

Abstract
Background
Single Nucleotide Polymorphisms (SNPs) are an increasingly important tool for genetic and biomedical research. Although current genomic databases contain information on several million SNPs and are growing at a very fast rate, the true value of a SNP in this context is a function of the quality of the annotations that characterize it. Retrieving and analyzing such data for a large number of SNPs often represents a major bottleneck in the design of large-scale association studies.
Description
SNPper is a web-based application designed to facilitate the retrieval and use of human SNPs for high-throughput research purposes. It provides a rich local database generated by combining SNP data with the Human Genome sequence and with several other data sources, and offers the user a variety of querying, visualization and data export tools. In this paper we describe the structure and organization of the SNPper database, we review the available data export and visualization options, and we describe how the architecture of SNPper and its specialized data structures support high-volume SNP analysis.
Conclusions
The rich annotation database and the powerful data manipulation and presentation facilities it offers make SNPper a very useful online resource for SNP research. Its success proves the great need for integrated and interoperable resources in the field of computational biology, and shows how such systems may play a critical role in supporting the large-scale computational analysis of our genome.
Background
Single Nucleotide Polymorphisms (SNPs) are an increasingly important tool for the study of the structure and history of our genome [1]. The most common application of SNPs is in association studies, that look for a statistically significant association between SNP alleles and phenotypes (usually diseases), in order to pinpoint candidate causative genes [2]. The power of association studies is a function of the number of SNPs used, and of their quality (defined here as the likelihood of the SNP locus actually being polymorphic in the population under study). For this reason, large databases of well-annotated SNPs have been developed, and are growing at an ever increasing rate.
In order to take advantage of the mass of known SNPs, now numbering almost five millions for the human genome alone, researchers need tools to easily and efficiently locate the desired SNPs, to evaluate their annotations, and to export them in formats suitable for subsequent analysis. This, in turn, requires large amounts of data from different sources to be integrated and analyzed. This is a challenging task even with the automated tools now at our disposal [3].
SNPper [4], developed by the Children's Hospital Informatics Program and the Innate Immunity PGA Project [5], aims at providing an integrated database of SNP-related data and a suite of online, user-friendly tools to query it. Since its initial public release [6], SNPper has grown from a simple repository of SNP data into a comprehensive resource for SNP analysis, through the integration of more information sources, better visualization tools and more powerful SNP evaluation methods. In this paper we focus on the organization, construction and content of the SNPper database, we describe the data retrieval, export and presentation features it offers, and we discuss how it may be used to support SNP-based research.
Construction and content
SNPper is built upon a relational database whose contents are the result of merging data from several well-established online resources, and of a post-processing step that produces additional annotations. In the following sections we will detail the data items that SNPper acquires from external databases, and we will describe how they are combined to provide an added value for the users.
Data sources
The main source of data for SNPper is dbSNP [7], currently the largest public repository of SNP data [8]. Out of the 7.7 million SNPs available in dbSNP build 118, the SNPper database contains the 4.8 million for which exact, unique position information is available in the latest release of the Human Genome assembly. The raw dbSNP data files are parsed to extract, for each SNP, its rs identifier, its alleles, its validation status, and the list of submissions. In dbSNP, a set of independent observations of the same polymorphism is represented by a RefSNP cluster, identified by a unique rs identifier. In turn, each independent submission of that SNP is described by a unique submission identifier (ss), a submitter handle, and a private SNP identifier. Knowing the list of submission for a SNP is especially important, since the number of independent observations of the same SNP is a good indicator of its quality. In particular, a SNP is considered validated by dbSNP if both of its alleles were observed a sufficient number of times by independent investigators using non-computational methods.
Even more important is the knowledge of the frequency of each allele of a SNP in different populations. The SNP Consortium [9] has recently made available a dataset providing frequency data for about 100,000 SNPs, in at least two different populations [10]. This data, too, is parsed and stored in the SNPper database, and is presented to the user when available.
