

Abstract
The innate cellular and molecular components required to mediate effective vaccination against weak tumor-associated antigens remain unclear. In this study we utilized polymeric cancer vaccines incorporating different classes of adjuvants to induce tumor protection, in order to identify dendritic cell subsets and cytokines critical to this efficacy. Three-dimensional, porous polymer matrices loaded with tumor lysates and presenting distinct combinations of GM-CSF and various TLR agonists effected 70–90% prophylactic tumor protection in B16-F10 melanoma models. In aggressive, therapeutic B16 models, the vaccine systems incorporating GM-CSF in combination with P(I:C) or CpG-ODN induced the complete regression of solid tumors (≤40mm2) resulting in 33% long-term survival. Regression analysis revealed that the numbers of vaccine-resident CD8(+) DCs and plasmacytoid DCs, along with local IL-12, and G-CSF concentrations correlated strongly to vaccine efficacy regardless of adjuvant type. Further, vaccine studies in Batf3−/− mice revealed that CD8(+) DCs are required to effect tumor protection, as vaccines in these mice were deficient in cytotoxic T cell priming, and IL-12 induction in comparison to wild-type. These studies broadly demonstrate that three-dimensional polymeric vaccines provide a potent platform for prophylactic and therapeutic protection, and can be used as a tool to identify critical components of a desired immune response. Specifically, these results suggest that CD8(+) DCs, plasmacytoid DCs, IL-12, and G-CSF play important roles in priming effective anti-tumor responses with these vaccines.
INTRODUCTION
The generation of immunity requires collaboration between dendritic cells (DCs) and T cells, as the priming of cytotoxic T lymphocyte (CTL) by DCs is a crucial event in the fight against infection and tumors1. DCs regulate immune responses by recognizing, processing, and decoding pathogen associated molecular patterns (PAMPs) and antigenic molecules2–6. PAMP recognition by pattern recognition receptors (PRRs) present intercellularly or at the DC surface signal the presence of infection and triggers signal transduction pathways ultimately resulting in DC activation4–6. Generally, activated DCs are characterized by enhanced expression of MHC and co-stimulatory molecules and pro-inflammatory cytokines, which enables DCs to translate pathogenic signals to naïve T cells and trigger adaptive immune responses2–8. Recent studies have demonstrated that DCs may act as a network of distinct subsets that each perform specialized functions to stimulate and polarize T cell responses in order to coordinate immune regulation9–15. Antigen processing and presentation to T cells is predominantly attributed to the conventional DC subset (cDCs), consisting of both CD8(−) DCs and CD8(+) DCs. CD8(+) DCs are especially adept at cross-presentation of exogenous antigen, IL-12 production and induction of cytotoxic T cell responses16–20. The plasmacytoid DC (pDC) subset has the capacity to produce significant amounts of type-1 interferons (IFNs) in response to microbial nucleic acids, particularly during viral infection, to facilitate T cell activation, growth and survival for disease clearance13–15, 21. Moreover, the processes mediated by pDC and CD8(+) DC subsets have been associated with priming T-helper 1 (Th1) effector cells for the control of infection and tumors. A balanced distribution of activated DC subsets is associated with the control of autoimmune disease and tumors, indicating that these cells may cooperate during the generation of protective immunity9,10,12,13.
Currently, cancer vaccines are designed to introduce antigen in combination with adjuvants to activate DCs either ex vivo prior to administration, or in situ7,8,21–24. A range of stimuli are used to trigger innate immunity resulting from DC maturation, including proinflammatory cytokines, PAMPs recognized by the toll-like receptor (TLR) family, and feedback signals from innate and adaptive immune cells. Discrete combinations of these stimuli and DC subsets may differentially control T cell activation and polarization, and these components may be optimized and exploited to generate effective immune responses that eradicate tumors or infectious agents. However, it is currently unclear what components and DC subsets should be included in cancer vaccines, partly because current techniques limit the cell types that can be cultured or targeted21–24. Standard DC-based protocols used in the clinic utilize monocyte-derived conventional DCs that are unable to cross-present antigens, or efficiently produce IL-12 or type-1 IFNs to prime CTL-mediated immune responses and tumor cell death22–24. There have been efforts to utilize type 1 differentiated DCs in combination with TLR agonists to boost CTL priming capacity, but this ex vivo maturation is accompanied by decreased migratory and stimulatory function upon implantation22.
Previously, we described macroporous polymer matrices that regulate the trafficking and activation of DCs in vivo by precisely controlling the presentation of GM-CSF and CpG-oligonucleotide (CpG-ODN) adjuvants25–26. When applied as cancer vaccines, these matrices led to CTL-mediated eradication of melanoma tumors26. Here, these matrices were modified to present 3 different classes of TLR agonists, CpG-ODN, monophosphoryl lipid A (MPLA), and polyinosinic:polycytidylic acid (P(I:C)), all in combination with GM-CSF. We hypothesized that PLG matrices presenting various combinations of adjuvants would modulate the distribution of DC subsets at the vaccine site, and influence the development of immunity to antigens loaded within these vaccine systems. This approach would then allow us to identify critical cellular and molecular hallmarks for effective vaccination or immunotherapy. Therefore, we first quantified the ability of various adjuvants to generate activated DC subsets and cytokines in vivo. The impact of DC induction on T cell-mediated immunity and cancer vaccine efficacy with models of B16-F10 melanoma was next assessed. These studies demonstrated that anti-tumor efficacy requires CD8(+) DCs and is strongly correlated with pDC numbers. Survival outcomes were also correlated to an array of inflammatory cytokines, which revealed a strong relationship between IL-12 and G-CSF production and antitumor efficacy. Altogether, these studies demonstrate that one can recruit and utilize various DC subsets for in situ vaccination, and provide important cellular and molecular insights into cancer vaccine design.
MATERIALS & METHODS
Cell Culture
B16-F10 melanoma cells were obtained from American Type Culture Collection (catalog: ATCC CRL-6475) in 2010 and 2012. Upon receipt, the cells were cultured to passage three, aliquoted and frozen in liquid nitrogen. For tumor experiments, B16-F10 cells were thawed and cultured in DMEM (Life Technologies, Inc.), containing 10% fetal bovine serum (Life Technologies, Inc.), 100 units/ml penicillin, and 100 µg/ml streptomycin. The cells were maintained at 37°C in a humidified 5% CO2/95% air atmosphere and early passage cells (between 4 and 9) were utilized for experiments. The cells have not been reauthenticated since receipt.
