement JF, Whitaker-Menezes D, Murphy GF, Uitto J. Targeted inactivation of the type VII collagen gene (Col7a1) in mice results in severe blistering phenotype: a model for recessive dystrophic epidermolysis bullosa. J Cell Sci. 1999;112:3641–48. doi: 10.1242/jcs.112.21.3641. [DOI] [PubMed] [Google Scholar]
- 29.Kiel MJ, Yilmaz OH, Morrison SJ. CD150− cells are transiently reconstituting multipotent progenitors with little or no stem cell activity. Blood. 2008;111:4413–14. doi: 10.1182/blood-2007-12-129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolar J, Ishida-Yamamoto A, Riddle M, et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113:1167–74. doi: 10.1182/blood-2008-06-161299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173:803–14. doi: 10.2353/ajpath.2008.070977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lara-Corrales I, Arbuckle A, Zarinehbaf S, Pope E. Principles of wound care in patients with epidermolysis bullosa. Pediatr Dermatol. 2010;27:229–37. doi: 10.1111/j.1525-1470.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 33.Tamai K, Yamazaki T, Chino T, et al. PDGFR{alpha}-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci USA. 2011;108:6609–14. doi: 10.1073/pnas.1016753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrof G, Abdul-Wahab A, Proudfoot L, Pramanik R, Mellerio JE, McGrath JA. Serum levels of high mobility group box 1 correlate with disease severity in recessive dystrophic epidermolysis bullosa. Exp Dermatol. 2013;22:433–35. doi: 10.1111/exd.12152. [DOI] [PubMed] [Google Scholar]
- 35.Bruckner-Tuderman L, von der Mark K, Pihlajaniemi T, Unsicker K. Cell interactions with the extracellular matrix. Cell Tissue Res. 2010;339:1–5. doi: 10.1007/s00441-009-0891-x. [DOI] [PubMed] [Google Scholar]
- 36.Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17:140–48. doi: 10.1016/j.molmed.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasmooij AM, Jonkman MF, Uitto J. Revertant mosaicism in heritable skin diseases: mechanisms of natural gene therapy. Discov Med. 2012;14:167–79. [PubMed] [Google Scholar]
- 38.Almaani N, Nagy N, Liu L, et al. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130:1937–40. doi: 10.1038/jid.2010.64. [DOI] [PubMed] [Google Scholar]
- 39.Pasmooij AM, Garcia M, Escamez MJ, et al. Revertant mosaicism due to a second-site mutation in COL7A1 in a patient with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130:2407–11. doi: 10.1038/jid.2010.163. [DOI] [PubMed] [Google Scholar]
- 40.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–15. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 41.Murauer EM, Gache Y, Gratz IK, et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:74–83. doi: 10.1038/jid.2010.249. [DOI] [PubMed] [Google Scholar]
- 42.Osborn MJ, Starker CG, McElroy AN, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21:1151–59. doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mavilio F, Pellegrini G, Ferrari S, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 44.Cavazzana-Calvo M, Lagresle C, Hacein-Bey-Abina S, Fischer A. Gene therapy for severe combined immunodeficiency. Ann Rev Med. 2005;56:585–602. doi: 10.1146/annurev.med.56.090203.104142. [DOI] [PubMed] [Google Scholar]
- 45.Rivat C, Santilli G, Gaspar HB, Thrasher AJ. Gene therapy for primary immunodeficiencies. Hum Gene Ther. 2012;23:668–75. doi: 10.1089/hum.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawkins R. The selfish gene. New York: Oxford University Press; 1976. [Google Scholar]
- 47.Moiani A, Paleari Y, Sartori D, et al. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J Clin Invest. 2012;122:1653–66. doi: 10.1172/JCI61852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Handel EM, Cathomen T. Zinc-finger nuclease based genome surgery: it’s all about specificity. Curr Gene Ther. 2011;11:28–37. doi: 10.2174/156652311794520120. [DOI] [PubMed] [Google Scholar]
- 49.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–46. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 50.Tolar J, Xia L, Riddle MJ, et al. Induced pluripotent stem cells from individuals with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:848–56. doi: 10.1038/jid.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Itoh M, Kiuru M, Cairo MS, Christiano AM. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci USA. 2011;108:8797–802. doi: 10.1073/pnas.1100332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tolar J, Xia L, Lees CJ, et al. Keratinocytes from induced pluripotent stem cells in junctional epidermolysis bullosa. J Invest Dermatol. 2013;133:562–65. doi: 10.1038/jid.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–15. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 54.Araki R, Uda M, Hoki Y, et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013;494:100–04. doi: 10.1038/nature11807. [DOI] [PubMed] [Google Scholar]