The second main data source used by SNPper is Goldenpath [11], the Human Genome browser developed at UCSC [12]. Goldenpath provides the entire contents of its annotation database, in addition to the full human genome sequence, as a collection of plain text files, in easy to parse formats. SNPper takes full advantage of this feature to populate tables containing the absolute chromosomal location of SNPs and known genes. The annotation available for each gene includes the exact position of all its exons and introns, and of its coding sequence. Combined with a local copy of the human genome, this data allows SNPper to retrieve the DNA sequence of any gene or portion of a gene, for example to automatically translate its coding sequence into the corresponding amino acid sequence.
Finally, other sources of information that are parsed to populate the database include LocusLink [13], to fill in some of the gene annotation fields, GeneOntology [14], to determine the set of all GeneOntology classes each gene belongs to, and SWISS-PROT [15] to gather information about protein domains. This data is all exploited to enrich SNP annotation as described in the next paragraph. Pointers to these and other databases (e.g. OMIM, Unigene) are also stored by SNPper to make it easier for the user to retrieve more information about any gene of interest.
Post-processing
Since data on genes and on SNPs is obtained from different sources, a post-processing step is used to integrate them, and to generate some additional annotations. The procedure starts by examining one gene at a time, and collecting all the SNPs that lie on or close to it (up to a maximum distance of 10,000 bp). The number of SNPs thus obtained is stored in the genes table, in order to allow the user to easily evaluate the SNP density of each gene. At the same time, a record is added to a separate table to detail the association between the SNP and the gene. Since a SNP may belong to more than one gene (due to overlapping genes, or more commonly to multiple isoforms of the same gene), there may be multiple records for the same SNP. In addition to the SNP and gene identifiers, this record stores the "role" of the SNP with respect to that gene (e.g. intronic, exonic, coding), its relative position (distance from the starting ATG of the gene) and, for coding SNPs, the amino acid change they cause. This last field is computed by translating the coding sequence for each gene obtained from Goldenpath, and applying the nucleotide change induced by each SNP it contains.
Some basic quality control tests are performed during this phase. For example, we check whether the base found in the Goldenpath genome sequence at the SNP position matches one of its two alleles, or whether the translation of the coding sequence is correct (approximately 10% of the genes in Goldenpath fails this check, due to the inaccurate positioning of their exons). In general, though, the agreement between SNPper's two main data sources (dbSNP and Goldenpath) is very good, which means that the annotations we derive from them are accurate and consistent. Further quality control procedures, including ensuring consistency with other SNP resources, are beyond the scope of this work.
User-supplied data
SNPper provides users with the ability to add new, private SNPs to its database. This is accomplished by specifying a gene name and uploading two files: the DNA sequence, in FASTA format, that the SNPs were located in, and a tab-delimited text file containing the alleles of each SNP and its position relative to the start of the reference sequence. SNPper will then determine the absolute position of each SNP, by aligning its flanking sequence with the sequence for the gene in question, and will store the new SNP records into its database. Once there, they will be displayed by SNPper together with the preexisting SNPs. SNPs added in this way are considered private, so that they are only visible to the user who submitted them, but can optionally be made public.
Utility and discussion
Queries
SNPper offers several different ways of retrieving SNPs, reflecting the different purposes the SNPs might be used for. In the simplest case, SNPs can be retrieved by specifying one or more rs or ss identifiers, if known. Another common query retrieves a set of contiguous SNPs belonging to a specific region of a chromosome, specified either through an absolute position range or through the name of a cytogenetic band. To support gene-oriented studies, SNPper can generate the set of SNPs on or around a gene (up to a user-specified maximum distance), or a set of genes. Genes can be specified using their HUGO name, or through their Genbank, Locuslink, OMIM, or Unigene identifiers. A set of genes of interest, in turn, can be specified by position (i.e., all the genes in a chromosome region), or through a GeneOntology class. This last feature makes it possible to study sets of SNPs that are potentially associated with a biological process of interest, rather than being linked by position.
