

Abstract
MicroRNAs play an essential role in diverse cellular processes, such as proliferation, differentiation, apoptosis, and stress response. Recent studies demonstrate that miRNAs are important for timing developmental decisions and fine-tuning cellular determination in vertebrate heart development. In an elegant set of experiments reported in this issue of Genes & Development, Liu et al. (3242–3254) demonstrate that miR-133a functions as an inhibitor of cardiomyocyte proliferation and a modifier of serum response factor (SRF)-dependent transcriptional signaling in the murine heart. Both targeted deletion and transgenic overexpression of miR-133a can result in the same cardiac phenotype, ventricular septal defect (VSD) and heart failure. The new data add another piece to the puzzle of regulatory networks that are implicated in cardiac disease. It will be interesting to see, if miR-133a is also involved in human heart diseases, especially VSD and dilated cardiomyopathy.
Keywords: microRNA, heart development, serum response factor, myocyte enhancer factor-2, cyclin D2
MicroRNAs (miRNAs) were first discovered in Caenorhabditis elegans by Lee et al. (1993) and represent a novel class of regulatory modules that enable cells to fine-tune even the most complex gene expression cascades. Encoded by distinct intronic or intergenic genomic regions, the transcribed noncoding RNAs, so-called pri-miRNAs are processed to short, 70- to 100-nucleotide (nt)-long stem–loop forming precursors called pre-miRNAs and finally to the mature miRNAs. In this canonical maturation process, several important proteins are involved. First, the “microprocessor complex” consisting of the nuclear ribonuclease III Drosha and dsRNA-binding protein DGCR8 (DiGeorge syndrome critical region gene 8; syn. Pasha) converts pri-miRNAs into pre-miRNAs and then the mature miRNAs are formed by the endonuclease Dicer to finally participate in the cytoplasmatic RNA-induced silencing complex (RISC) (Bernstein et al. 2001; Denli et al. 2004). Mature miRNAs are ∼18–26 nt in length and inhibit either protein translation from mRNA transcripts through imperfect base pairing with their 3′-untranslated region (UTR) or by inducing mRNA degradation (Bartel 2004; Rodriguez et al. 2004; Valencia-Sanchez et al. 2006). So far, >700 human miRNAs have been catalogued in the miRBase online database (http://microrna.sanger.ac.uk; Griffiths-Jones et al. 2008). Based on sequence analysis, miRNAs are predicted to target up to 30% of all mRNA transcripts encoded in our genome (Berezikov et al. 2005; Li et al. 2006).
Today, the overall biological importance of miRNAs is well demonstrated in a wide range of cellular processes, such as proliferation, differentiation, apoptosis, stress response, oncogenesis, and tumor suppression (Xu et al. 2003; Calin et al. 2005, 2006; Cimmino et al. 2005; Dresios et al. 2005; Zhao et al. 2005). Several studies denote miRNAs as particularly important for fine-tuning cellular transcriptomes by eliminating fluctuations in mRNA levels and removal of inappropriate transcripts (Stark et al. 2005; Lee et al. 2007). Furthermore, complexity is added to miRNA-regulated networks by the fact that, on the one hand, a specific miRNA can potentially target dozens of mRNAs, and on the other hand, a specific mRNA can be targeted by different miRNAs.
miRNAs in cardiac development and function
The heart is one of the first organs to function in the developing embryo. During heart development, a series of molecular events finally results in an organ of a multifaceted cellular composition with complex morphological and functional units. During the last decade, several key elements of the transcriptional network that orchestrates vertebrate heart development were successfully identified, although little is known so far about the role of post-transcriptional control mechanisms in this process. However, in recent studies, several miRNAs were identified as new regulators and modifiers, not only of cardiac development, but also of adaptive or maladaptive responses of the adult heart, such as cardiomyocyte growth.
