

Abstract
A chiral copper-hydride catalyst for the asymmetric conjugate reduction of α,β-unsaturated carbonyl compounds has been used for the reduction of substrates containing β-nitrogen substituents. A new set of reaction conditions has allowed for a variety of β-azaheterocyclic acid derivatives to be synthesized in excellent yields and with high degrees of enantioselectivity. In addition, the effect that the nature of the nitrogen substituent has on the rate of the conjugate reduction reaction has been explored.
Many β-amino acids and their derivatives are of interest as structural components and because of their biological activity (1–3). The asymmetric synthesis of these compounds has been realized through a variety of methods (1, 4, 5), most notably asymmetric Mannich reactions (6–8), diastereoselective or enantioselective conjugate additions (9–13), and the reduction of β-amido α,β-unsaturated esters or nitriles (6, 14–18). Although the reduction of α,β-unsaturated esters has proven to be a particularly powerful tool for this transformation, the various methods used are generally limited to substrates that contain a primary amido group in the β position.
We recently described a versatile system for the asymmetric conjugate reduction of a wide variety of α,β-unsaturated carbonyl compounds (19–27). Appropriately substituted acyclic α,β-unsaturated esters, cyclopentenones, lactones, and lactams could all be reduced in high yield and with excellent levels of enantioselectivity. However, to our knowledge there have been no successful conjugate reductions of substrates containing β-heteroatoms. One possible explanation for this is that deactivation of the enoate can occur by the interaction of a lone pair of electrons on the heteroatom with the conjugated π system of the enoate. We therefore reasoned that suitable substrates for this reaction that contained β-heteroatoms would possess functional groups in which this interaction is minimized. We report here a method for the asymmetric conjugate reduction of α,β-unsaturated carbonyl compounds substituted with a nitrogen in the β position. This method allows for the preparation of a variety of azaheterocycles and derivatives not available by other methods of asymmetric reduction.
Methods
Preparation of Substrates. Various β-iodo enoates were effectively coupled with azaheterocycles by using a modified protocol derived from our recently reported copper-catalyzed vinylation of amides and carbamates (Scheme 1) (28–31). For small quantities of material, an oven-dried Schlenk flask equipped with a Teflon-coated magnetic stir bar was allowed to cool to room temperature under nitrogen and then was charged with Cu(I) iodide (0.10 mmol), potassium phosphate (1.5 mmol), and (if a solid) the nitrogen nucleophile (1.5 mmol). The flask was then capped with a rubber septum, evacuated, and backfilled with nitrogen; this process was repeated one time. Toluene (0.50 ml) was added, followed by the diamine (0.20 mmol), the nitrogen nucleophile (if a liquid) (1.5 mmol), and the vinyl iodide (1.0 mmol) as a solution in toluene (0.50 ml). The septum was then replaced with a Teflon screw cap under a positive pressure of nitrogen, and the flask was sealed and placed in a 65°C oil bath with stirring. On complete conversion of the vinyl iodide (as judged by gas chromatography), the reaction mixture was allowed to cool to room temperature. The reaction solution was partitioned between water and ethyl acetate, the phases were separated, and the aqueous phase was extracted three additional times with ethyl acetate. The combined organic layers were then dried over magnesium sulfate and filtered, and the solvent was removed with the aid of a rotary evaporator. The crude residue was then purified by flash chromatography on silica gel to give the desired compound. When larger quantities of material were desired, the same procedure was followed with the exception that a flame-dried 25-ml round-bottom flask was used rather than the Schlenk flask. Physical characterization data for all compounds are available in Supporting Methods, which is published as supporting information on the PNAS web site.
Scheme 1.

