

Abstract

Three lactams having respectively ~20 kcal/mol, ~10 kcal/mol, and 0 kcal/mol of resonance energy have been subjected to electrospray ionization mass spectrometry (ESI/MS) as well as to attempted reaction with dimethyldioxirane (DMDO). The ESI/MS for all three lactams are consistent with fragmentation from the N-protonated, rather than the O-protonated tautomer. Each exhibits a unique fragmentation pathway. DFT calculations are employed to provide insights concerning these pathways. N-Ethyl-2-pyrrolidinone and 1-azabicyclo[3.3.1]nonan-2-one, the full- and half-resonance lactams, are unreactive with DMDO. The “Kirby lactam” (3,5,7-trimethyl-1-azaadamantan-2-one), has zero resonance energy and reacts rapidly with DMDO to generate a mixture of reaction products. The structure assigned to one of these is the 2,2-dihydroxy-N-oxide, thought to be stabilized by intramolecular hydrogen bonding and buttressing by the methyl substituents. A reasonable pathway to this derivative might involve formation of an extremely labile N-oxide, in a purely formal sense an example of the hithertounknown amide N-oxides, followed by hydration with traces of moisture.
Introduction
Unstrained amides and lactams, such as N-ethyl-2-pyrrolidinone (1, R = C2H5), have 15–20 kcal/mol of stabilization (resonance) energy and this is reflected in the high rotational barriers of the amides.1–6 Protonation on oxygen rather than on nitrogen is favored by 10–15 kcal/mol. 7–14 In spite of this selectivity, protonation on nitrogen does play the key role in acid-catalyzed N-H proton exchange in unstrained primary and secondary amides, including peptides and proteins.15 In contrast, the bridgehead bicyclic lactam 2 (“2-quinuclidone”)16–22 and its derivatives as well as 3,5,7-trimethyl-1-azaadamantan-2-one (3, “Kirby lactam”)23–26 have orthogonal amide linkages and zero resonance energy, and as such behave more like amino ketones rather than lactams. R. B. Woodward and co-workers actually considered the 2-quinuclidone system and predicted ketone-like properties over seven decades ago.27 Indeed Woodward and co-workers performed a calorimetric study28 on penicillin that strongly suggested the β-lactam structure later proven by Crowfoot and co-workers.29 Protonation on nitrogen is favored by ca 20 kcal/mol in these species such as 2 and 3.10,11 Distortion of the amide linkage is quantified by three independent parameters:30 twist angle about the OC-N bond (τ), pyramidalization at nitrogen (χN) and pyramidalization at the carbonyl carbon (χC). Since this last parameter is usually quite small,30 a three-dimensional plot of energy versus τ and χN is quite useful.5 There can be a delicate balance between protonation on oxygen or nitrogen depending on these parameters. For example, 1-azabicyclo-[3.2.2]nonan-2-one favors methylation on nitrogen (4), while slightly less distorted 1-azabicyclo-[3.3.2]decan-2-one favors methylation on oxygen (5).12,31 1-Azabicyclo[3.3.1]nonan-2-one (6), has roughly half the resonance energy of an unstrained amide and is the only known example in which both N-protonated (6-NH+) and O-protonated (6-OH+) tautomers co-exist in comparable concentrations.32,33
The gas-phase proton affinity of 1-azabicyclo[2.2.2]octan-2-one (2) is 964.2 kJ/mol (230.4 kcal/mol), some 15–20 kcal/mol higher (more basic) than typical amides and lactams,22 and slightly lower than the published value for the corresponding amine, 1-azabicyclo[2.2.2]-octane (975 kJ/mol or 233.1 kcal/mol).34 Such increased localization of the nitrogen lone pair, resulting from N-CO twisting and/or pyramidalization at the amide nitrogen, should increase the propensity for other electron-pair sharing reactions beyond protonation. One intriguing possibility is the formation of hitherto unknown amide N-oxides (e.g. 7). While no amide N-oxides have been isolated or observed spectroscopically, the intermediacy of a urea N-oxide has been postulated.35 So too has the intermediacy of a carbamate N-oxide.36 While formation of an N-oxide from an unstrained amide or lactam (e.g. 7) would entail a large resonance-loss penalty, no such loss would be associated with formation of 8. Of course, it can be cogently argued that 8 is actually an alpha-keto tertiary amine N-oxide rather than an amide N-oxide. Tertiary amine oxides are well known stable compounds. While Cope eliminations of amine N-oxides to form alkenes and hydroxylamines are well-known reactions, 8 lacks β-hydrogens and even the as-yet-unknown unsubstituted compound would be unlikely to undergo this reaction for steric reasons. 1-Azabicyclo[3.3.1]nonan-2-one (6) is, however, most certainly a lactam in its chemical and spectroscopic properties. Formation of the amide (lactam) N-oxide 9 might be feasible since roughly 10 kcal/mol, rather than 20 kcal/mol, of resonance energy would be lost. Indeed similar logic (reduced rotational barrier associated with reduced energy) was invoked to rationalize intermediacy of the carbamate N-oxide,36 and this principle would seem to apply to ureas35 as well. An earlier published series of HF ab intio calculations (6-31G*) predicted that (gas-phase) reactions of planar lactams (e.g. 1) with H2O2 are endothermic (ΔE = +4 to +6 kcal/mol) and mildly exothermic for the orthogonal bridgehead lactams 2 and 3 (ΔE = −8.7 kcal/mol and −8.8 kcal/mol).37 These calculations predicted mild exothermicities for reactions of unstrained lactams with dimethyldioxirane (e.g. ΔE = −6.6 kcal/mol for 1) and significant exothermicities for 2 and 3 (−19.2 kcal/mol and −19.3 kcal/mol).37 Although energetically attractive, the predicted lengthening of the CO-N bonds (e.g. 0.11 Å upon conversion of 1 to 7 and 0.06 Å upon conversion of 3 to 8),37 suggest very enhanced lability.
Despite the fact that an MP2/6-31G** study found O-protonated formamide to be ca 60 kJ/mol (ca 14 kcal/mol) more stable than its N-protonated tautomer, gas phase chemical ionization using CH4 as reagent gas produced only NH4+, corresponding to CO loss, reflecting fragmentation of the N-protonated tautomer.38 Only when much more exothermic proton transfers (e.g. H2 as reagent gas) were explored, were H2O and NH3 loss, both originating from the O-protonated tautomer, observed in addition to loss of CO from the N-protonated tautomer.38 The origins of this non-statistical behavior were attributed to a) simultaneous dynamic formation of both N- and O-protonated tautomers, b) a much higher pair of barriers to decomposition of the O-protonated tautomer (284 and 289 kJ/mol {ca 68 and 69 kcal/mol} respectively), and c) non-statistical partitioning of the transition state connecting the O-protonated to the N-protonated tautomer, on the one hand, and formation of HCO+ with loss of NH3 on the other. 38 This experimental (ca 3 × 10−7 Torr) and computational study quite naturally explored only unimolecular decomposition pathways for the two protonated formamide tautomers.38
An electrospray ionization tandem mass spectrometry study39 of 2-pyrrolidinone reported elimination of NH3 and formation of the unsaturated acylium ion depicted in Scheme 3 (the calculated relative energies in parentheses compare well with the published energies39). The proposed mechanism involved initial O-protonation followed by isomerization to the N-protonated tautomer (calculated to be 9.5 kcal/mol higher in energy), cleavage of the weakened N-CO bond followed by further fragmentation and shifting of H from C to N.39 A detailed comparison of calculated gas-phase proton affinities for 2-pyrrolidinone (O-protonated), using a range of DFT and ab initio methods, with the experimental value was published in a follow-up study.40 The solution-phase result for 2-pyrrolidinone resembles the gas-phase result for the much simpler molecule formamide only in so far as the lower energy CI decomposition of formamide (CH4 as reagent gas) also derives from the less stable, N-protonated tautomer. One might have imagined distinct fragmentation pathways for O-protonated versus N-protonated lactams or amides as a means for distinguishing protonation site in “borderline” bridgehead bicyclic lactams such as the [3.2.2] and [3.3.2] systems cited earlier. However, it appears that the pathways proceed via N-protonated tautomers even if these are present in vanishingly low concentrations. Before describing the present experimental study, it is worthwhile noting that electrospray ionization-induced fragmentations of benzamides41 and fatty acid primary amides (RCONH2)42 appear to be well explained by their initially-formed, more stable O-protonated tautomers. This is because alternative rearrangement and fragmentation pathways are very accessible via the initial O-protonated MH+ ions derived from these molecules.
Scheme 3.