A series of elgant studies by the group of Eric N. Olson (van Rooij et al. 2007) has helped to unravel several miRNA functions in the vertebrate heart. To date, a number of miRNAs have been found to be expressed in a muscle-specific pattern; however, only miR-208, which is encoded within intron 27 of the murine α-myosin heavy-chain gene (α-MHC), is specifically expressed in the heart. miR-208-deficient mice are viable and without obvious cardiac abnormalities directly after birth (van Rooij et al. 2007). Shortly afterward, miR-208-deficient mice develop progressive heart failure accompanied by the up-regulation of fast skeletal muscle and stress responsive genes. Interestingly, when miR-208-deficient mice are exposed to an increased cardiac afterload by transverse aortic constriction (TAC) virtually no cardiomyocyte hypertrophy or myocardial fibrosis can be observed. One mRNA target of miR-208 predicted by computational analysis and successfully validated by Luciferase reporter assays is the Thyroid Hormone Receptor-Associated Protein-1 (THRAP1). As predicted, THRAP1 expression is significantly increased in miR-208-deficient mice, which enhances the activity of the Thyroid Receptor-Associated Protein complex (TRAP) to suppress β-MHC expression regulated by negative thyroid response elements (TRE). Hence, this elegant study by van Rooij et al. (2007) from the Olson laboratory demonstrates that miR-208 is a cardiac-specific regulator of β-MHC expression in response to stress and thyroid signaling and highlights for the first time the importance of miRNAs especially in the context of myocardial hypertrophy.
Some miRNAs can be found in a bicistronic position in the mammalian genome and hence are transcribed together. miR-1 and miR-133a are even encoded by duplicated bicistronic genetic loci (miR-1-1/miR133a-2 and miR-1-2/miR133a-1) with identical sequences of the mature miRNAs. Furthermore, another bicistronic cluster encoding the nearly identical miR-206/miR-133b is expressed in skeletal muscle tissue, whereas miR-1 and miR-133a are expressed in both heart and skeletal muscle cells. The transcription of miR-1 and miR-133a is regulated by critical myocyte differentiation factors including serum response factor (SRF), Myocardin, Myogenic Differentiation-1 (MyoD) and Myocyte Enhancer Factor-2 (Mef2) (Zhao et al. 2005).
Overexpression of miR-1 in the developing heart decreases the pool of proliferating cardiomyocytes by directly targeting Hand2 (Heart and Neural Crest Derivatives Expressed-2), a helix–loop–helix transcription factor that normally promotes ventricular cardiomyocyte expansion. As a result, miR-1 transgenic mice have thin-walled hearts and develop heart failure. Overexpression of miR-1 in cultured cardiomyocytes revealed Ras GTPase-activating protein (RasGAP), Cyclin-dependent kinase 9 (Cdk9), and other growth-related factors as additional targets of miR-1 (Sayed et al. 2007).
Targeted deletion of miR-1-2 unmasked further functions of this particular miRNA in the mammalian heart. Loss of miR-1-2 results in ventricular septal defects (VSDs) and the death of approximately half of the animals immediately after birth. Surviving littermates show diverse cardiac phenotypes. Hearts of animals that reach adulthood, are either dilated and go into failure, show increased numbers of cardiomyocytes (cardiac hyperplasia) and increased weights, or electrophysiological abnormalities with lower heart rates, shortened PR and prolonged QRS intervals and a high incidence of sudden cardiac death (Zhao et al. 2007).
Interestingly, both miR-1 and miR-133a were found to be down-regulated in ventricular tissue of mice after TAC or phenylephrine treatment as well as in heart tissue of humans suffering from hypertrophic cardiomyopathy. Accordingly, overexpression of miR-133a in cultured cardiomyocytes blunts hypertrophic responses to agonist treatment (Care et al. 2007). To further examine miR-133a function in vivo, Care et al. (2007) infused mice with cholesterol-conjugated ssRNA antisense oligonucleotides (antagomirs) targeting miR-133a. The antagomir-treated animals develop hypertrophic cardiomyopathy, whereas injection of control antagomirs did not lead to a cardiac phenotype. Targets of miR-133a that were identified and can potentially explain the hypertrophic phenotype are Ras Homolog Gene Family Member A (RhoA) and Cell Division Cycle 42 (Cdc42).
Although the existing studies revealed many important roles for miR-1 and miR-133a in heart development and function, until now a lack of genetic deletion models for miR-133a prevented an even deeper understanding of the function of the miR-1/miR-133a clusters in the mammalian heart.
microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart
Liu et al. (2008) now add one more piece to the puzzle of the miR-1/miR-133a cluster. In this issue of Genes & Development, the investigators extensively characterize genetically engineered mice deficient for either miR-133a-1 or miR-133a-2, or both, as well as mice overexpressing miR-133a with surprising results.