Copper-Catalyzed Asymmetric Conjugate Reduction. A 1-dram vial equipped with a Teflon-coated magnetic stir bar was charged with Cu(II) acetate monohydrate (0.05 eq) and (S)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl [(S)-BINAP)] (0.05 eq). The vial was capped with a screw-on top with a Teflon center, through which a glass pipette filled with calcium sulfate was inserted. Tetrahydrofuran (THF) was then added to the vial via syringe, and this was allowed to stir for ≈5 min. Then polymethylhydrosiloxane (PMHS) (4.0 eq) was added to the vial, and this was again allowed to stir for 5 min. Finally, a solution of the substrate (0.33 M in the total volume of THF) and t-BuOH (4.0 eq) in THF was added to the vial and allowed to stir for the time indicated. On complete conversion of the starting material, as judged by gas chromatography or thin layer chromatography, the reaction was worked up in one of two ways. For the first, the reaction mixture was diluted with ethyl acetate and then partitioned between water and ethyl acetate. The phases were separated, and the aqueous layer was extracted three times with ethyl acetate. The combined organic layers were dried over magnesium sulfate and filtered, and the solvent was removed with the aid of a rotary evaporator. The crude residue was then purified by flash chromatography over silica gel. Alternatively, the reaction mixture was loaded directly onto a silica gel column and purified by flash chromatography. Physical characterization data for all compounds are available in Supporting Methods.
Results and Discussion
With a means to access a variety of β-amino-substituted α,β-unsaturated esters in hand, we applied our previously reported reaction protocol. This entailed the use of Cu(I) chloride, (S)-p-tol-BINAP, sodium tert-butoxide (1 eq relative to CuCl), and PMHS, a mild and inexpensive hydride source, in toluene (19–24). Although our initial results were promising, further optimization was required. Examination of various Cu(I) and Cu(II) salts indicated that a number were effective precatalysts. Interestingly, we found that Cu(OAc)2·H2O was effective as a precatalyst for the reaction even in the absence of added tert-butoxide. Several solvents or solvent mixtures can be used; however, the use of THF provided faster reaction rates than that observed with other solvents. This rate enhancement is likely due to greater solubility of the copper precatalyst in THF. An additional increase in rate was observed, as in previous systems (19, 32–36), when sterically hindered alcohols were added to the reaction mixture. Further, when reactions were performed under an atmosphere of air, the reaction rates were faster than those carried out under inert atmosphere (19, 37). It should be noted that in all of these studies the enantiomeric excess of the product was unchanged. The combination of these findings led to the protocol for the reaction of 3-aza-2-enoates, the scope of which was examined, and the results of which are summarized in Table 1.
Table 1. Asymmetric conjugate reduction.
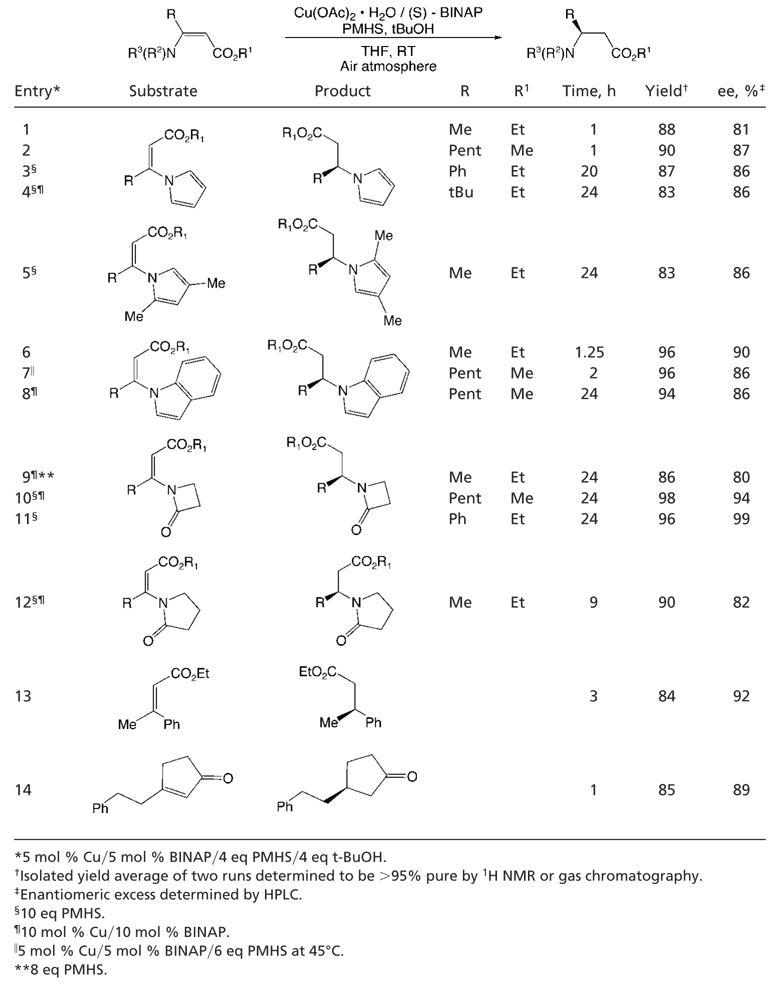 |