Mice
C57BL/6 mice (6–8-week-old female; Jackson Laboratories), B6.129S2-Cd8atm1Mak/J (CD8 T cell KO; 6–8-week-old female; Jackson Laboratories), B6.129P2(SJL)-Myd88tm1.1Defr/J (MyD88 KO; 6–8-week-old female; Jackson Laboratories), C57BL/6J-Ticam1Lps2/J (TRIF KO; 6–8-week-old female; Jackson Laboratories)and Batf3−/− knockout mice (B6.129S(C)-Batf3tm1.1Kmm/J; 6–8-week-old female; Jackson Laboratories) were cared for in accordance with the American Association for the Accreditation of Laboratory Animal Care International regulations. B6.129S(C)-Batf3tm1.1Kmm/J and B6.129S2-Cd8atm1Mak/J congenic mice were backcrossed with C57BL/6 mice for at least 11 and 13 generations, respectively. Experiments were all approved by the Harvard University Institutional Animal Care and Use Committee.
Matrix Fabrication
A 85:15, 120 kD copolymer of D,L-lactide and glycolide (PLG) (Alkermes, Cambridge, MA) was utilized in a gas-foaming process to form porous PLG matrices27. In brief, PLG microspheres encapsulating GM-CSF were first made using standard double emulsion28. The double emulsion process was also utilized to fabricate PLG microspheres containing MPLA (Avanti Polar Lipids, Alabaster, AL) as an adjuvant41. PLG microspheres were then mixed with 150 mg of the porogen, sucrose (sieved to a particle size between 250 µm and 425 µm), and compression molded. The resulting disc was allowed to equilibrate within a high-pressure CO2 environment, and a rapid reduction in pressure causes the polymer particles to expand and fuse into an interconnected structure27. The sucrose was leached from the scaffolds by immersion in water, yielding scaffolds that were 80–90% porous.
To incorporate tumor lysates into PLG scaffolds, biopsies of B16-F10 tumors that had grown subcutaneously in the backs of C57BL/6J mice (Jackson Laboratory, Bar Harbor Maine), were digested in collagenase (250 U/ml) (Worthington, Lakewood, NJ) and suspended at a concentration equivalent to 107 cells per ml after filtration through 40 µm cell strainers. The tumor cell suspension was subjected to 4 cycles of rapid freeze in liquid nitrogen and thaw (37°C) and then centrifuged at 400 rpm for 10 min. The supernatant (1ml) containing tumor lysates was collected, incubated with the PLG microspheres and lyophilized. The resulting mixture was used to make PLG scaffold-based cancer vaccines.
To incorporate CpG-ODNs and P(I:C) into PLG scaffolds, CpG-ODN 1826, 5’-tcc atg acg ttc ctg acg tt-3’, (Invivogen, San Diego, CA) or P(I:C) (high molecular weight; Invivogen, Sand Diego, CA) was first condensed with poly(ethylenimine) (PEI, Mn ~60,000, Sigma Aldrich) molecules by dropping CpG-ODN 1826 or P(I:C) solutions into a PEI solution, while vortexing the mixture29. The charge ratio between PEI and CpG-ODN (NH3+:PO4−) was kept constant at 7 during condensation. The charge ratio between PEI and P(I:C) (NH3+:PO4−) was kept constant at 3 during condensation. The condensate solutions were then vortexed with 60 µl of 50% (wt/vol) sucrose solution, lyophilized and mixed with dry sucrose to a final weight of 150 mg. The sucrose containing PEI-CpG-ODN condensate was then mixed with blank, GM-CSF and/or tumor lysate loaded PLG microspheres to make PLG cancer vaccines.
In Vitro Release Studies
To determine the efficiency of GM-CSF incorporation and the kinetics of GMCSF release from PLG scaffolds, 125I-labeled hr-GM-CSF (Perkin Elmer) was utilized as a tracer. Scaffolds were prepared with iodinated GM-CSF and placed in 3 ml of Phosphate Buffer Solution (PBS) in an incubator (37°C). At various timepoints, the PBS release media was collected and replaced with fresh media. The amount of 125I-hr-GMCSF released from the scaffolds was determined at each time point by counting the radioactivity of the removed media in a gamma counter and normalizing the result to the total 125I-GM-CSF incorporated into the scaffolds. The total amount of CpG-ODN incorporated into PLG scaffolds and released into PBS over time was determine by using an oligreen assay (Invitrogen).
In addition, the MPLA incorporation and release was determined by dissolving the matrix in a 2:1 chloroform:methanol mixture. The solution was vigorously shaken for 10 minutes and evaporated under a stream of N2. One milliliter of 3N methanolic HCl, 1ml of hexane, and 25µl of a 10mg/ml pentadecanoic acid internal standard stock solution was added to each reaction vessel. The vessels were capped tightly and heated at 50°C for 3hours. The samples were allowed to cool for 10 minutes and then 1ml each of water and hexane was added. The reaction vessels were vigorously shaken for 5 minutes and allowed to phase separate. One microliter of the hexane layer was sampled for the gas chromatography-mass spectrometry analysis. The MPLA content was then quantified by detecting the total methyl ester derivative of myristic acid.
In situ identification of DCs and T cells
GM-CSF loaded PLG matrices and matrices containing GM-CSF in combination with either 100ug of CpG-ODN, MPLA, or P(I:C) were implanted into subcutaneous pockets on the back of 7–9 week old male C57BL/6J mice. To analyze DC recruitment, scaffolds were excised at various time-points and the ingrown tissue was digested into single cell suspensions using a collagenase solution (Worthington, 250 U/ml) that was agitated at 37°C for 45 minutes. The cell suspensions were then poured through a 40µm cell strainer to isolate cells from scaffold particles and the cells were pelleted and washed with cold PBS and counted using a Z2 coulter counter (Beckman Coulter). To assess, DC infiltration and activation, subsets of the total cell population isolated from PLG matrices were then stained with primary antibodies (BD Pharmingen, San Diego, CA) conjugated to fluorescent markers to allow for analysis by flow cytometry. APC-conjugated CD11c (dendritic cell marker), FITC-conjugated MHCII and PE-conjugated CD86 (B7, costimulatory molecule) stains were conducted for DC recruitment and activation analysis. To further delineate the presence of specific DC subsets, cells were also stained with APC-conjugated CD11c and PE-conjugated PDCA-1 (plasmacytoid DC marker) or APC-conjugated CD11c and PE-conjugated CD8 (CD8 DCs). DC subsets were classified as CD8(+)CD11c(+) DCs (CD8+ DCs) and CD11c(+)CD8(−)PDCA-1(+) (pDCs). To track dendritic-cell emigration from the implant site, FITC was incorporated into the scaffolds25,31. To assess T cell infiltration PE-Cy7 conjugated CD3 stains were performed in conjunction with APC-conjugated CD8a (CD8 T cells), and PE-conjugated FoxP3 (Treg) and analyzed with flow cytometry. Cells were gated according to single positive FITC, APC and PE stainings and using isotype controls. The percentage of cells staining positive for each surface antigen was recorded.
Tumor growth assays, protective cytokines and Trp2 pentamer analysis
PLG scaffolds with melanoma tumor lysates and GM-CSF in combination with CpG-ODN, MPLA, or P(I:C) were implanted subcutaneously into the lower left flank of C57BL/6J mice. For prophylactic vaccinations, animals were challenged 14 days later with a subcutaneous injection of 105 B16-F10 melanoma cells (ATCC, Manassas, NJ) in the back of the neck. Animals were monitored for the onset of tumor growth (approximately 1mm3) and sacrificed for humane reasons when tumors grew to 20 – 25 mm (longest diameter).