While miR-133a-1 and miR-133a-2 seem to have redundant functions and do not cause obvious cardiac abnormalities when deleted individually, targeted deletion of both miRNAs results in cardiac malformations and embryonic or postnatal lethality. MiR-133a double knockout mice die from two distinct phenotypes: (1) animals that die shortly after birth and exhibit hearts with large VSDs, dilated right ventricles, and atria; and (2) mice that survive to adulthood have no VSDs but develop dilated cardiomyopathy (DCM), cardiac fibrosis, and heart failure. The knockout strategy used for miR-133a deletion affects neither expression of “Mind Bomb-1” (Mib-1), which intron hosts the miR-1-2/miR-133a-1 gene cluster, nor miR-1-1 or miR-1-2 levels, respectively. This result is important, because as outlined above, decreased expression of miR-1-1 or miR-1-2 by itself would result in a cardiac phenotype similar to that observed in miR-133a-deficient mice. Surprisingly, miR-133a deficiency does not lead to hypertrophic cardiomyopathy, as one would have expected from previous studies, where miR-133a-antagomir treatment induced cardiac hypertrophy in mice (Care et al. 2007). These significant differences in the cardiac phenotypes of miR-133a-antagomir treated and genetically miR-133a depleted mice might be explained either by the fact that antagomir treatment is transient and usually does not lead to complete loss of the targeted miRNA, or by the fact that the antagomir may target rather nonspecifically other miRNAs, thereby provoking a different phenotype.
Histological and immunohistochemical analysis of miRNA-133a-deficient hearts revealed enhanced cardiomyocyte proliferation throughout ventricles and atria in addition to increased apoptosis and fibrosis near the basal and apical interventricular septum, findings potentially explaining the development of lethal VSDs in the miR-133a knockouts (Hoffman et al. 2004). Echocardiographic and histological analyses of surviving animals without VSDs reveal dilated hearts with thinned ventricular walls, severely reduced contractile function, intraventricular thrombi formation, and marked myocardial fibrosis. These surprising findings immediately raise questions concerning the molecular mechanisms by which miR-133a regulates proliferation and apoptosis of cardiomyocytes and why deficient animals either develop hearts with large VSDs or DCM. To answer this question, Liu et al. (2008) analyzed the cardiac transcriptome of miR-133a-deficient hearts and identified a variety of dysregulated transcripts. Several genes involved in cardiomyocyte cell cycle control, such as Cyclin D1 (CCND1), Cyclin D2 (CCND2), and Cyclin B1 (CCNB1) were found to be significantly up-regulated in miR-133a-deficient hearts. Interestingly, Cyclin D2 carries a miR-133a seed sequence in its 3′-UTR and is directly targeted by miR-133a as confirmed by Luciferase reporter assays. Surprisingly, Rho-A and Caspase-9 were found to be not regulated on either the transcriptional and protein level in miR-133a-deficient hearts. These two miR-133a targets were identified previously in cell culture experiments (Xu et al. 2007). These controversial findings could be explained by differences in the models used (cardiomyocyte culture vs. knockout animals).
In a further attempt to dissect the effects of miR-133a on cardiomyocyte proliferation, Liu et al. (2008) overexpressed miR-133a under the control of the cardiac β-MHC promoter. Astoundingly, transgenic animals died by embryonic day 15.5 (E15.5) due to VSDs and diminished cardiomyocyte proliferation resulting in ventricular walls only consisting of two to three cell layers, unable to fulfil the hemodynamic needs of the developing mouse. Ventricular thinning is most likely due to reduced cardiomyocyte proliferation, since TUNEL staining did not reveal increased cardiomyocyte apoptosis in miR-133a transgenic animals.
In addition to the dysregulation of growth and cell cycle-related factors, smooth muscle genes, such as smooth muscle α-Actin, Transgelins, Calponin I, and Caldesmon are also up-regulated in miR-133a-deficient hearts. Interestingly, the myogenic transcription factor SRF, which binds to CArG boxes in the regulatory region of a variety of important muscle-specific growth and differentiation factors, is also negatively regulated by miR-133a and hence increased in miR-133a-deficient hearts, thereby potentially explaining the elevated levels of smooth muscle gene transcripts (Norman et al. 1988; Wang et al. 2004). Together, the ectopic expression of smooth muscle genes and loss of inhibition of the miR-133a targets SRF and Cyclin D2 could cause aberrant cardiomyocyte proliferation, apoptosis, and development of DCM (Zhang et al. 2001).