- 55.Saleh MA, Ishii K, Kim YJ, et al. Development of NC1 and NC2 domains of type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. J Dermatol Sci. 2011;62:169–75. doi: 10.1016/j.jdermsci.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Amabile G, Welner RS, Nombela-Arrieta C, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–64. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bilousova G, Chen J, Roop DR. Differentiation of mouse induced pluripotent stem cells into a multipotent keratinocyte lineage. J Invest Dermatol. 2011;131:857–64. doi: 10.1038/jid.2010.364. [DOI] [PubMed] [Google Scholar]
- 58.Wu SM, Hochedlinger K. Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat Cell Biol. 2011;13:497–505. doi: 10.1038/ncb0511-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tolar J, Grewal SS, Bjoraker KJ, et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant. 2008;41:531–35. doi: 10.1038/sj.bmt.1705934. [DOI] [PubMed] [Google Scholar]
- 60.Tolar J, Blazar BR, Wagner JE. Concise review: transplantation of human hematopoietic cells for extracellular matrix protein deficiency in epidermolysis bullosa. Stem Cells. 2011;29:900–06. doi: 10.1002/stem.647. [DOI] [PubMed] [Google Scholar]
- 61.Nagy N, Almaani N, Tanaka A, et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131:1771–74. doi: 10.1038/jid.2011.85. [DOI] [PubMed] [Google Scholar]
- 62.Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol Ther. 2009;17:939–46. doi: 10.1038/mt.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uitto J. Cell-based therapy for RDEB: how does it work? J Invest Dermatol. 2011;131:1597–99. doi: 10.1038/jid.2011.125. [DOI] [PubMed] [Google Scholar]
- 64.Ito K, Sawamura D, Goto M, et al. Keratinocyte-/fibroblast-targeted rescue of Col7a1-disrupted mice and generation of an exact dystrophic epidermolysis bullosa model using a human COL7A1 mutation. Am J Pathol. 2009;175:2508–17. doi: 10.2353/ajpath.2009.090347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexeev V, Uitto J, Igoucheva O. Gene expression signatures of mouse bone marrow-derived mesenchymal stem cells in the cutaneous environment and therapeutic implications for blistering skin disorder. Cytotherapy. 2010;13:30–45. doi: 10.3109/14653249.2010.518609. [DOI] [PubMed] [Google Scholar]
- 66.Fritsch A, Loeckermann S, Kern JS, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest. 2008;118:1669–79. doi: 10.1172/JCI34292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180:2581–87. doi: 10.4049/jimmunol.180.4.2581. [DOI] [PubMed] [Google Scholar]
- 68.Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murata H, Janin A, Leboeuf C, et al. Donor-derived cells and human graft-versus-host disease of the skin. Blood. 2007;109:2663–65. doi: 10.1182/blood-2006-07-033902. [DOI] [PubMed] [Google Scholar]
- 70.Khan FM, Sy S, Louie P, et al. Nasal epithelial cells of donor origin after allogeneic hematopoietic cell transplantation are generated at a faster rate in the first 3 months compared with later posttransplantation. Biol Blood Marrow Transplant. 2010;16:1658–64. doi: 10.1016/j.bbmt.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 71.Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 72.Park D, Sykes DB, Scadden DT. The hematopoietic stem cell niche. Front Biosci. 2012;17:30–39. doi: 10.2741/3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wagers AJ. The stem cell niche in regenerative medicine. Cell Stem Cell. 2012;10:362–69. doi: 10.1016/j.stem.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 74.Watt FM, Huck WT. Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013;14:467–73. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 75.Burdick JA, Watt FM. High-throughput stem-cell niches. Nat Methods. 2011;8:915–16. doi: 10.1038/nmeth.1745. [DOI] [PubMed] [Google Scholar]
- 76.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu CP, Polak L, Rocha AS, et al. Identification of stem cell populations in sweat glands and ducts reveals roles in homeostasis and wound repair. Cell. 2012;150:136–50. doi: 10.1016/j.cell.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hsu YC, Pasolli HA, Fuchs E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell. 2011;144:92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bonfanti P, Claudinot S, Amici AW, Farley A, Blackburn CC, Barrandon Y. Microenvironmental reprogramming of thymic epithelial cells to skin multipotent stem cells. Nature. 2010;466:978–82. doi: 10.1038/nature09269. [DOI] [PubMed] [Google Scholar]
- 80.Bruckner-Tuderman L. Systemic therapy for a genetic skin disease. N Engl J Med. 2010;363:680–82. doi: 10.1056/NEJMe1004319. [DOI] [PubMed] [Google Scholar]
- 81.Pasteur L. Dans les champs de l’observation le hasard ne favorise que les esprits préparés. Lecture. University of Lille; France: 1854. in French. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.