To assess PLG vaccine efficacy in the therapeutic setting, C57/BL6J mice were challenged with a subcutaneous injection of 5×105 B16-F10 melanoma cells (ATCC, Manassas, NJ) in the back of the neck. At day 9 after tumor challenge PLG vaccines loaded with 3000ng GM-CSF in combination with 100 µg of CpG-ODN, MPLA or P(I:C), and tumor lysates were implanted subcutaneously into the lower left flank of C57BL/6J mice. A subset of mice were vaccinated again 10 days after the initial vaccination (Day 19).
To determine in vivo concentrations of inflammatory cytokines at the matrix implant site, adjacent tissue was excised and digested with tissue protein extraction reagent (Pierce). After centrifugation, the concentrations of cytokines in the supernatant were then analyzed with ELISA (R&D systems) and Bio-Plex Pro™ Mouse Cytokine 23-plex Assay (Biorad), according to the manufacturers instructions. Local cytokine analysis at the vaccine site was performed in wild-type C57BL/6J mice, Batf3−/− mice, and CD8 T cell knockout mice.
To study the generation of TRP-2-specific cytotoxic T lymphocytes, single cell suspensions were prepared from the spleens of mice immunized with PLG vaccines [Antigen + 3000ng GM-CSF + 100µg (CpG or MPLA or P(I:C)] at various timepoints. These cells were initially stained with PE-H-2Kb/TRP2 pentamers (Sigma Aldrich), and subsequently stained with FITC-anti-CD8 and PE-CY7 CD3 mAb (mAb (BD Pharmingen, San Diego) before being analyzed using flow cytommetry.
TIL characterization
On the indicated days, B16-F10 tumors were removed from mice, and digested in 1 mg/mL collagenase II (250 U/ml) (Worthington, Lakewood, NJ) and 0.1 mg/mL DNase for 1 hour at 37°C. Dissociated cells were filtered through a 40-µm filter, and directly stained with antibodies for phenotype characterization by fluorescence-activated cell sorting (FACS) analysis. APC-anti-CD8 and PE-Cy7-anti CD3 were used to identify T cells isolated from the B16F10 tumors. These TILs were also costained with FITC-anti-IFNγ, and PE-anti-CD107a. All antibodies were obtained from eBioscience, San Diego, CA.
Statistical analysis
All values in the present study were expressed as mean ± S.D. Statistical significance of differences between the groups were analyzed by a Student’s t test and a P value of less than 0.05 was considered significant.
RESULTS
Controlled GM-CSF and TLR Agonist Presentation
Macroporous, poly-lactide-co-glycolide (PLG) matrices (Fig 1A) were designed to quickly release GM-CSF25. Approximately 60% of the protein was released by day 10 (Fig 1B–D), to induce the recruitment of DCs or their precursors. GM-CSF loaded PLG scaffolds were also modified to present TLR-activating, CpG-ODN, MPLA and P(I:C) molecules, as danger signals. The in vitro release kinetics of GM-CSF were similar in all conditions, irrespective of which TLR agonist was included in the vaccine (Fig 1B–1D). TLR agonists were more stably associated with scaffolds than was GM-CSF, as only 20–30% of incorporated CpG-ODN, P(I:C) and MPLA were released over the first 10 days in vitro, followed by slow and sustained release of danger signals over the next 14 days. Presentation of the TLR agonists in this system was designed to provide a long-term, local signal to activate DCs. Importantly, the relatively high molecular weight and composition of the particular PLG chosen to fabricate scaffolds results in slow scaffold degradation31, allowing for long-term analysis of the vaccine site and its regulation over DC activation and T cell immunity.
Figure #1. SEM of scaffolds, and in vitro release kinetics of various TLR agonists from GM-CSF loaded PLG scaffolds.

(A) Photomicrographs of top surface of macroporous PLG scaffold (scale bar – 3mm), and SEM micrograph of a scaffold cross-section (scale bar – 50µm). Cumulative release of (B) CpG-rich oligonucleotides (CpG 1826), (C) P(I:C) or (D) MPLA in combination with the cumulative release of GM-CSF from each of the PLG scaffold types. Values represent mean and standard deviation (n=5 or 6).
Controlled DC generation and activation in vivo
To examine the ability of PLG matrices to recruit and activate dendritic cells in vivo, matrices delivering GM-CSF in combination with danger signals were implanted subcutaneously into the backs of C57BL/6J mice. The magnitude of DC infiltration and activation into the matrices was determined by FACS analysis of cell populations subsequently isolated from the polymeric material after 7 days. Control matrices delivering GM-CSF alone contained 2.4 ± 0.2 × 105 CD11c(+) (Fig. 2A& S1) cells, with relatively low expression levels of the activation markers, MHCII (2.6% of total CD11c(+) cells) and CD86 (4.6% of CD11c(+) cells). Inclusion of TLR-activating danger signals into PLG matrices significantly enhanced dendritic cell generation and activation in situ. Presentation of CpG-ODN, MPLA and P(I:C) enhanced the total number of recruited DCs by 2.5, 1.9, and 2.2 fold, respectively (Fig S1), as compared to GM-CSF delivery alone. Analysis of the activation state of matrix-resident DCs revealed that local TLR induction produced significant percentages of activated DCs, as CD11c(+) cells positive for MHCII(+) and CD86(+) comprised approximately 30%, 19%, and 28% of the total cells recruited to CpG, MPLA and P(I:C) loaded matrices, respectively. Matrices presenting TLR agonists mediated approximately 15 (MPLA) to 20 (P(I:C)) to 23-fold (CpG-ODN) increases in the total number of activated DCs at the implant site, relative to control matrices devoid of this signaling (Fig S1).
Figure #2. DC recruitment, activation, and LN homing is regulated by TLR agonist presentation at vaccine site.

(A) FACS histograms and plots representing scaffold infiltrating dendritic cells in GM-CSF loaded scaffolds (Con) or scaffolds loaded with GM-CSF in combination with CpG-ODN (CpG), MPLA (MPLA), and P(I:C) (P(IC)) at day 7 post-implantation in mice. Histograms indicate the relative frequency of CD11c(+) dendritic cells infiltrating the indicated scaffold formulation. Density plots indicate cells stained for CD11c(+) in combination with activated, DC markers, CD86(+) and MHCII(+). Numbers in the upper right quadrant of FACS plots indicate the percentage of CD11c(+) dendritic cells positive for activation markers. (B) The number of FITC(+)CD11c(+) dendritic cells present in the draining, inguinal lymph nodes at 2, 4, and 7 days after implantation of FITC-loaded matrices incorporating, GM-CSF (Control) and matrices loaded with GM-CSF in combination with CpG-ODN (CpG), MPLA (MPLA), and P(I:C) (P(IC)). Values represent mean and standard deviations (n=6). * P<0.05 ** P<0.01, as compared to GM-CSF loaded matrices (Con).