MiR-133a—a novel candidate gene for human heart disease
The main cardiac phenotypes provoked by changes in miR-133a expression resemble those found in two important cardiovascular diseases in humans; namely, congenital muscular VSD and DCM. Muscular VSDs occur in up to 5% of live births, often associated with other malformations of the cardiovascular system (Roguin et al. 1995). Although VSDs represent a common disease, the genetic programs that control ventricular septation are only poorly understood and only few genes including the transcription factors T-box 5 (TBX5) and NKX2.5 are shown to be causative for isolated VSDs (Basson et al. 1994; Benson et al. 1999). DCM is a heart disease characterized by dilation of the left ventricle and impaired systolic function (Richardson et al. 1996). The annual incidence of DCM is five to eight in 100,000, which contributes to a significant amount of morbidity, mortality, and healthcare costs in western countries (Gillum 1994). Although a large number of genes were identified as causes of familial or sporadic DCM and several molecular mechanisms leading to impairment of contractile function have been elucidated (Rottbauer et al. 2005; Bendig et al. 2006; Karkkainen and Peuhkurinen 2007), there are also many familial cases in which a distinct mutation within known disease genes could not be identified. Based on the results of Liu et al. (2008), it will be interesting to evaluate the role of miR-133a in human congenital VSD and DCM.
Perspective
In summary, the study by Liu et al. (2008) implicates again a central role of miRNAs in the mammalian heart (Fig. 1). Therapeutic manipulation of miRNAs in cardiac disease thus appears compelling, but seems to be complicated in the light of the data discussed here. Further deciphering of the complex regulatory networks modulated by miRNAs in the healthy and diseased heart will possibly guide future molecular drug development strategies. Based on antisense approaches, synthetic anti-miR oligonucleotides (AMOs), miR sponges and cholesterol-conjugated ssRNA analogs (antagomirs) are examples of promising strategies to specifically knock down specific miRNAs in vivo (Krutzfeldt et al. 2005; Weiler et al. 2006; Care et al. 2007). Different modes of delivery for miRNA interfering molecules already have been evaluated. Lentiviral-mediated antisense expression or intravenous application of AMOs or antagomirs seem promising strategies (Krutzfeldt et al. 2005; Scherr et al. 2007). The cholesterol conjugation of antagomirs permits efficient entry into the cell without toxic side effects and was used recently for blocking miR-133a in the heart of living mice (Care et al. 2007). In addition, in a recent in vivo study in a mouse neuroblastoma model, antagomirs directed against miR-17-5p dramatically inhibited tumor growth leading to complete regression of tumor mass in 30% of the treated animals (Fontana et al. 2008).
Figure 1.
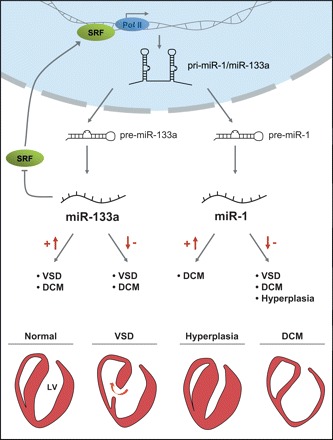
Cardiac phenotypes of miR-133a and miR-1 knockout and transgenic mice. The bicistronic miRNAs miR-1 and miR-133a are transcribed by RNA Polymerase II (Pol II), processed from pri- to pre-miRNAs, and finally into mature miRNAs. The expression of miR-133a and miR-1 is regulated by SRF. MiR-133a directly targets SRF mRNA, forming a negative regulating feedback loop. The double knockout of miR-133a-1 and miR-133a-2 induces either lethal VSDs or DCM (DCM). Overexpression of miR-133a also leads to VSDs and cardiac hypoplasia. Similarly, overexpression of miR-1 results in hypoplastic hearts and consecutive heart failure, whereas the knockout of miR-1-2 leads to diverse phenotypes, including VSDs, DCM, or cardiac hyperplasia.