Various N-vinyl pyrroles (Table 1, entries 1–4) were reduced in excellent yield with high levels of enantioselectivity, including the hindered substrate 4,4-dimethyl-3-pyrrol-1-yl-pent-2-enoic acid ethyl ester (Table 1, entry 4) (38, 39). α,β-Unsaturated esters containing an indole moiety (Table 1, entries 6–8) in the β position were also effectively transformed. In addition, substrates that possessed lactams in the β position were also efficiently reduced. Both β-lactam-containing (Table 1, entries 9–11) and pyrrolidinone-containing substrates (Table 1, entry 12) gave excellent yields and enantioselectivities under the reduction conditions used.
Hindered substrates [containing one or two large substituents at the β position (Table 1, entries 3, 4, 5, and 8)] required long reaction times, likely a result of unfavorable steric interactions. As a result of the lower reaction rates, it was necessary to use 6–10 eq of PMHS, because the reaction of the silane with trace moisture in the atmosphere was competitive. Interestingly, when attempting to reduce 3-indol-1-yl-oct-2-enoic acid methyl ester, it was found that the reaction could either be performed under the standard reaction conditions (room temperature) by using a slightly higher catalyst loading and longer reaction time (Table 1, entry 8) or run with a shorter reaction time and with less catalyst by performing the reaction in a sealed tube under nitrogen at 45°C (Table 1, entry 7). These results suggest that both the efficiency and the enantioselectivity of the system are essentially independent of temperature over this range.
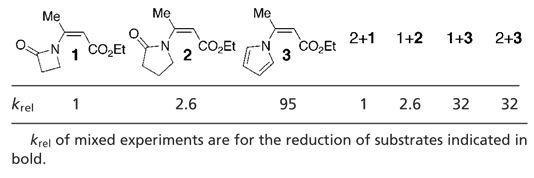
Although substrates with either a heterocycle or a lactam in the β position were viable under the reaction conditions, their reaction rates varied. Typically, substrates containing a lactam in the β position required longer reaction times than those with a β-pyrrole or -indole substituent. A more detailed examination of the reaction rates of several substrates was performed, and the results are summarized in Table 2. Interestingly, when an equimolar amount of substrates 1 and 3 were mixed and subjected to the usual reaction conditions, a decrease in the rate of reduction of 3 was observed. Similar results were obtained when a mixture of 2 and 3 were submitted to the reaction conditions as well. This leads us to propose that the carbonyl group of the lactam moiety likely coordinates to the copper in a nonproductive manner, giving rise to lower reaction rates of substrates 1 and 2 than substrate 3. Additionally, when equal molar amounts of substrates 1 and 2 were mixed and subjected to the reaction conditions, their rates were identical to those run in the absence of the second substrate. This suggests that although the coordination of the lactam to copper does inhibit the reaction, the more significant effect on the rate is the interaction of the lone pair on nitrogen with the π system. The observed higher relative rate for the δ-lactam relative to the β-lactam is presumably due to the greater N C character in the former. That substrate 3 is the most reactive is consistent with this notion, because the interaction between the nitrogen's lone pair of electrons and the π system in this substrate should be minimal.
C character in the former. That substrate 3 is the most reactive is consistent with this notion, because the interaction between the nitrogen's lone pair of electrons and the π system in this substrate should be minimal.