In all cases, the result of a query is a SNPset, a data structure that holds a collection of SNPs. Different types of SNPset exist, according to the type of query that produced them, and different operations are defined on them. For example, for a SNPset containing contiguous SNPs it makes sense to measure the SNP density, and SNPper provides a function to reduce the number of SNPs it contains while maintaining a uniform spacing. For a SNPset generated from a set of genes, this operation would not make sense (since the SNPs may be spread on different chromosomes) and is therefore not available. In general, a SNPset represents a set of SNPs that was produced by a single query, and that can be manipulated and analyzed as a whole. SNPsets are only visible to the user that generated them, and are persistent data structures: they are stored in the server as long as they are used, and are automatically removed after a sufficient amount of time has passed since they were last accessed. The user may also choose to save a SNPset, in which case it will be available in future sessions.
SNP visualization
SNPper provides multiple ways of displaying SNPs through its web interface. To start, each SNP is described individually in a page that displays general data (SNP identifiers, position, alleles, validation status), its list of submitters, the list of genes it belongs to, its frequency in different populations (consisting of the sample size and the major and minor allele frequencies) if available, and the list of protein domains the SNP falls in, if any. SNPs that belong to a gene may be displayed in the context of the corresponding DNA sequence or (for coding SNPs) amino acid sequence. In both cases, the SNP position is highlighted and a popup window is used to display information about it (name, position, alleles). Figure 1 shows an annotated gene sequence containing SNPs, and Figure 2 shows the detailed information page about one of the SNPs in that sequence.
Figure 1.

An annotated gene sequence containing SNPs. A portion of the sequence for gene F13B (on chromosome 1) is shown. Black areas represent exons, while introns are in gray. The nucleotide positions on the left are relative to the human genome assembly provided by Goldenpath. SNPs are indicated by bold, underlined nucleotides, and their dbSNP identifier appears to the right of the sequence. A pop-up window displays additional information about individual SNPs: in this example, SNP rs6003 is shown to be a validated, non-synonymous coding SNP.
Figure 2.
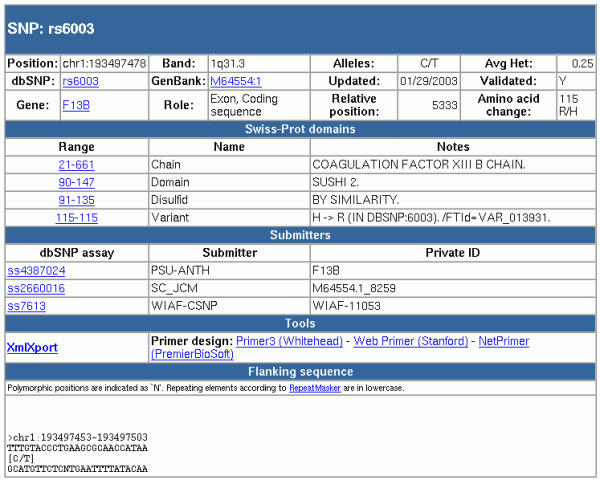
Detailed information page for an individual SNP. The page is divided into five sections. The first one contains general information about the SNP, including its identifier, its alleles, and its position in the gene (or genes) it belongs to. The second portion describes the protein domains that this SNP belongs to, according to SWISS-PROT. The next section lists all submissions for this SNP, and provides the dbSNP submission identifier, the submitter's name and the private SNP identifier. The "Tools" section contains links to various PCR primer design programs, and to a facility to export the data shown in this page in XML format. Finally, the flanking sequence for this SNP is shown, up to a maximum of 10,000 nucleotides.
SNPsets are displayed using tables that list the name of each SNP, its position, its alleles, and other information dependent on the specific SNPset type. SNPsets that contain a large number of SNPs are optionally displayed in abbreviated form for performance reasons. The table also contains links to perform the different commands available on a SNPset, such as saving it, exporting it (see next section) and refining it. The SNPset refinement operation, in particular, allows the user to "filter" the SNPs it contains according to several different criteria: general properties (e.g. validation), position relative to a gene (e.g. to select only exonic SNPs, or promoter SNPs), frequency or heterozygosity, submitters (to select only SNPs from specified submitters, or with a minimum number of distinct submitters), or average distance (in order to reduce the number of SNPs while maintaining a uniform spacing). In all cases, the result is to hide those SNPs that don't satisfy the user's criteria: while still part of the SNPset, these SNPs are no longer visible, and will not be considered in any analysis, display or export operation.