Because miR-1 inhibits cardiomyocyte growth and proliferation, therapeutic repression of miR-1 could be beneficial for myocardial regeneration—for instance, after myocardial infarction—while overexpression of miR-1 could be used for the treatment of cardiac hypertrophy. However, it should be noted that genetic deletion of miR-1-2 results in electrophysiological abnormalities and a high incidence of sudden cardiac death while overexpression of miR-1 in normal or infarcted rat hearts also exacerbates arrhythmias because of slowed conduction and depolarization of the cytoplasmic membrane by post-transcriptional repression of KCNJ2 (potassium channel subunit Kir2.1) and GJA1 (Connexin 43) (Yang et al. 2007). One could consider that up-regulation of miR-133a might be an interesting strategy to prevent cardiac growth, e.g., in patients with hypertrophic cardiomyopathy or in response to increased cardiac afterload. However, similar to the case with miR-1, increased levels of miR-133a also are accompanied by electrophysiological abnormalities (Xiao et al. 2007). Although further problems, such as reaching a high enough target specificity or targeting only a selected cell population are still unresolved, hope remains that miRNA-based therapies will help to combat cardiovascular disease.
Acknowledgments
Work in Wolfgang Rottbauer’s laboratory was supported by NIH grants 5R01HL49579, 5R01DK55383, and 1RO1HL63206 (to M.C.F); Deutsche Forschungsgemeinschaft (DFG) Ro2173/1-1 and Ro2173/2-1 (to W.R.); and Bundesministerium für Bildung und Forschung 01GS0108, 01GS0420, and NGFNplus, as well as a Klaus-Georg and Sigrid Hengstberger Stipendium (to W.R.).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1753508.
References
- Bartel D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Basson C.T., Cowley G.S., Solomon S.D., Weissman B., Poznanski A.K., Traill T.A., Seidman J.G., Seidman C.E. The clinical and genetic spectrum of the Holt-Oram syndrome (heart–hand syndrome) N. Engl. J. Med. 1994;330:885–891. doi: 10.1056/NEJM199403313301302. [DOI] [PubMed] [Google Scholar]
- Bendig G., Grimmler M., Huttner I.G., Wessels G., Dahme T., Just S., Trano N., Katus H.A., Fishman M.C., Rottbauer W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes & Dev. 2006;20:2361–2372. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D.W., Silberbach G.M., Kavanaugh-McHugh A., Cottrill C., Zhang Y., Riggs S., Smalls O., Johnson M.C., Watson M.S., Seidman J.G., et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E., Guryev V., de van Belt J., Wienholds E., Plasterk R.H., Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Bernstein E., Caudy A.A., Hammond S.M., Hannon G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Calin G.A., Ferracin M., Cimmino A., Di Leva G., Shimizu M., Wojcik S.E., Iorio M.V., Visone R., Sever N.I., Fabbri M., et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- Calin G.A., Garzon R., Cimmino A., Fabbri M., Croce C.M. MicroRNAs and leukemias: How strong is the connection? Leuk. Res. 2006;30:653–655. doi: 10.1016/j.leukres.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Care A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N.D., et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- Cimmino A., Calin G.A., Fabbri M., Iorio M.V., Ferracin M., Shimizu M., Wojcik S.E., Aqeilan R.I., Zupo S., Dono M., et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli A.M., Tops B.B., Plasterk R.H., Ketting R.F., Hannon G.J. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Dresios J., Aschrafi A., Owens G.C., Vanderklish P.W., Edelman G.M., Mauro V.P. Cold stress-induced protein Rbm3 binds 60S ribosomal subunits, alters microRNA levels, and enhances global protein synthesis. Proc. Natl. Acad. Sci. 2005;102:1865–1870. doi: 10.1073/pnas.0409764102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L., Fiori M.E., Albini S., Cifaldi L., Giovinazzi S., Forloni M., Boldrini R., Donfrancesco A., Federici V., Giacomini P., et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One. 2008;3:e2236. doi: 10.1371/journal.pon.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillum R.F. Idiopathic dilated cardiomyopathy. Epidemiology. 1994;5:383–385. doi: 10.1097/00001648-199405000-00026. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman J.I., Kaplan S., Liberthson R.R. Prevalence of congenital heart disease. Am. Heart J. 2004;147:425–439. doi: 10.1016/j.ahj.2003.05.003. [DOI] [PubMed] [Google Scholar]