Mechanism proposed by Crotti et al39 for formation of m/z 69 ion in the ESI/MS study of 2-pyrrolidinone. The calculated (B3LYP/6-31G*) relative energies in kcal/mol in parentheses agree well with the published values;39 (calculated relative standard scaled free energies are in brackets).
The present study compares ESI mass spectra for three tertiary lactams (1, 3 and 6) differing markedly in their structures and thermodynamic stabilization (resonance) energies. There are no published CI or electrospray studies of significantly twisted lactams or amides. However, Ly et al observed the fragmentation of the N-protonated tetrafluoroborate salt of 2 using collision induced dissociation (CID) and observed loss of 44 mass units (vinyl alcohol).22
Computational Methodology
Calculations were performed using Spartan-10 and Gaussian 09W with DFT (B3LYP/6-31G*, enthalpies and free energies were calculated using a scale factor of 0.9804, or B3LYP/6-31+G**, scale factor: 0.964).50–51 Model 1H- and 13C-NMR spectra were calculated in Spartan 10 at the EDF2/6-31G* level of theory. The RMS error between experimental and calculated 13C chemical shifts is 1.8 ppm for Spartan 10.50 Images were created from Spartan and Gaussian files using CYLview.52
Results and Discussion
Electrospray Mass Spectrometry
Figure 1.a. is the ESI mass spectrum of N-ethyl-2-pyrrolidinone. Prominent features include the M+1 (MH) ion (protonated N-ethyl-2-pyrrolidinone), the proton-bridged dimer (m/z 227) and a small m/z 86 peak corresponding to loss of C2H4. The MS/MS (MS2) spectrum of the m/z 114 ion (Figure 1.b) shows prominent m/z at 97, 86, 69 and 55. The m/z 86 ion is likely derived from the concerted rearrangement depicted in Scheme 4. Since m/z 86 corresponds to the M+1 observed for 2-pyrrolidinone itself, the fragmentation to m/z 69 (loss of NH3) is consistent with the results of Crotti et al.39 These authors do not explicitly report an m/z 55 ion, although one may envision loss of CH3NH2 from the m/z 86 ion. Interestingly, ESI of N-methyl-2-pyrrolidinone does not include an MH-17 (m/z 83) ion but does include m/z 69 and m/z 58 ions assigned by the authors to [MH-CH3NH2]+ and [MH-CH3NCH]+ ions.53 Although it might be tempting to postulate the m/z 97 to arise from loss of OH from the more stable O-protonated m/z 114 ion, this homolytic cleavage requires in excess of an estimated34 100 kcal/mol and is clearly prohibited. Instead, this appears to correspond to loss of NH3 as observed for 2-pyrrolidinone itself.39 Perhaps it is initiated by rearrangement of ethyl from N to O starting from the N-protonated m/z 114 ion. Scheme 5 depicts an alternative mechanism to that in Scheme 3 for loss of NH3 from N-protonated 2-pyrrolidinone (relative energies in parentheses). The cyclopropyloxocarbenium ion postulated in Scheme 5 is stable in super acid at −80 °C.54 However, the rearrangement step to generate this ion is calculated to have a very high barrier.
Figure 1.

a. ESI/MS of N-ethyl-2-pyrrolidinone. b. MS/MS of m/z 114 ion.
Scheme 4.

Postulated mechanism for loss of C2H4 from N-ethyl-2-pyrrolidinone in ESI/MS [Calculated (B3LYP/6-31G*) relative total energies are in parentheses and relative standard scaled free energies are in brackets].
Scheme 5.