Because LN-homing of DCs is a critical factor in generating immunity we investigated the ability of TLR agonists to promote DC emigration from PLG matrices to the draining lymph nodes. As utilized in previous reports, fluorescein isothiocyanate (FITC) was incorporated into PLG scaffolds as a model antigen, as dendritic cells recruited to the scaffold will ingest this label30. The label can be later used to identify these cells following their trafficking to the inguinal lymph nodes. At day 2, MPLA stimulation led to a significantly higher overall number of scaffold-derived DCs in the inguinal lymph nodes as compared to controls and scaffolds containing CpG-ODN or P(I:C) as a stimulant (2B). However, at later timepoints, day 4 and 7 after implantation, the number of DCs induced to home to LN by MPLA subsided to control levels (Fig 2B), whereas CpG-ODN or P(I:C) stimulation resulted in a significantly higher number of DCs. At day 7 after implantation, CpG and P(I:C) loaded matrices resulted in more than a 10-fold increase in LN DCs over control conditions and MPLA stimulation (Fig 2B), and these results were consistent with the DC activation observed at the vaccine site (Fig S1 and S2). These data indicate that CpG-ODN or P(I:C) agonists, as presented from PLG matrices, were able to promote greater and sustained lymph node homing as compared to MPLA or control conditions.
Interestingly, stimulation of the cells which infiltrated PLG matrices with CpG-ODN, MPLA or P(I:C) enriched the numbers of CD11c(+)PDCA-1(+) pDCs and CD11c(+)CD8(+) cDCs (Fig. 3A) relative to controls. The danger signals increased the numbers of pDCs at the implant site by approximately 4-fold relative to control matrices, with an average pDC number of 140,000 cells residing in the scaffolds presenting any of the TLR agonists (Fig 3A). CD8(+) DCs were also present at the implant site at approximately 5-fold higher levels with MPLA and P(I:C) presentation, and at a 9-fold higher number when utilizing CpG-ODN as a stimulant. Strikingly, the local delivery of TLR-activating agents promoted the local production of IL-12 (200–400 ng/ml) at the implant site (Fig 3B), with CpG and P(I:C) inducing the highest levels. The IL-12 concentration correlated with the increased numbers of activated DCs and DC subsets in these conditions (Fig. 3A). Additionally, the concentrations of a panel of candidate inflammatory cytokines were assayed at the vaccine site (Table S1 & S2; Fig S3). Elevated levels of IFN-α (Fig S3) resulted from CpG-ODN and P(I:C) presentation, while MPLA had no effect on IFN-α concentration. However, MPLA led to 4-fold higher levels of TNF-α (Table S1 & Fig S4). Interestingly, IL-12 concentrations at MPLA loaded matrices were 2-fold lower than found in CpG-ODN and P(I:C) loaded matrices. TNF-α may inhibit monocyte and DC derived IFN-γ, IL-12 and T cell priming32 and the aforementioned cytokine profiles, suggesting that MPLA loaded matrices might be less efficient at stimulating anti-tumor T cell responses compared to matrices incorporating CpG-ODN and P(I:C).
Figure #3. CD8(+) and pDC subsets, and IL-12 concentrations at vaccine site.

(A) The total numbers of CD11c(+)CD8(+) DCs and pDCs at scaffold site at day 7, and (B) the local IL-12 concentration after implantation of GM-CSF loaded scaffolds (Con) and scaffolds loaded with GM-CSF in combination with CpG-ODN (CpG), MPLA (MPLA), and P(I:C) (P(IC)). Values represent mean and standard deviations (n=6). * P<0.05 ** P<0.01, as compared to GM-CSF loaded matrices (Con).
Prophylactic vaccination and correlates to its efficacy
Since PLG matrices presenting TLR agonists generate distinct and activated DC populations in situ and potent cytokine production, the anti-tumor efficacy of these systems was tested in the poorly immunogenic, B16-F10 melanoma model. B16 tumor lysates were used as a source of tumor antigen in vaccine formulations. Prophylactic PLG-based vaccines presenting both B16-F10 tumor lysates and GM-CSF resulted in 10% of the vaccinated mice surviving, tumor-free (Fig4A), after an otherwise lethal cell challenge at day 14 post-vaccination. Importantly, antigen loaded matrices with GM-CSF in combination with TLR agonists produced significant, and long-term tumor protection. CpG-ODN, MPLA and P(I:C) presentation from PLG vaccines resulted in 90, 80 and 70% survival rates (Fig. 5B). Regression analysis was subsequently performed to determine whether induction of long-term survival was related to pDC, CD8(+) (Fig 4A&B), and cytokine levels (Table S2) at the vaccine site: previously published data using various doses of CpG-ODN26 were included in the analysis. Strikingly, animal survival rates were strongly correlated with the numbers of pDCs, CD8(+) DCs and with endogenous IL-12 and G-CSF generated by PLG vaccines (Fig 4A–D; Table S2). These findings suggest that the important parameters potentiating vaccine efficacy include cross-presentation by CD8(+) DCs, and cooperative mechanisms promoted by pDCs and the cytokines, IL-12 and G-CSF.
Figure #4. Prophylactic vaccination, and survival correlation to CD8(+) and pDC subsets and IL-12 concentrations at vaccine site.

Mice were vaccinated with PLG vaccines 14 days prior to B16-F10 melanoma tumor challenge (105 cells). (A) A comparison of survival in untreated mice (Control) and mice treated with GM-CSF loaded PLG scaffolds (GM-CSF) or with PLG scaffolds loaded with GM-CSF in combination with CpG-ODN (CpG), P(I:C), or MPLA. Plots of the normalized magnitude of (B) CD11c(+)CD8(+) DC infiltration, (C) pDC infiltration at the vaccine site, and (D) local IL-12 concentration versus the percent of animals surviving at Day 100 following B16-F10 melanoma tumor challenge (survival data taken from experimental conditions in (A; red data points) and previously reported data with this system26). r values in B–C represent the linear correlation coefficient between y-axis variables (normalized by baseline levels in control scaffolds) and survival percentage.
Figure 5. Therapeutic Vaccination and anti-tumor T cell activity.