Table 2. Relative rates in conjugate reduction reaction.
 |
The reaction conditions described here are more convenient than those we reported earlier for asymmetric conjugate reductions, eliminating the use of air- and moisture-sensitive CuCl and hygroscopic sodium tert-butoxide. In addition to being effective for the reduction of β-heteroatom-containing α,β-unsaturated esters, these new conditions were also successful in the reduction of simple α,β-unsaturated ketones and esters (Table 1, entries 13 and 14), affording yields and enantioselectivities comparable to those obtained by using previous systems (19–24).
In conclusion, we have developed a method for the asymmetric conjugate reduction of α,β-unsaturated esters containing β-heteroatoms. We found that this system tolerated the presence of both lactams and azaheterocycles in the β position of various enoates. This method has led to the asymmetric synthesis of a number of interesting β-amino acid derivatives.
Supplementary Material
Acknowledgments
We thank Mr. Valdas Jurkauskas for conducting preliminary experiments. We thank the National Institutes of Health (Grant GM46059) for funding. Pfizer, Merck, Bristol-Myers Squibb, and Novartis are acknowledged for additional unrestricted support.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BINAP, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; PMHS, polymethylhydrosiloxane; THF, tetrahydrofuran.
References
- 1.Juaristi, E. (1997) Enantioselective Synthesis of β-Amino Acids (Wiley-VCH, New York).
- 2.Gellman, S. H. (1998) Acc. Chem. Res. 31, 173–180. [Google Scholar]
- 3.Seebach, D. & Matthews, J. L. (1997) Chem. Commun., 2015–2022.
- 4.Ma, J. A. (2003) Angew. Chem. Int. Ed. Engl. 42, 4290–4299. [DOI] [PubMed] [Google Scholar]
- 5.Liu, M. & Sibi, M. P. (2002) Tetrahedron 58, 7991–8035. [Google Scholar]
- 6.Wenzel, A. G. & Jacobsen, E. N. (2002) J. Am. Chem. Soc. 124, 12964–12965. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi, S., Matsubara, R. & Kitagawa, H. (2002) Org. Lett. 4, 143–145. [DOI] [PubMed] [Google Scholar]
- 8.Córdova, A., Watanabe, S., Tanaka, F., Notz, W. & Barbas, C. F., III (2002) J. Am. Chem. Soc. 124, 1866–1867. [DOI] [PubMed] [Google Scholar]
- 9.Sibi, M. P. & Chen, J. (2002) Org. Lett. 4, 2933–2936. [DOI] [PubMed] [Google Scholar]
- 10.Sammis, G. M. & Jacobsen, E. N. (2003) J. Am. Chem. Soc. 125, 4442–4443. [DOI] [PubMed] [Google Scholar]
- 11.Myers, J. K. & Jacobsen, E. N. (1999) J. Am. Chem. Soc. 121, 8959–8960. [Google Scholar]
- 12.Krause, N. & Hoffmann-Röder, A. (2001) Synthesis, 171–196.