Postulated alternative mechanism (see Scheme 3) for loss of NH3 from 2-pyrrolidinone in ESI/MS [calculated (B3LYP/6-31G*) relative energies in kcal/mol shown in parentheses and relative standard scaled free energies in brackets].
Although ESI/MS is regarded as a “soft technique”, relative to electron impact (e.g. 70 eV) for example, it is worthwhile noting that the internal energies of ions using this technique may also be significant. While the specifics vary considerably with instrument conditions, internal energies in the range of 1.3 to 2.2 eV (30–50 kcal/mol) are not uncommon.55 This allows for sizeable activation energies and endothermicities in rearrangements and fragmentations.
The ESI/MS of 1-azabicyclo[3.3.1]nonan-2-one is shown in Figure 2.a. It is a very simple spectrum showing primarily the M+1 ion and the hydrogen-bridged dimer (m/z 279). Figure 2.b. is the MS2 spectrum for the M+1 ion (m/z 140). The very clean fragmentation to m/z 96 is attributed to loss of vinyl alcohol analogous to the loss of vinyl alcohol from N-protonated 1-azabicyclo[2.2.2]octan-2-one and attributed to McLafferty rearrangement.22 The corresponding postulated mechanism for 1-azabicyclo[3.3.1]nonan-2-one, also concluding with a McLafferty rearrangement, is depicted in Scheme 6. For this molecule, although N- and O-protonated tautomers are of comparable stability, fragmentation also originates entirely from the N-protonated tautomer. The calculations for the corresponding extrusion of vinyl alcohol from the [2.2.2] system are depicted in Scheme 7. The energies, including the activation parameters, for the two postulated McLafferty rearrangements are quite similar. A seeming oddity in Schemes 6 and 7 is the calculated result of slightly lower total energies for the McLafferty transition states than for the products. This is certainly, in part, an artifact of enhanced ion-dipole attraction in the transition state relative to the separated ion and molecule in the gas phase.55a,b In each case the predicted free energies are significantly lower than those of the transition states due to dissociation into an ion and a molecule.
Figure 2.

a. ESI/MS of 1-azabicyclo[3.3.1]nonan-2-one. b. MS2 of the m/z 140 ion.
Scheme 6.

Postulated ESI fragmentation pattern of 1-azabicyclo[3.3.1]nonan-2-one with loss of vinyl alcohol via McLafferty rearrangement as postulated for N-protonated 1-azabicyclo[2.2.2]octan-2-one.22 [Calculated (B3LYP/6-31G*) relative energies in kcal/mol in parentheses and relative standard scaled free energies in brackets].
Scheme 7.

Ly et al22 postulated pathway for fragmentation of N-protonated 1-azabicyclo[2.2.2]-octan-2-one in their collision-induced dissociation (CID) study of the tetrafluoroborate salt. The relative calculated (B3LYP/6-31G*) energies (kcal/mol) are in parentheses and the calculated relative scaled free energies are in brackets.
Figure 3.a. is the ESI/MS of Kirby lactam. The MS2 fragmentation of the M+1 ion (m/z 194) loses 28 mass units. This is fully consistent with the N-protonated M+1 ion losing CO as the initial step to yield m/z 166. From this ion are derived m/z 149 (loss of NH3) and m/z 135 (loss of CH3NH2?). Once again, and unsurprisingly, it is the N-protonated lactam (this time it is the O-protonated lactam is present in vanishingly small concentrations) that determines the fragmentation pathway (Scheme 8).
Figure 3.

a. is the ESI/MS of Kirby lactam. 3.b. MS2 of the m/z 194 ion.
Scheme 8.
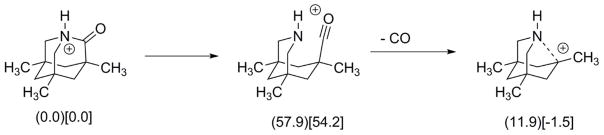
Postulated initial steps in the fragmentation pathway for the ESI of Kirby lactam. (Calculated relative energies in kcal/mol in parentheses and relative standard scaled free energies in brackets).
Reaction with Dimethydioxirane
Table 1 lists (gas phase) standard free energies of reaction and free energies of activation for the three lactams investigated in this study for their reactions with dimethyldioxirane to yield the lactam N-oxide and acetone. This is of course, a critical question concerning the chemical reactivities of three lactams, calculations were performed with and without diffuse functions (B3LYP/6-31G* and B3LYP/6-31+G**). The N-oxidation of N-methyl-2-pyrrolidinone is calculated to be only mildly exergonic with the highest activation. N-oxidation of 1-azabicyclo[3.3.1]nonan-2-one is slightly more exergonic and faster. N-oxidation of Kirby lactam is calculated (B3LYP/6-31+G**) to be considerably more exergonic with an activation barrier 9 kcal/mol lower than that of N-methyl-2-pyrrolidinone.
Table 1.
Calculated (B3LYP/6-31+G**) standard free energies of reaction (ΔG°) and scaled activation (ΔG‡) in kcal/mol for oxidation of lactams with dimethyldioxirane to produce lactam N-oxides and acetone.
| Lactam | B3LYP/6-31G* | B3LYP/6-31+G** | ||
|---|---|---|---|---|
| ΔG° | ΔG‡ | ΔG° | ΔG‡ | |
|
| ||||
| N-Methyl-2-pyrrolidinone (1, R = CH3) | −4.0 | 35.8 | −7.8 | 34.9 |
|
| ||||
| 1-Azabicyclo[3.3.1]nonan-2-one (6) | −7.5 | 32.0 | −13.0 | 29.8 |
|
| ||||
| 3,5,7-Trimethyl-1-azaadamantan-2-one (3) (“Kirby lactam”) | −17.2 | 27.5 | −22.6 | 26.1 |
Mixing N-methyl-2-pyrrolidinone with DMDO/CDCl3 under ambient conditions did not lead to a reaction- NMR spectra were identical to those of the starting materials. The same observation was made for N,N-dimethylacetamide. 1-Azabicyclo[3.3.1]nonan-2-one also did not react with DMDO/CDCl3. In contrast, 3,5,7-trimethyl-1-azaadamantan-2-one reacted rapidly with DMDO/CDCl3. Figure 4 displays the 1H-NMR spectrum of the crude solid product obtained from mixing a roughly 2:1 ratio of this lactam to DMDO/CDCl3. The lactam was used in excess to try to minimize over-oxidation as well as to retain some unreacted started material for spectroscopic comparison. The resonances (ppm, after 0.11 ppm offset correction) at 3.00 (4H), 1.79 (2H), 1.65 (1H), 1.55 (3H), 1.07 (3H), and 0.86 (6H) correspond to unreacted Kirby lactam.23–25 The upfield methyl singlets suggest at least two other related reaction products present in significant quantity. The resonances (0.10 ppm offset corrected) at ca 3.6 ppm and 3.8 ppm suggest methylene protons on carbons attached to a N-O functionality or to an O-H or an O-R functionality.
Figure 4.