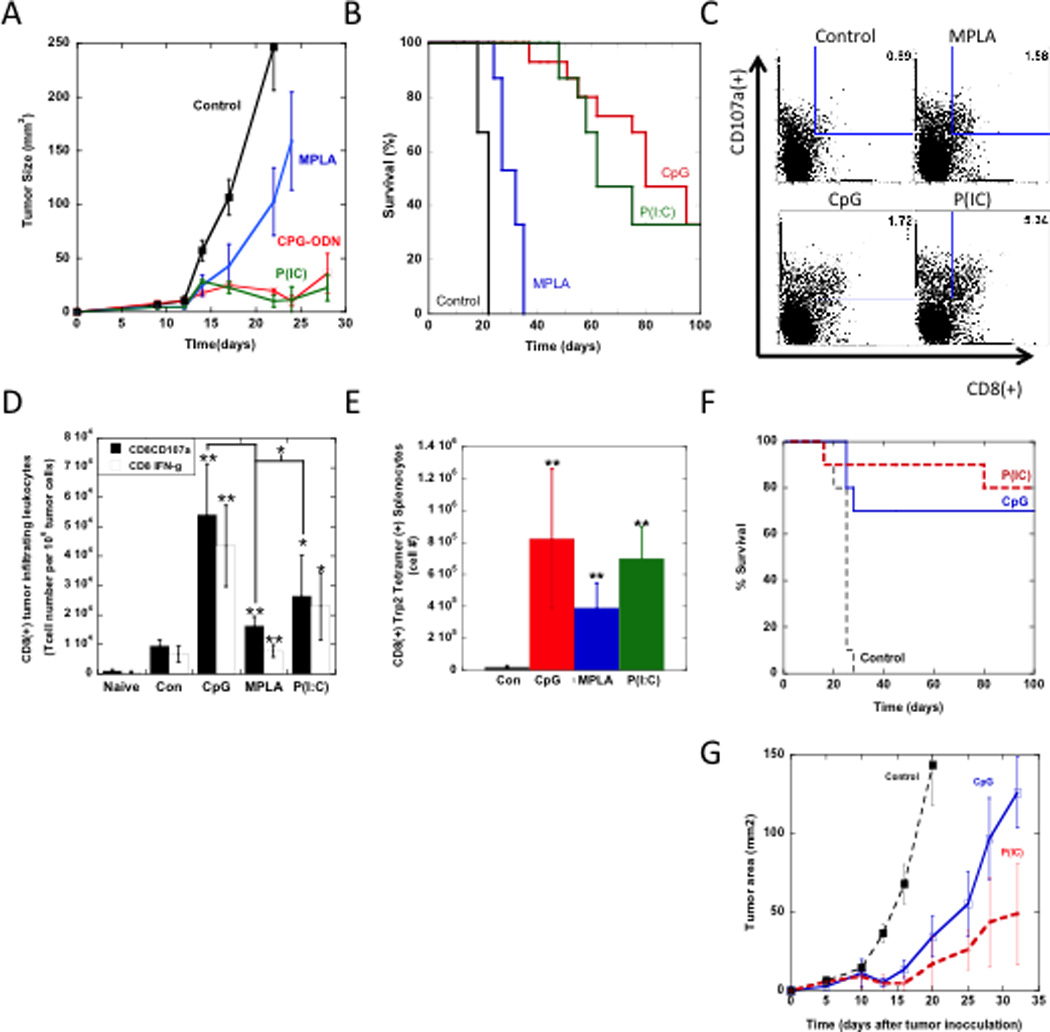
A comparison of the (A) tumor size and (B) overall survival in mice bearing established melanoma tumors (inoculated with 5×105 B16-F10 cells and allowed to develop for 9 days) that were treated with either GM-CSF loaded matrices (Con) or matrices loaded with GM-CSF in combination with CpG-ODN (CpG), MPLA (MPLA), and P(I:C) (P(IC)). Mice were vaccinated twice, at days 9 and 19. (C) FACS plots representing tumor infiltrating leukocytes in tumors of mice that were vaccinated once with either GM-CSF loaded matrices (Control) or matrices loaded with GM-CSF in combination with CpG-ODN (CpG), MPLA (MPLA), and P(I:C) (P(IC)) at Day 9 after tumor inoculation. Single cell suspensions were prepared from tumors at Day 18 and stained for activated, cytotoxic T cell markers, CD8(+) and CD107a. Numbers in FACS plots indicate the percentage of the cell population positive for both markers. (D) The numbers of CD8(+), tumor-infiltrating T cells positive for both IFNγ and CD107a in untreated mice (naïve) or mice vaccinated with various treatments. (E) The total numbers of Trp2-specific cytotoxic T cells in splenocytes of vaccinated mice. (F and G) Prophylactic and therapeutic vaccine efficacy in the lewis lung carcinoma (LLC) model. (F) A comparison of survival in untreated mice (Control) and mice treated with PLG vaccines loaded with GM-CSF in combination with CpG-ODN (CpG), or P(I:C). Mice were vaccinated with PLG vaccines 14 days prior to LLC tumor cell challenge (105 cells). (G) A comparison of the tumor size in mice bearing established LLC tumors (inoculated with 5×105 LLC cells and allowed to develop for 9 days) that were untreated (Con) or treated with PLG vaccines loaded with CpG-ODN (CpG) or P(I:C) (P(IC)). (N=10). Data in represent the mean and standard deviation (D and E) or standard error of the mean (B and G).* P<0.05 ** P<0.01, as compared to control matrices (loaded with GM-CSF), unless otherwise noted.
Moreover, TLR-MyD88 and TLR-TRIF are critical signaling pathways regulating cytokine production and immune responses33, and a complete loss of prophylactic vaccine efficacy was observed when utilizing vaccines in TRIF and MyD88 knockout mice, regardless of the TLR agonist (Fig S5). Although P(I:C) is thought to be TRIF-dependent (TLR3), CpG MyD88 dependent (TLR9), and MPLA (TLR4) can activate via TRIF and MyD88, it is likely that TLR agonists presented within tumor cell lysate and polymeric formulations require both pathways to promote the appropriate cytokine production and cellular regulation that actuates effective vaccination.
Therapeutic vaccination and anti-tumor cytotoxic T cell activity
As specific vaccine formulations containing various TLR agonists produced significant numbers of activated DCs and conferred prophylactic immunity, it was hypothesized that these would lead to superior therapeutic responses and cytotoxic T-cell responses. To test this hypothesis, mice challenged with 5 × 105 B16-F10 melanoma cells were subsequently vaccinated at days 9 and 19, after tumors were established. All tumor-bearing mice implanted with control PLG matrices demonstrated rapid tumor growth and required euthanasia by Day 24, as expected (Fig. 5A & B). PLG vaccines presenting MPLA as an adjuvant decreased the rate of tumor progression (Fig. 5A), and a slight increase in mean survival time (~1.5 fold increase) over controls was found (Figure 5A and 5B). Complete tumor regression (Tumors ≤ 36 mm2) and long term survival of mice (33% survival) was achieved in the subset of mice vaccinated with PLG vaccines exploiting P(I:C) and CpG-ODN as an adjuvant.