- 13.Sibi, M. P. & Manyem, S. (2000) Tetrahedron 56, 8033–8061. [Google Scholar]
- 14.Drexler, H. J., You, J., Zhang, S., Fischer, C., Baumann, W., Spannenberg, A. & Heller, D. (2003) Org. Process Res. Dev. 7, 355–361. [Google Scholar]
- 15.Davies, H. M. L. & Venkataramani, C. (2002) Angew. Chem. Int. Ed. Engl. 41, 2197–2199. [PubMed] [Google Scholar]
- 16.Nelson, S. G. & Spencer, K. L. (2000) Angew. Chem. Int. Ed. Engl. 39, 1323–1325. [DOI] [PubMed] [Google Scholar]
- 17.Zhou, Y. G., Tang, W., Wang, W. B., Li, W. & Zhang, X. (2002) J. Am. Chem. Soc. 124, 4952–4953. [DOI] [PubMed] [Google Scholar]
- 18.Hodous, B. L. & Fu, G. C. (2002) J. Am. Chem. Soc. 124, 1578–1579. [DOI] [PubMed] [Google Scholar]
- 19.Hughes, G., Kimura, M. & Buchwald, S. L. (2003) J. Am. Chem. Soc. 125, 11253–11258. [DOI] [PubMed] [Google Scholar]
- 20.Jurkauskas, V., Sadighi, J. P. & Buchwald, S. L. (2003) Org. Lett. 14, 2417–2420. [DOI] [PubMed] [Google Scholar]
- 21.Jurkauskas, V. & Buchwald, S. L. (2002) J. Am. Chem. Soc. 124, 2892–2893. [DOI] [PubMed] [Google Scholar]
- 22.Moritani, Y., Appella, D. H., Jurkauskas, V. & Buchwald, S. L. (2000) J. Am. Chem. Soc. 122, 6797–6798. [Google Scholar]
- 23.Appella, D. H., Moritani, Y., Shintani, R., Ferreira, E. M. & Buchwald, S. L. (1999) J. Am. Chem. Soc. 121, 9473–9474. [Google Scholar]
- 24.Yun, J. & Buchwald, S. L. (2001) Org. Lett. 3, 1129–1131. [DOI] [PubMed] [Google Scholar]
- 25.Lipshutz, B. H. & Servesko, J. M. (2003) Angew. Chem. Int. Ed. Engl. 42, 4789–4792. [DOI] [PubMed] [Google Scholar]
- 26.Czekelius, C. & Carreira, E. M. (2003) Angew. Chem. Int. Ed. Engl. 42, 4793–4795. [DOI] [PubMed] [Google Scholar]
- 27.Lipshutz, B. H., Caires, C. C., Kuipers, P. & Chrisman, W. (2003) Org. Lett. 5, 3085–3088. [DOI] [PubMed] [Google Scholar]
- 28.Jiang, L., Job, G. E., Klapars, A. & Buchwald, S. L. (2003) Org. Lett. 5, 3667–3669. [DOI] [PubMed] [Google Scholar]
- 29.Piers, E., Wong, T., Coish, P. D. & Rogers, C. (1994) Can. J. Chem. 72, 1816–1819. [Google Scholar]
- 30.Rossi, R., Bellina, F. & Mannina, L. (1997) Tetrahedron 53, 1025–1044. [Google Scholar]
- 31.Lebedev, A. Y., Izmer, V. V., Kazyul'kin, D. N., Beletskaya, I. P. & Voskoboynikov, A. Z. (2002) Org. Lett. 4, 623–626. [DOI] [PubMed] [Google Scholar]
- 32.Lipshutz, B. H. (2002) in Modern Organocopper Chemistry, ed. Krause, N. (Wiley-VCH, Weinheim, Germany), pp. 175–179.
- 33.Stryker, J. M., Mahoney, W. S., Daeuble, J. F. & Brestensky, D. M. (1992) in Catalysis in Organic Synthesis, ed. Pascoe, W. E. (Dekker, New York), pp. 29–44.
- 34.Daeuble, J. F. & Stryker, J. M. (1995) in Catalysis of Organic Reactions, eds. Scaros, M. & Prunier, M. L. (Dekker, New York), pp. 235–247.
- 35.Chen, J. X., Daeuble, J. F., Brestensky, D. M. & Stryker, J. M. (2000) Tetrahedron 56, 2153–2166. [Google Scholar]
- 36.Chen, J. X., Daeuble, D. M. & Stryker, J. M. (2000) Tetrahedron 56, 2789–2798. [Google Scholar]
- 37.Sirol, S., Courmarcel, J., Mostefai, N. & Riant, O. (2001) Org. Lett. 3, 4111–4113. [DOI] [PubMed] [Google Scholar]
- 38.Kashima, C., Maruyama, T., Fujioka, Y. & Harada, K. (1989) J. Chem. Soc. Perkins Trans. 1, 1041–1046.
- 39.Convery, M. A., Davis, A. P., Dunne, C. J. & MacKinnon, J. W. (1995) Tetrahedron Lett. 36, 4279–4282. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.