1H-NMR (CDCl3, offset upfield by 0.10 ppm) of the crude solid product derived from mixing roughly a 2:1 ratio of 3,5,7-trimethyl-1-azaadamantan-2-one, 3 (in CDCl3), with DMDO/CDCl3. Unreacted Kirby lactam is present along with at least two products (16 is tentatively assigned).
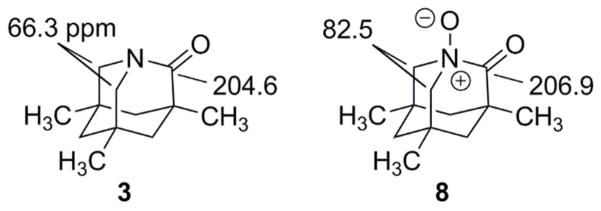
Thin layer chromatography (silica gel) [95% ethanol] led to clean separation of two fractions: Rf 0.50 and Rf 0.75. The Rf 0.50 fraction consisted of essentially pure Kirby lactam (1H- and 13C-NMR spectra). Although arguably less polar than the oxidized compounds in the Rf 0.75 fraction, Kirby lactam has basicity comparable to, if slightly weaker than, an amine. Figure 5.a. is the 13C-NMR spectrum (CDCl3) of the Rf 0.75 fraction. Figure 5.b. shows the expansion of the 10–70 ppm section of this 13C-NMR spectrum. The downfield peaks at 187.7 ppm and 108.8 ppm in Figure 5.a. are noteworthy. The downfield resonance is consistent with a carboxyl or a carboxylate carbon. The resonance at 108.8 ppm is consistent with a gem-diol carbon. The resonance at 52.2 ppm (Figure 5.b.) is consistent with the value reported for the N-CH2 carbons of the amino acid derived from simple hydrolysis of the lactam (see 14 below; 52.5 ppm, D2O-CD3CN) and is consistent with the 187.7 ppm peak (190.0 ppm, D2O-CD3CN).25 The two resonances at 66.4 and 67.4 ppm are consistent with some C-N-O as well as some C-O functionalities. (There are also two smaller resonances in this region). Although Kirby lactam itself has resonances at 65.97 and 52.2 ppm,25 this starting material was cleanly separated from the reaction products by TLC. It is interesting to note the calculated (gas-phase) 13C chemical shifts upon conversion of Kirby lactam to its N-oxide (Scheme 9). The predicted change in downfield shift of the carbonyl carbon (C2) is only 2.3 ppm, while the predicted change for C8,9 is 16.3 ppm, apparently a much more sensitive probe.
Figure 5.


a. 13C-NMR (CDCl3) spectrum of the TLC fraction (Rf 0.75) of the reaction products of Kirby lactam with DMDO in CDCl3. b. Expanded scale of the NMR spectrum of 5.a.
Scheme 9.
Calculated (vacuum) chemical shifts for Kirby lactam and the corresponding N-oxide
How well do the calculations reproduce experiment? Table 2 lists experimental solution (and calculated gas phase) 13C chemical shift values for a series of compounds in this series. It appears that the chemical shifts are well reproduced with calculated C2 values typically 3–7 ppm downfield from the experimental value and C8,9 typically 0–5 ppm downfield from the experimental value.
Table 2.
Comparison of selected experimental (solution) and calculated (gas-phase) 13C chemical shifts for C2 and C8,9 for relevant models compounds. Note that for ease in comparison the numbering scheme has been retained for the amino acid (14) and for the N-hydroxyamino acid (17).
| Molecule | C2 (exp’t) | C2 (calc’d) | C8,9 (exp’t) | C8,9 (calc’d) |
|---|---|---|---|---|
| Kirby Lactam (CDCl3) (3) | 200.0 | 204.6 | 66.0 | 66.3 |
| Kirby Lactam, N-Protonated (10) (CD3CN) | 179.1 | 182.4 | 62.3 | 66.5 |
| Kirby Lactam, N-Methylated (11) | 178.2 | 185.4 | 72.3 | 74.6 |
| Kirby Lactam Acetal (12) (CDCl3) | 105.1 | 109.0 | 58.7 | 60.4 |
| Kirby Lactam, N-Protonated Hydrate (13) (CDCl3) | 107.5 | 111.7 | 57.1 | 62.1, 61.4 |
| 1-Azabicyclo[3.3.1]nonan-2-one (6) | 185.0 | 182.6 | C8: 51.5; C9: 52.8 | C8: 52.0 C9: 53.4 |
| 1-Azabicyclo[3.3.1]nonan-2-one, N-Protonated (6-NH+) | 178.6 | 179.6 | C8 or C9 ca 56.0 or 56.5 |
C8: 54.2 C9: 57.7 |
| Amino Acid (Zwitterion) (14) | 190.0 | 52.5 | ||
| Kirby Lactam N-Oxide (8) | ------- | 206.9 | ------ | 82.5 |
| Kirby Lactam Hydroxylamine Lactone (15) | ------- | 184.2 | ------ | 63.7 |
| Kirby Lactam N-Oxide-2,2-Diol (16) | ------ | 110.7 | ------ | 72.3 |
| Kirby Lactam N-Hydroxyamino Acid (17) | ------ | ------ |
The downfield 13C chemical shift at 108.8 ppm (Figure 5.a.) suggests a gem-diol. At this point, reference is made to the ESI mass spectra in Figures 6.a–c. Figure 6.a. is the ESI/MS (m/z range 160–260) of the Rf 0.75 fraction dissolved in ethanol. Figure 6.b. is the MS/MS (MS2) derived from m/z 228. Figure 6.c. is the MS3 derived from m/z 210. The major pathway suggests loss of water from the O-protonated species (18) derived from 16, followed by loss of carbon monoxide (Scheme 10). Table 3 compares calculated (vacuum) chemical shifts for the N-oxide-2,2-diol 16 with possible assignments from the experimental 13C-NMR and 1H spectra. Scheme 10 depicts the structure calculated for 16. It is important to note that 16 is calculated in the present study to be 15 kcal/mol higher in energy than 17 in vacuum although this value is calculated to be only 5 kcal/mol in dichloromethane, acetone or water. It should be noted that a series of proton transfers to form 17, some of which are energetically-demanding, may also explain why 16 is formed preferentially. While the 1.58 Å N-C2 bond calculated for 16 is quite long and a potential source of instability, intramolecular hydrogen bonding (Figure 7) as well as the aforementioned buttressing by the methyl groups26 provide stability. Although it is tempting to invoke 17 as a product in the mixture, one might have expected an additional carboxyl carbon in the 13C-NMR spectrum. As noted earlier, the carboxylate carbon (187 ppm) is attributed to 14. While an m/z 184 is observed in Figure 6.a., the MS2 of m/z 228 (Figure 6.b.) shows no evidence of m/z 184 corresponding to expected loss of CO2 from 17. Such negative evidence is not, of course, very strong. While N-hydroxylamines are typically five orders of magnitude weaker bases than the corresponding amines,57 simple N-hydroxyamino acids are zwitterionic.58 The product (16) would appear to be kinetically-determined (relative to 17).
Figure 6.

a. The MS/MS (MS2) derived from m/z 228 of the Rf 0.75 fraction dissolved in ethanol. b. The MS3 derived from m/z 210.
Scheme 10.