To further characterize the therapeutic response, mice were vaccinated at 9 days after tumor challenge and FACs analysis was used to determine the induction of B16-F10 tumor-infiltrating leukocytes (TILs). Strikingly, a one-time dose of vaccines loaded with TLR-agnoists produced significant numbers of tumor-infiltrating CD8(+) CTLs, as compared with control animals (Fig. 5C–D). CD8(+) T cell infiltrates were further characterized for co-expression of IFNγ and CD107a, a marker for cytotoxic-associated cell degranulation. These cell populations were markedly enhanced in vaccine treated animals (Fig 5C–D). Vaccines featuring CpG-ODN, P(I:C) and MPLA signaling resulted in approximately 6.1, 3.1 and 1.4 fold increases in IFNγ(+), CD107a(+) TILs in comparison to controls. Moreover, CpG loaded vaccines resulted in significantly higher numbers of activated TILs in comparison to their P(I:C) and MPLA counterparts (Fig 5D).
The activation of systemic CTL responses was also monitored by staining splenocytes with MHC class I/TRP2 peptide pentamers to identify CTLs with specificity to tyrosinase-related protein (TRP)-2. This is a main antigenic target of melanoma vaccines in mice and humans. A significant expansion of TRP2 specific CTLs was observed in the spleens of mice vaccinated with CpG-ODN, MPLA and P(I:C) loaded vaccines, in comparison to controls devoid of TLR agonists (Fig. 5E). Analysis of cells infiltrating the vaccines incorporating TLR agonists also revealed a significant, local CD3(+)CD8(+) T cell response (Fig. S6), likely in response to sustained tumor antigen presentation.
Vaccination of transgenic mice lacking CD8(+) T lymphocytes resulted in decreased levels of IL-12 and IFN-γ (Fig. S7 and S8), in comparison to wild-type. The IL-12-IFNγ pathway is a positive feedback mechanism, with each cytokine augmenting production of its counterpart32,34; these data suggest that the immune responses induced by this vaccine system may be amplified by cytokine mediated cross talk between DCs and primed CTLs.
To investigate whether the potent anti-tumor efficacy induced by P(I:C) and CpG could translate to other tumor settings, we tested these systems in the lewis lung carcinoma (LLC) model. The P(I:C) and CpG agonists were used because these agonists induced the highest survival rates in B16 melanoma models. Similar to the results in the B16 prophylactic model, P(I:C) and CpG vaccines loaded with LLC tumor cell lysates prophylactically promoted 80 and 70% survival (Fig 5G). Also, in therapeutic models mice were challenged with 5 × 105 LLC cells and subsequently vaccinated at day 7. A single vaccination with either the P(I:C) or CpG vaccine formulations were able to attenuate the growth rate of LLC tumors by approximately 3-fold (Fig 5F) and double median survival times.
Vaccine efficacy is impaired in mice lacking CD8(+) DC
Since PLG vaccines incorporating TLR agonists were capable of generating CD8(+) DC populations in situ, which correlated to potent anti-tumor CTL responses and survival (Figure 4 and 5), we next examined whether these cells were required to confer anti-tumor immunity in vivo. Batf3−/− transgenic mice were used in these experiments, as they lack CD8(+) DCs, without exhibiting abnormalities in other hematopoietic cell types or tissue architecture20. Wild type and Batf3−/− mice were vaccinated with CpG-ODN loaded PLG vaccines and challenged with B16-F10 cells 14 days later. Vaccination of wild type mice promoted complete protection against tumor growth and long-term survival (100% survival), as expected, but vaccinated Batf3−/− were not protected and tumor growth rates were similar to unvaccinated, wild-type animals (Fig 6A). Moreover, vaccinated Batf3−/− failed to produce the local CTL responses observed in wild-type mice, and a 3-fold decrease in TRP2 specific cytotoxic T cells in these mice coincided with higher ratios of FoxP3(+) T regulatory (Tregs) cells at the vaccine site in this condition (Fig 6B). These results indicate that a lack of CD8(+) DCs resulted in limited cytotoxicity, and allowed regulatory pathways mediated by Tregs to potentially extinguish the vaccine mediated, immune response. Surprisingly, the vaccine produced significantly lower numbers of local plasmacytoid DCs and activated DCs in Batf3−/− mice (Figure S9) compared to WT mice, suggesting that CD8(+) DCs play a role in generating or maintaining these cells and this effect likely contributed to the loss of vaccine efficacy. The systemic production of anti-tumor CTLs was also impaired in Batf3−/−, as a 3 fold reduction in Trp2 specific CTLs was measured in the spleens of these mice in comparison to wild-type controls (Fig 6D). These results indicate that vaccine efficacy in this system is critically regulated by CD8 DCs, potentially via their ability to cross present tumor antigens, to produce Th-1 induction factors such as IL-12, and to generate and interact with CTLs.
Figure 6. Vaccine efficacy is impaired in mice lacking CD8(+) DC.

(A) Survival times of untreated mice (control), wild type C57BL/6J mice (WT) and Batf3−/− mice (CD8 DC KO), vaccinated with PLG vaccines 14 days prior to B16-F10 melanoma tumor challenge (105 cells). (B) Analysis of cytotoxic and regulatory T cells at PLG vaccine site of WT and CD8 DC KO mice at day 10 post implantation. FACS dot plots indicate scaffold-infiltrating cells stained for CD3(+) and Trp2(+) tetramer. Numbers in the upper right quadrant of FACS plots indicate the percentage Trp2 specific cytotoxic T cell, and numbers in lower right quadrant represent the rest of the T cell population at the vaccine site. Graphs indicate the total numbers of Trp2-specific cytotoxic T cells at implant site and the ratio of CD8(+) cytotoxic T cells to regulatory T cells. (C) Fold increase in IL-12 concentration at vaccine site and (D) Trp(2)-specific cytotoxic T cells in spleens of vaccinated, WT and CD8 DC KO mice. CpG-ODN was the adjuvant utilized in vaccines. Data represent mean and standard deviation, (n=5)* P<0.05 ** P<0.01.
Because our regression analysis identified IL-12, G-CSF and IL-6 as strong correlates to survival (Table S2), we investigated whether the presence of CD8(+) DCs effected induction of these cytokines by PLG vaccines. Wild type mice were able to induce the production of the T cell growth factor, IL-12, at the vaccine site at 5-fold higher levels than found in vaccinated Batf3−/− mice (Fig 6C). The partial loss of IL-12 production in CD8(+) DC knock-out mice suggests that these cells are important producers or mediators of this Th1-polarizing cytokine. Similarly, knocking out the CD8(+) DC population resulted in approximately 3-fold decreases in the levels of IL-6 and G-CSF at the vaccine site (Fig. S10 and S11). These cytokines are associated with priming innate immunity and danger signal activation, including TLR signaling, suggesting these mechanisms of DC stimulation are compromised in Batf3−/− mice.
DISCUSSION
To address the limitations of current cancer vaccines, we utilized a PLG matrix controlling the presentation of tumor lysate, GM-CSF and TLR agonists to create a vaccine node that recruits and activates multiple DC subsets in situ. This tool was used to investigate the contribution of DC subsets to vaccine efficacy and demonstrated that effective tumor cell killing required the participation of CD8(+) DCs, with strong correlations to pDC number and IL-12, G-CSF, and IL-6 concentrations. This study expands on previous reports, by demonstrating that these components were critical to vaccine efficacy, regardless of the type of stimulant incorporated within the scaffolds.