- Karkkainen S., Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann. Med. 2007;39:91–107. doi: 10.1080/07853890601145821. [DOI] [PubMed] [Google Scholar]
- Krutzfeldt J., Rajewsky N., Braich R., Rajeev K.G., Tuschl T., Manoharan M., Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs.’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee C.T., Risom T., Strauss W.M. Evolutionary conservation of microRNA regulatory circuits: An examination of microRNA gene complexity and conserved microRNA-target interactions through metazoan phylogeny. DNA Cell Biol. 2007;26:209–218. doi: 10.1089/dna.2006.0545. [DOI] [PubMed] [Google Scholar]
- Li S.C., Pan C.Y., Lin W.C. Bioinformatic discovery of microRNA precursors from human ESTs and introns. BMC Genomics. 2006;7:164. doi: 10.1186/1471-2164-7-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Bezprozvannaya S., Williams A.H., Qi X., Richardson J.A., Bassel-Duby R., Olson E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes & Dev. 2008 doi: 10.1101/gad.1738708. (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman C., Runswick M., Pollock R., Treisman R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 1988;55:989–1003. doi: 10.1016/0092-8674(88)90244-9. [DOI] [PubMed] [Google Scholar]
- Richardson P., McKenna W., Bristow M., Maisch B., Mautner B., O'Connell J., Olsen E., Thiene G., Goodwin J., Gyarfas I., et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–842. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Griffiths-Jones S., Ashurst J.L., Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguin N., Du Z.D., Barak M., Nasser N., Hershkowitz S., Milgram E. High prevalence of muscular ventricular septal defect in neonates. J. Am. Coll. Cardiol. 1995;26:1545–1548. doi: 10.1016/0735-1097(95)00358-4. [DOI] [PubMed] [Google Scholar]
- Rottbauer W., Just S., Wessels G., Trano N., Most P., Katus H.A., Fishman M.C. VEGF-PLCγ1 pathway controls cardiac contractility in the embryonic heart. Genes & Dev. 2005;19:1624–1634. doi: 10.1101/gad.1319405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed D., Hong C., Chen I.Y., Lypowy J., Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- Scherr M., Venturini L., Battmer K., Schaller-Schoenitz M., Schaefer D., Dallmann I., Ganser A., Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A., Brennecke J., Bushati N., Russell R.B., Cohen S.M. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Valencia-Sanchez M.A., Liu J., Hannon G.J., Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes & Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- van Rooij E., Sutherland L.B., Qi X., Richardson J.A., Hill J., Olson E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Wang Z., Wang D.Z., Hockemeyer D., McAnally J., Nordheim A., Olson E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428:185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- Weiler J., Hunziker J., Hall J. Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13:496–502. doi: 10.1038/sj.gt.3302654. [DOI] [PubMed] [Google Scholar]
- Xiao J., Luo X., Lin H., Zhang Y., Lu Y., Wang N., Zhang Y., Yang B., Wang Z. MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J. Biol. Chem. 2007;282:12363–12367. doi: 10.1074/jbc.C700015200. [DOI] [PubMed] [Google Scholar]
- Xu P., Vernooy S.Y., Guo M., Hay B.A. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr. Biol. 2003;13:790–795. doi: 10.1016/s0960-9822(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Xu C., Lu Y., Pan Z., Chu W., Luo X., Lin H., Xiao J., Shan H., Wang Z., Yang B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007;120:3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- Yang B., Lin H., Xiao J., Lu Y., Luo X., Li B., Zhang Y., Xu C., Bai Y., Wang H., et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007;13:486–491. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- Zhang X., Azhar G., Chai J., Sheridan P., Nagano K., Brown T., Yang J., Khrapko K., Borras A.M., Lawitts J., et al. Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H1782–H1792. doi: 10.1152/ajpheart.2001.280.4.H1782. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Samal E., Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Ransom J.F., Li A., Vedantham V., von Drehle M., Muth A.N., Tsuchihashi T., McManus M.T., Schwartz R.J., Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]