Proposed ESI fragmentation scheme of the proposed N-oxide-2,2-diol 16.
Table 3.
Calculated 13C- and 1H-NMR chemical shifts (vacuum) with experimental chemical shifts (CDCl3) in the TLC Rf fraction 0.75 tentatively assigned to the N-oxide 2,2-diol 16.
| 13C Chemical Shift (Calculated) | 13C Chemical Shift (Experimental) | 1H Chemical Shift (Calculated) | 1H Chemical Shift (Experimental) | ||
|---|---|---|---|---|---|
| C2: | 110.7 | 108.8 | 8,9-Heq: | 3.25 | 3.3 |
| C8,9: | 72.3 | 67.4 | 8,9-Hax: | 2.54 | 2.6 |
| C6: | 48.0 | 48.6 or 48.3 | 4,10-Heq | 2.23 | 2.35 |
| C4,10: | 45.2 | 45.5 | 6-Heq | 1.33 | (1.3–1.4) |
| C3: | 36.3 | 37.2 | 6-Hax | 1.30 | (1.3–1.4) |
| C5,7: | 31.1 | 31.2 | 4,10-Hax | 1.09 | (1.1–1.15) |
| C5,7-Me | 27.5 | 26.0, 28.7 or 28.9 | 3-CH3 | 1.17 | 1.1 |
| C3-Me | 24.3 | 25.6 or 26.0 | 5,7-CH3 | 0.90 | 0.9 |
Figure 7.

Selected calculated (B3LYP/6-31G*) bond lengths for the 2,2-dihydroxy-N-oxide of Kirby lactam (16), emphasizing the hydrogen bonds that stabilize the ion despite an elongated (1.58 Å) N-C2 bond.
It is worthwhile recalling the behavior of Kirby lactam under conditions of low, medium and high pH as published by Kirby (Scheme 11).25 At pH 3.30, only the N-protonated diol 13 is present. At pH 7.45, only the zwitterionic amino acid 14 is present. At pH 12.5, only the anionic 18 is observable. However, at pH 4.28, both 13 and 14 are observed separately at room temperature. Warming the solution at this pH causes coalescence at 60–63 °C (Rate = 280 s−1). In order to exchange 13 and 14, loss or gain of a proton is required.
Scheme 11.

Structures of species derived from 1-azaadamantan-2-one in aqueous media at different pH published by Kirby and co-workers.25
The N-oxide-2,2-diol 16 is a neutral analogue of 13, the N-protonated hydrate of Kirby lactam observed at low pH (3.30). The neutral N-oxide is stabilized by intramolecular hydrogen bonding. Unlike most carbonyl hydrates, 13 has been isolated in pure form,25 so it is not unreasonable that 16 can also be isolated.
There remain considerable uncertainties in other assignments in the TLC Rf 0.75 fraction. One may consider rearrangement of an initially-formed N-oxide to the hydroxylamine lactone 15. Concerted rearrangements of hypothetical acyclic amide N-oxides to such esters are calculated to be both exothermic and extremely rapid.37,59 However, the geometry for concerted rearrangement requires approach of the oxy anion via a trajectory essentially perpendicular to the plane of the carbonyl group. This is unlikely for 8, which is calculated to have a very high (>35 kcal/mol) barrier to this rearrangement.59 Alternatively, one might imagine N-C2 cleavage in 8 followed by rapid formation of 15. However, the IR spectrum of the Rf 0.75 TLC fraction (Supporting Information, S2.b.) shows no evidence of a carbonyl group when compared to the dry Kirby lactam (Supporting Information, S2.a). The possibility of N-C2 cleavage followed by immediate loss of CO (analogous to Scheme 7) suggests other possible products including 19 and 20. The IR spectrum is consistent with C-O stretching frequencies and the m/z 166 (loss of CO from the m/z 194 ion) in the ESI/MS of the Rf 0.75 fraction could be consistent with N-protonated 19 (see also Table 2). The sharp band at 729 cm−1 and broad bands between 1600 and 1700 cm−1 (Supporting Information, S2.b) might even suggest the possibility of nitrite60 species such as 21 or 22. One might imagine formation of the nitroso compound 23 by analogy to rearrangement of similarly-substituted species61,62 not as sterically-constrained as 21. However, the downfield chemical shifts calculated for a nitroso carbon (ca 90–110 ppm) are absent and there is no evidence for an oxime rearrangement product in the 13C-NMR spectrum of the Rf 0.75 TLC fraction. To date there is insufficient information for definitive assignments of all of the products of reaction between Kirby lactam and DMDO. Low temperature NMR analysis was performed, however, the results were not definitive. The results are included in the supporting information (S1)
Assuming that a major product of the reaction between Kirby lactam and DMDO, in the presence of a trace of water, is the 2,2-dihydroxy-N-oxide 16, a key question is the pathway. Does it proceed via oxidation by DMDO to the sought-after N-oxide 8 followed by hydration to 16, or does hydration to 25 precede oxidation (see Scheme 13). The calculated free energies suggest that initial oxidation to 8 is favored. This is intuitively reasonable because the Kirby lactam nitrogen is a strong nucleophile and DMDO is a strong electrophile. A desired comparison of activation barriers is limited at this level of calculation. Combination of a single molecule of water with Kirby lactam provides a complex in which the water molecule hydrogen bonds simultaneously with the lactam’s nitrogen and oxygen atoms. The problem is one in which solvation and dynamics should furnish a more reasonable approach. It is interesting that the calculated energy of activation and scaled free energy of activation for attack of DMDO on hydrate 25 (0.5 kcal/mol and 13.7 kcal/mol) are much lower than the corresponding activation barriers for attack of DMDO on Kirby lactam itself. This reflects the incipient hydrogen bonding present in the product 16. It is well to remember that in aqueous solution, the product of reaction of water with Kirby lactam is the amino acid (14) rather than 25. These are different solution conditions than a trace of water in CDCl3 solution.
Scheme 13.