As described previously, GM-CSF delivery was utilized to promote DC recruitment to the vaccine site where these cells are subsequently activated by TLR-agonist presentation. We determined that vaccines were able to maintain elevated levels of GM-CSF for at least 14 days regardless of the TLR agonist incorporated into the system. Interestingly, GM-CSF concentrations at the vaccine site at day 14 were approximately 80–110 fold higher than would be expected solely from release from the vaccine, as determined by in vitro release kinetics. The absolute concentrations were also lower than control matrices with only GM-CSF (Table S1). The assay is not able to distinguish between exogenous and endogenous GM-CSF and cellular infiltrates may differentially produce and consume GM-CSF at the vaccine site in response to local vaccine activity (Table S1).
In this report, we now describe methods to incorporate different classes of TLR agonists into biomaterial vaccine systems and control their presentation. Inclusion of TLR stimulation was required to significantly upregulate DC co-expression of MHCII and CD86 (Fig 2), enhancing their capacity to propagate antigen-specific T cell populations. Appropriate TLR signaling increased the generation of CD8(+) and pDC subsets at the vaccine site, and stimulated the production of IFNs and the potent T cell growth factor, IL-12. Moreover, removal of TLR agonists from the system resulted in decreased numbers of Trp2 specific, cytotoxic CD8(+) T cells locally at the vaccine site and systemically in spleens and tumors, and this coincided with reduced survival in vaccine studies.
Interestingly, a recent report described the sequestration of tumor specific, CD8(+) T cells at the site of vaccines with persistent antigen presentation. In that study, this resulted in dysfunctional immune responses and local T cell apoptosis relative to relatively non-persistent vaccine formulations34. In contrast to those peptide-based vaccines, the system in this report was fabricated to present whole tumor cell lysates within controlled inflammatory microenvironments to confer an immuno-stimulatory and tumor-mimicking microenvironment, which produced significant, antigen specific T cell infiltration at both the vaccine site and the tumor environment (fig 5C, 5D, 5E, 6B and S6). For example, at 10 days post CpG-mediated vaccination the tumor and the PLG vaccine site were similarly enriched with CD8(+) T cells (~5% of total cells; >106 antigen specific Tcells in tumors) with potent anti-tumor effects. A double dose of PLG vaccines presenting P(I:C) or CpG-ODN induced potent tumor rejection in therapeutic models of B16-F10 melanoma, causing complete tumor regression in over a third of vaccinated animals and eradicating tumors reaching 35mm2 in size. The tumor sites in vaccinated animals demonstrated intense and activated CD8(+), cytotoxic T cell infiltrates, as determined by CD107a and IFN-γ expression, likely effecting tumor-cell killing. Although the PLG vaccines were able to induce prophylactic and therapeutic survival in B16 models, unlike peptide in mineral oil formulations, the findings by Hailemichael et al.34 suggest that we could further improve efficacy by utilizing more degradable PLG formulations that may reduce the duration of antigen presentation to focus the induced T cell response on tumors.
These systems outperformed the preclinical, historical results of current vaccine modalities extensively studied in the clinic, including irradiated tumor cell vaccines and DC-based vaccines7,8,26,36,37,. Also, vaccines containing CpG-ODN and P(I:C) stimulation were able to confer efficacious anti-tumor responses in both prophylactic and therapeutic LLC models indicating that these results are transferrable to other tumor settings.
Interestingly, vaccines presenting MPLA signaling slowed tumor growth rates but did not cause tumors to completely regress. The current vaccine design is able effectively exploit CpG-ODN and P(I:C) signaling, as compared to MPLA, to promote higher average levels of cell surface activation markers on DCs and cytokine profiles that promote Th-1 polarization and CTL responses. Additionally, MPLA was a strong inducer of TNF-α, in comparison to CPG-ODN and P(I:C) (Fig S2), and this may inhibit IL-12, and IFN pathways priming CTL responses32,34. It is important to note that these results do not definitively address the potency of TLR4 antagonism relative to TLR3 and TLR9. Other design variables such as MPLA dose, and alternative TLR4 agonists should be tested in the future. However, the data does indicate that the induction of activated DCs and cytotoxic T cell is dependent on TLR stimulation and is consistent with survival rates.
Strikingly, all of the vaccinated Batf3−/− mice generated tumors in prophylactic models whereas 90% of wild-type mice were protected and survived long-term. This finding expands upon our correlative measures by proving that CD8(+) DCs are, in fact, a critical component of cancer vaccine efficacy in these systems. Further, cytokine analysis of the vaccine site of Batf3−/− mice revealed that local IL-12 levels, and CTL responses were markedly reduced, suggesting that CD8(+) DCs are an important source or mediator of local production of IL-12, and Th-1 polarization. CD8(+) DC participation, vaccination not only resulted in reduced cytotoxic, CD8(+) T cell activity (at tumor site and spleen), it also allowed the progression of Treg activity. High FoxP3(+) Treg to CD8(+) Tcell ratios indicates unbalanced immunosuppression that could extinguish vaccine efficacy and promote tumor growth38,39. CD8(+) DCs and IL-12 can cause Treg inhibition or their conversion to IFNγ producing, effector T cells40–42, and these mechanisms are potentially critical to the efficacy of these material-based cancer vaccines. Surprisingly, another cytokine, G-CSF, was found to be a strong correlate of vaccine efficacy (Table S2) even though it has been associated with suppression of T cell proliferation and cytolytic effectors43–45. G-CSF, however is a strong mobilizing factor for mononuclear cells and DCs, and its elevated endogenous production may combine with vaccines presenting TLR ligands to enhance DC differentiation and activation, and anti-tumor responses46,47.
An interesting aspect of this vaccine system is CTL homing to the vaccine site (Fig. S3), likely due to long term antigen presentation. Knocking out CD8(+) T cells also resulted in a significant reduction of local levels of IFNγ and IL-12 (Fig. S4 & S5). Others have shown that T cell derived IFNγ enhances DC expression of IL-12 and co-stimulatory molecules, creating a feedback loop that amplifies CTL-mediated responses to infection. In the current study, after vaccine priming, T cells that home back to the antigen-presenting vaccine site may be important vaccine components themselves, as they may sustain and amplify CTL responses via IFNγ-mediated DC activation and IL-12 production.
These findings provide evidence that CD8(+) DCs, pDCs and IL-12, or their equivalent functionally play key roles in material-based cancer vaccines. The relation between pDC, IL-12, G-CSF and vaccine efficacy are currently correlative and further studies should be undertaken to critically examine these relationships. Finally, the methods described here may be adapted to identify cellular and molecular components that may be utilized to design immunotherapeutic applications for other clinical indications, such as infectious and autoimmune disease.