Comparison of two alternative pathways to form the 2,2-hydroxy-N-oxide of Kirby lactam: Oxidation followed by hydration or hydration followed by oxidation. Calculated relative total energies (kcal/mol) are in parentheses and calculated relative free energies are in brackets.
Conclusions
The three lactams investigated in this study have ca 20 kcal/mol, ca 10 kcal/mol and 0 kcal/mol of stabilization (resonance) energy respectively. The first, N-ethyl-2-pyrrolidinone, protonates virtually exclusively on oxygen; the third, 3,5,7-trimethyl-1-azaadamantan-2-one (Kirby lactam) protonates virtually exclusively on nitrogen. The second, 1-azabicyclo[3.3.1]nonan-2-one, forms both N- and O-protonated species in equilibrium. Despite these major differences, the ESI/MS for all three indicate that fragmentation originates from the N-protonated tautomer.
N-ethyl-2-pyrrolidinone, N,N-dimethylacetamide, and 1-azabicyclo[3.3.1]nonan-2-one do not react with dimethyldioxirane (DMDO) at ca 20–25 °C. Kirby lactam (3,5,7-trimethyl-1-azaadamantan-2-one) does react rapidly with dimethyldioxirane. In the presence of a trace of water, there is some hydrolysis of the lactam to its amino acid. The lactam also forms at least two products involving oxidation at the bridgehead nitrogen. On the basis of 13C- and 1H-NMR as well as ESI/MS, one product is identified as the N-oxide-2,2-diol (16), possibly formed by transient formation of the N-oxide, followed by addition of water to form the gem-diol. This interesting structure is an analogue to the structure of the N-protonated gem-diol of the Kirby lactam that is the exclusive species observed by Kirby in aqueous solution at pH 3.30. Although the N-C2 bond in this molecule is calculated to be quite long (1.58 Å), intramolecular hydrogen bonding and steric buttressing by the methyl substituents should stabilize this structure. Although other oxidation products are clearly present, they are not separated and it is difficult to assign structures on the basis of limited spectroscopic data.
It is possible that the postulated unstable N-oxide of Kirby lactam was observed by NMR at −50 °C and this would be consistent with initial formation of 8 followed by addition of water to form 16. However, it is clear that even in this most favored case (i.e. 8), amide N-oxides are exceedingly unstable and readily hydrate in the presence of a trace of water and may also rapidly lose CO. While, constitutionally speaking, the N-oxide of Kirby lactam (8) is formally an “amide N-oxide”, it is in reality a tertiary alpha-ketoamine oxide. Future studies will include more strictly anhydrous conditions and use of the more reactive methyltrifluoromethyldioxirane in place of DMDO in order to probe the potential reactivity of the 1-azabicyclo[3.3.1]nonan-2-one, calculated to have half the stabilization energy of an unstrained amide or lactam.
Experimental
N-Ethyl-2-pyrrolidone and N,N-dimethylacetamide (Sigma-Aldrich) were used as supplied. 1-Azabicyclo[3.3.1]nonan-2-one43–47 was synthesized according to published procedure43 and the spectroscopic properties of the neutral and protonated compounds matched those reported.32 3,5,7-Trimethyl-1-azaadamantan-2-one was synthesized according to published procedure23–25 and its 1H- and 13C-NMR as well as its IR spectra were identical to those of the published compound. Dimethyldioxirane (DMDO) was prepared using the method of Murray and Singh48 and was extracted into CDCl3 using a published procedure.49 Addition of DMDO in CDCl3 to lactam in CDCl3 was performed at ambient temperatures (20–25 °C). Prior to exposure to DMDO, the Kirby lactam/CDCl3 solution was dried using anhydrous sodium sulfate. The studies were performed using a 0.5 molar ratio of the lactam to DMDO (regardless of solvent) and yielded a mixture. 1H NMR (400 MHz, CDCl3, see Figure 4, assignments are tentative) – δ: 3.69-3.63 (dm, 1H, J=12.5 Hz, unassigned), 3.27-3.20 (m, 2H, 16), 3.03-2.99 (dm, 1H, J = 9.2 Hz, unassigned), 2.94, 2.98 (AB(ABXY), 4H, JAB = 13.4 Hz, JAY = 2.18 Hz, JBX= 4.21 Hz, A does not couple to X and B does not couple to Y, 3), 2.70-2.59 (m, 3H), 2.55-2.49 (dm, 1.5H, J = 13.6 Hz, 16), 2.28-2.24 (dm, 1H, J = 8.63 Hz, 16), 2.0-1.93 (dm, 1.5H, J = 13.0 Hz, unassigned), 1.77-1.72 (dm (X), J = 12.5 Hz, 2H, 3), 1.62 (dt (Y), J = 12.5, 2.21 Hz, 1H, 3), 1.51–1.57 (dm, J = 12.85 Hz, 3H, 3), 1.34-1.10 (m, 5H, unassigned), 1.06-0.91 [m including: 1.03 (s, 3H, 3), 0.97 (s, 3H, 16)], 0.89-0.73 [m, including: 0.86 (s), 0.84 (s, 6H, 3), 0.83 (s), 0.81 (s), 0.77 (s, 5H, 16)]. 13C NMR (101 MHz, CDCl3) δ 207.26, 200.35, 187.88, 109.03, 98.39, 67.60, 66.57, 66.20, 57.73, 57.36, 52.43, 48.84, 48.56, 46.97, 46.86, 46.44, 45.75, 44.64, 44.32, 43.76, 37.42, 35.16, 34.88, 31.38, 31.17, 30.72, 29.96, 29.88, 29.10, 28.87, 26.18, 25.77, 25.47, 23.45, 22.12. Product after column performed: 13C NMR (101 MHz, CDCl3) δ 187.68, 108.81, 67.39, 66.36, 57.51, 57.14, 52.22, 48.62, 48.34, 48.02, 46.75, 46.63, 45.81, 45.53, 44.43, 44.11, 37.20, 34.95, 34.66, 31.15, 29.74, 29.66, 28.88, 28.65, 25.96, 25.56, 25.25, 21.90.
A low-temperature reaction was performed by adding a DMDO/CDCl3 solution, stored overnight at −20 °C, and cooled in a dry ice/acetone bath just prior to use, to a pre-cooled (dry ice/acetone) solution of Kirby lactam in CDCl3 in an NMR tube. The solution was immediately mixed and inserted into the NMR probe, pre-cooled to −50 °C. 13C NMR (126 MHz, CDCl3) δ 181.41, 176.01, 106.89, 94.73, 87.41, 67.05, 66.31, 64.98, 64.52, 64.23, 59.75, 58.51, 56.32, 53.74, 52.19, 47.86, 47.42, 45.63, 45.24, 45.13, 44.99, 44.83, 43.22, 43.18, 42.71, 42.64, 37.88, 35.52, 34.72, 34.26, 33.53, 33.51, 32.95, 32.52, 31.97, 31.40, 31.38, 30.87, 30.49, 30.11, 29.80, 29.43, 28.66, 28.54, 28.32, 27.93, 27.87, 25.91, 22.96, 22.81, 18.36, 15.37, 14.38, 14.23.
Electrospray ionization mass spectrometry experiments (ESI-MS) were performed on a ThermoFisher (San Jose, CA, USA) LTQ mass spectrometer equipped with a TriVersa Nanomate (Advion, Ithaca, NY, USA) nanoelectrospray source. For electrospray ionization mass spectrometry experiments, samples were dissolved in ethanol/isopropanol for direct infusion. All interrogated ions were proton adducts [M+H]+. For all direct infusion experiments, signal was averaged over multiple scans to achieve suitable spectrum quality. In general, spray voltages on the Nanomate were typically set to 1.4–1.8 kV with nitrogen gas pressure set to 0.3–0.4 psi; this was generally sufficient to generate a spray current between 10–200 nA. For the LTQ, capillary temperature was set to 180 °C. Capillary voltage and tube lens voltage were set to 49 V and 125 V, respectively, and were tuned to maximize intensity of the precursor ion while minimizing in-source fragmentation. For fragmentation experiments, activation Q was set to 0.250 and activation time was 30 ms., which are default values. Normalized collision energy was set to 35%. Isolation widths were typically set to 2 m/z to capture complete precursor isotopic envelopes.
Supplementary Material
Scheme 1.

Scheme 2.

Scheme 12.

Acknowledgments
The authors gratefully acknowledge the contributions of Logan Rawlins and Monique Pichon, Xavier University of Louisiana, in the synthesis of the Kirby lactam, Brian Sliter, University of New Hampshire, for synthesis of 1-azabicyclo[3.3.1]nonan-2-one, Dr. Patricia Stone-Wilkinson, University of New Hampshire, for assistance with low-temperature NMR studies, and Dr. Vernon Reinhold, University of New Hampshire, for access to the Glycomics Center ESI/MS facility. Professor Kathleen M. Morgan gratefully acknowledges Awards SC3 GM084738 and R15 GM59037-01A1 from the National Institute of General Medical Sciences.
Footnotes
Select NMR, IR and MS spectra, images of McLafferty rearrangement calculated transition states, and Cartesian coordinates/calculated energies for all ground state and transition state molecules are located in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Pauling L. The Nature of the Chemical Bond. Cornell University Press; Ithaca: 1939. [Google Scholar]
- 2.Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials science. J. Wiley & Sons; New York: 2003. [Google Scholar]
- 3.Wiberg KB. In: The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials science. Greenberg A, Breneman CM, Liebman JF, editors. J. Wiley & Sons; New York: 2003. p. 33. [Google Scholar]
- 4.Mucsi Z, Tsai A, Szori M, Chass GA, Viscolcz B, Csizmadia IG. J Phys Chem A. 2007;111:13245. doi: 10.1021/jp0759325. [DOI] [PubMed] [Google Scholar]
- 5.Glover SA, Rosser AA. J Org Chem. 2012;77:5492. doi: 10.1021/jo300347k. [DOI] [PubMed] [Google Scholar]
- 6.Szostak M, Aubé J. Chem Rev. 2013 doi: 10.1021/cr4000144. full/10.1021/cr4000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birchall T, Gillespie RJ. Can J Chem. 1963;41:2642. [Google Scholar]
- 8.Olah GA, White AM. Chem Rev. 1970;70:561. [Google Scholar]
- 9.Klumpp DA, Rendy R, Zhang Y, Gomez A, McElrea A. Org Lett. 2004;6:1789. doi: 10.1021/ol049512z. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg A, Venanzi CA. J Am Chem Soc. 1993;115:6951. [Google Scholar]
- 11.Greenberg A, Moore DT, DuBois TD. J Am Chem Soc. 1996;118:8658. [Google Scholar]
- 12.Werstiuk NH, Brown RS, Wang Q. Can J Chem. 1996;74:524. [Google Scholar]
- 13.Mujika JI, Mercero JM, Lopez X. J Phys Chem A. 2003;107:6099. [Google Scholar]
- 14.Mujika JI, Mercero JM, Lopez X. J Am Chem Soc. 2005;127:4445. doi: 10.1021/ja044873v. [DOI] [PubMed] [Google Scholar]
- 15.Perrin CL. Acc Chem Res. 1989;22:268. [Google Scholar]
- 16.Pracejus H. Chem Ber. 1959;92:988. [Google Scholar]
- 17.Pracejus H, Kehlen M, Kehlen H, Matschiner H. Tetrahedron. 1965;21:2257. [Google Scholar]
- 18.Blackburn GM, Skaife CJ, Kay IT. J Chem Res, Synop. 1980:294. [Google Scholar]
- 19.Somayaji V, Brown RS. J Org Chem. 1986;51:2676. [Google Scholar]
- 20.Brown RS, Bennet AJ, Slebocka-Tilk H. Acc Chem Res. 1992;25:481. [Google Scholar]
- 21.Tani K, Stoltz BM. Nature. 2006;441:731. doi: 10.1038/nature04842. [DOI] [PubMed] [Google Scholar]
- 22.Ly T, Krout M, Pham DK, Tani K, Stoltz BM, Julian RR. J Am Chem Soc. 2007;129:1864. doi: 10.1021/ja067703m. [DOI] [PubMed] [Google Scholar]
- 23.Kirby AJ, Komarov IV, Wothers PD, Feeder N. Angew Chem Int Ed. 1998;37:785. doi: 10.1002/(SICI)1521-3773(19980403)37:6<785::AID-ANIE785>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 24.Kirby AJ, Komarov IV, Feeder N. J Am Chem Soc. 1998;120:7101. [Google Scholar]
- 25.Kirby AJ, Komarov IV, Feeder N. J Chem Soc Perkin Trans 2. 2001:522. [Google Scholar]
- 26.Morgan KM, Rawlins ML, Montgomery MN. J Phys Org Chem. 2005;18:310. [Google Scholar]
- 27.Doering WE, Chanley JD. J Am Chem Soc. 1946;68:586. doi: 10.1021/ja01208a017. reference 12. We thank an anonymous reviewer of this paper for directing our attention to this reference. [DOI] [PubMed] [Google Scholar]
- 28.Woodward RB, Neuberger A, Trenner NR. In: The Chemistry of Penicillin. Clarke HT, Johnson JR, Robinson R, editors. Princeton University Press; Princeton, NJ: 1949. pp. 415–439. [Google Scholar]
- 29.Crowfoot D, Bunn CW, Rogers-Low BW, Turner-Jones A. In: The Chemistry of Penicillin. Clarke HT, Johnson JR, Robinson R, editors. Princeton University Press; Princeton, NJ: 1949. pp. 310–367. [Google Scholar]