Supplementary Material
Footnotes
Conflict of Interest: O.A., G.D., and D.J.M. are inventors on both a patent and a filed application for a patent on the technologies described in this manuscript. The other authors disclosed no potential conflicts of interest.
REFERENCES
- 1.Lanzavecchia A, Sallusto F. Regulation of T Cell Immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 4.Sansonetti PJ. The innate signaling of dangers and the dangers of innate signaling. Nat. Immunol. 2006;7:1237–1242. doi: 10.1038/ni1420. [DOI] [PubMed] [Google Scholar]
- 5.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 6.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Gilboa E. Dendritic cell based vaccines. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banchereau J, Steinman RM. Taking dendritic cells into medicine. Nature. 2007;49:419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 9.Naik SH, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 10.O'Garra1 A, Trinchieri G. Are dendritic cells afraid of commitment? Nat Immunol. 2004;5:1206–1208. doi: 10.1038/ni1204-1206. [DOI] [PubMed] [Google Scholar]
- 11.D'Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;2:293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 13.Liu YJ. Dendritic cell subsets and lineages and their functions in innate and adaptive immunity. Cell. 2001;106:259–262. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 14.Jego G, Palucka KA, Blank JP, Chalouni P, Banchereau J. Plasmacytoid Dendritic Cells Induce Plasma Cell Differentiation through Type I interferoun and Interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 15.Randolph GJ, Ochando J, Partida-Sanchez S. Migration of dendritic cell subsets and their precursors. Annu. Rev. Immunol. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 16.Schnorrer P. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. PNAS. 2006;28:10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skokos D, Nussenzweig MC. CD8– DCs induce IL-12–independent Th1 differentiation through Delta 4 Notch-like ligand in response to bacterial LPS. J Exp Med. 2007;204:1525–1531. doi: 10.1084/jem.20062305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Den Haana JM, Lehara SM, Bevan MJ. CD8+ but Not CD8− Dendritic Cells Cross-prime Cytotoxic T Cells In Vivo. J Exp Med. 2000;12:1685, 1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat. Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- 20.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 22.Hansen M, Hjortø GM, Donia M, Met O, Larsen NB, Andersen MH, Thor Straten P, et al. Vaccine. 4. Vol. 31. Elsevier Ltd; 2013. Comparison of clinical grade type 1 polarized and standard matured dendritic cells for cancer immunotherapy; pp. 639–646. [DOI] [PubMed] [Google Scholar]
- 23.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 24.Curiel TJ, Curiel DT. Tumor immunotherapy: inching toward the finish line. J Clin Invest. 2002;109:311–312. doi: 10.1172/JCI15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection mimicking materials to program dendritic cells in situ. Nat Mater. 2009;2:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali OA, Emerich D, Dranoff G, Mooney DJ. In Situ regulation of DC subsets and T Cells mediates tumor regression in mice. Sci Transl Med. 2009;1:8–19. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris LD, Kim BS, Mooney DJ. Open pore biodegradable matrices formed with gas foaming. J. Biomed. Mater. Res. 1998;42:396–402. doi: 10.1002/(sici)1097-4636(19981205)42:3<396::aid-jbm7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 28.Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled delivery systems for proteins based on poly(lactic/glycolic acid) microspheres. Pharm. Res. 1991;8:713–720. doi: 10.1023/a:1015841715384. [DOI] [PubMed] [Google Scholar]
- 29.Huang YC, Connell M, Park Y, Mooney DJ, Rice KG. Fabrication and in vitro testing of polymeric delivery system for condensed DNA. J. Biomed. Mater. Res. A. 2003;67:1384–1392. doi: 10.1002/jbm.a.20036. [DOI] [PubMed] [Google Scholar]
- 30.Thomas WR, Edwards AJ, Watkins MC, Asherson GL. Distribution of immunogenic cells after painting with the contact sensitizers fluorescein isothiocyanate and oxazolone. Different sensitizers form immunogenic complexes with different cell populations. Immunobiol. 1980;39:21–27. [PMC free article] [PubMed] [Google Scholar]
- 31.Houchin ML, Topp EM. Chemical Degradation of Peptides and Proteins in PLGA: A Review of Reactions and Mechanisms. J Pharm Sci. 2008;97(7):2395–2404. doi: 10.1002/jps.21176. [DOI] [PubMed] [Google Scholar]
- 32.Hodge-Dufour J, Marino MW, Horton MR, Jungbluth A, Burdick MD, Strieter RM, Noble PW, Hunter CA, Puré E. Inhibition of interferon γ induced interleukin 12 production: a potential mechanism for the anti-inflammatory activities of tumor necrosis factor Proc. Natl. Acad. Sci. USA. 1998;95:13806–13811. doi: 10.1073/pnas.95.23.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, Akira S. Sem. in Immunol. TLR signaling pathways. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Yun PL, DeCarlo AA, Collyer C, Hunter N. Modulation of an interleukin-12 and gamma interferon synergistic feedback regulatory cycle of T-cell and monocyte cocultures by Porphyromonas gingivalis lipopolysaccharide in the absence or presence of cysteine proteinases. Infect. Immun. 2002;70(10) doi: 10.1128/IAI.70.10.5695-5705.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hailemichael Y, Dai Y, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dora Estremera SM, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med. 2013;19(4):465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006 Jun;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, Macrae S, Nelson M, Canning C, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. U.S.A. 2008;105:3005–3010. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei G, Wei L, Zhu J, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Bailey-Bucktrout SL, Jeker LT, et al. Instability of the transcriptionfactor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oldenhove G, Bouladoux N, Wohlfert EA, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitabayashi A, Hirokawa M, Hatano Y, Lee M, Kuroki J, Niitsu H, Miura AB. Granulocyte colony-stimulating factor downregulates allogeneic immune responses by posttranscriptional inhibition of tumor necrosis factor α production. Blood. 1995;86:2220. [PubMed] [Google Scholar]
- 44.Rutella S, Rumi C, Lucia MB, Sica S, Cauda R, Leone G. Serum of healthy donors receiving granulocyte colony stimulating factor induces T cell unresponsiveness. Exp. Hematol. 1998;26:1. [PubMed] [Google Scholar]
- 45.Mielcarek M, Graf L, Johnson G, Torok-Storb B. Production of interleukin-10 by granulocyte colony stimulating factor mobilized blood products: a mechanism for monocyte-mediated suppression of T-cell proliferation. Blood. 1998;92:215. [PubMed] [Google Scholar]
- 46.Pulendran B, et al. Flt3-Ligand and Granulocyte Colony-Stimulating Factor Mobilize Distinct Human Dendritic Cell Subsets In Vivo. J of Immunol. 2000;165:566–572. doi: 10.4049/jimmunol.165.1.566. [DOI] [PubMed] [Google Scholar]
- 47.Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, Bogunovic M, Gautier EL, Miller J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;29(36(6)):1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.