- 30.Dunitz JD, Winkler RK. Acta Crystallogr, Sect B. 1975;31:251. [Google Scholar]
- 31.Werstiuk NH, Muchall HM, Roy CD, Ma J, Brown RS. Can J Chem. 1998;76:672. [Google Scholar]
- 32.Sliter B, Morgan J, Greenberg A. J Org Chem. 2011;76:2770. doi: 10.1021/jo200195a. [DOI] [PubMed] [Google Scholar]
- 33.Morgan J, Greenberg A. J Phys Org Chem. 2012;25:1422. [Google Scholar]
- 34.Lias SG, Bartmess JE, Liebman JF, Holmes JL, Levin RD, Mallard WG. Gas- Phase Ion and Neutral Thermochemistry. J Phys Chem Ref Data. 1988;17:321. Supplement No 1. [Google Scholar]
- 35.Coşkun N, Çetin M. Tetrahedron Lett. 2004;45:8973. [Google Scholar]
- 36.Annese C, D’Accolti L, De Zotti M, Fusco C, Toniolo C, Williard PG, Curci R. J Org Chem. 2010;75:4812. doi: 10.1021/jo100855h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg A, DuBois TD. J Molec Struct. 2001;567–568:303. [Google Scholar]
- 38.Lin H-Y, Ridge DP, Uggerud E, Vulpius T. J Am Chem Soc. 1994;116:2996. [Google Scholar]
- 39.Crotti AEM, Fonseca T, Hong H, Staunton J, Galembeck SE, Lopes NP, Gates PJ. Int J Mass Spectrom. 2004;232:271. [Google Scholar]
- 40.Vessecchi R, Galembeck SE. J Phys Chem A. 2008;112:4060. doi: 10.1021/jp800427q. [DOI] [PubMed] [Google Scholar]
- 41.Tu Y-P. Rapid Commun Mass Spectrom. 2004;18:1345. [Google Scholar]
- 42.Divito EB, Davic AP, Johnson ME, Cascio M. Anal Chem. 2012;84:2388. doi: 10.1021/ac203158u. [DOI] [PubMed] [Google Scholar]
- 43.Hall HK, Jr, Shaw RG, Deutschmann A. J Org Chem. 1980;45:3722. [Google Scholar]
- 44.Hall HK, Jr, El-Shekeil A. Chem Rev. 1983;83:549. [Google Scholar]
- 45.Buchanan GL. J Chem Soc, Chem Commun. 1981:814. [Google Scholar]
- 46.Steliou K, Poupart MA. J Am Chem Soc. 1983;105:7130. [Google Scholar]
- 47.Brehm R, Ohnhäuser D, Gerlach H. Helv Chim Acta. 1987;70:1981. [Google Scholar]
- 48.Murray RW, Singh M. Organic Syntheses. 1997;74:91. [Google Scholar]
- 49.Adam W, Zhao C, Saha-Möller CR, Jakka K. Oxidation of Organic Compounds by Dioxiranes. John Wiley & Sons; New York: 2009. p. 35. [Google Scholar]
- 50.Spartan 10, 1.2.0. Wavefunction, Inc; Irvine, CA: 2008. For a description of the improved calculation of 13C-NMR chemical shifts in Spartan 10 see: Spartan ’10 for Windows, Macintosh and Linux- Tutorial and User’s Guide. Wavefunction, Inc; Irvine, CA: 2011. pp. 480–487.
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian, Inc; Wallingford CT: 2010. See also: Foresman JB, Frisch A. Exploring Chemistry with Electronic Structure Methods. 2. Gaussian, Inc; Pittsburgh, PA: 1996.
- 52.CYLview, 1.0b. Legault, C. Y: Université de Sherbrooke; 2009. ( http://www.cylview.org) [Google Scholar]
- 53.Headley JV, Peru KM, Friesen DA, Neu T. J Chromatogr A. 2001;917:159. doi: 10.1016/s0021-9673(01)00687-2. [DOI] [PubMed] [Google Scholar]
- 54.Prakash GKS, Rasul G, Liang G, Olah GA. J Phys Chem. 1996;100:15805. [Google Scholar]
- 55.Gabelica V, De Pauw E. Mass Spectrom Rev. 2005;24:566. doi: 10.1002/mas.20027. [DOI] [PubMed] [Google Scholar]
- 56.a Chandrasekhar J, Smith SF, Jorgensen WL. J Am Chem Soc. 1985;107:154. [Google Scholar]; b Jorgensen WL. Acc Chem Res. 1989;22:184. [Google Scholar]
- 57.Bartmess JE. In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Part 1. Rappoport Z, Liebman JF, editors. Vol. 2. John Wiley & Sons; West Essex, UK: 2011. pp. 115–143. [Google Scholar]
- 58.Buehler E, Brown GB. J Org Chem. 1967;32:265. [Google Scholar]
- 59.Paroline H, Morgan JP, Greenberg A. Unpublished results [Google Scholar]
- 60.Nakanishi K, Solomon PH. Infrared Absorption Spectroscopy. 2. Holden-Day, Inc; San Francisco: 1977. pp. 45–46. [Google Scholar]
- 61.Bailey WF, Bobbitt JM, Wiberg KB. J Org Chem. 2007;72:4504. doi: 10.1021/jo0704614. [DOI] [PubMed] [Google Scholar]
- 62.Canterbury DP, Frontier AJ, Um JM, Cheong PHY, Goldfeld DA, Huhn RA, Houk KN. Org Lett. 2008;10:4597. doi: 10.1021/ol8019154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murray RW, Jeyaraman R, Pilley MK. J Org Chem. 1987;52:746. [Google Scholar]
- 64.Adam W, Chan YY, Cremer D, Gauss J, Scheutzuw D, Schindler M. J Org Chem. 1987;52:2800. [Google Scholar]
- 65.Gibert M, Ferrer M, Sánchez-Baeza F, Messeguer A. Tetrahedron. 1997;53:8643. [Google Scholar]
- 66.Dong Y, Vennerstrom JL. J Heterocycl Chem. 2001;38:463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.