Finally, Figure 3 shows the output of a Java applet that can be used to display SNPsets in graphical form. The structure of the gene is shown using bars of different colors and thickness; SNPs are identified by squares (if frequency information is available) or circles (otherwise), and are colored in green if validated, black otherwise. The applet provides commands to scroll the display left or right, and to zoom the display in or out.
Figure 3.
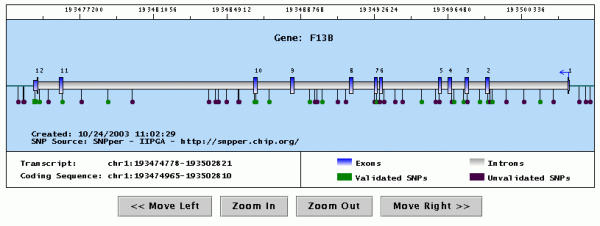
Graphical representation of a gene with the SNPs it contains. The picture shows the output of a Java applet displaying the gene structure (exons are in blue, introns in gray) and all SNPs in the same region (represented by dots or squares). The user may scroll or zoom the display using the supplied buttons, or recenter the display by clicking on the top portion of the picture. Clicking on a SNP opens a pop-up window containing information about it.
Data export
One of the most important design goals of SNPper is to provide easy ways of exporting SNP data in a variety of common formats. The SNPset export page, shown in Figure 4, allows the user to choose any number of fields from the available annotations (including flanking sequences, frequency information, validation status, amino acid change), and to specify the desired output format (XML, tab-delimited text, HTML, or BED) and destination (the data can be displayed in the browser window or sent by email to a user-supplied address). XML is gradually becoming a standard format for data exchange in biomedical applications, and is provided in SNPper to support interoperability with other programs. Tab-delimited text represents the simplest structured data format, and is therefore supported by most applications, such as spreadsheets or PCR primer design programs. HTML output allows users to display the data on a different website, for example to provide supplementary information for a publication. Finally, the BED format is used to exploit Goldenpath's "custom tracks" feature: the resulting file can be uploaded to the Goldenpath site, and the SNPset data will be displayed by the genome browser in its track display window.
Figure 4.
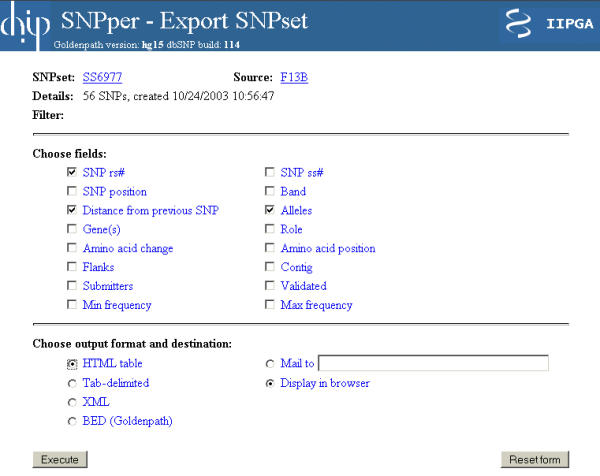
The SNPset export page. The first part of the form allows the user to select the set of annotations to be exported, out of the ones offered by SNPper. All combinations of annotations may be selected, although some fields are only applicable to a subset of all SNPs (e.g., Amino acid change), and some fields might contain multiple values (e.g., Gene). The second section is used to determine the format of the exported data, and its destination. The user may choose between exporting the data as an HTML table, as a tab-delimited text file, as an XML document, or as a BED file. This last option is used to upload the exported data to the Goldenpath genome browser, as a custom track. Finally, the user may choose to display the resulting file in the browser window, or to have it sent by email to a specified address.
SNPper also provides an alternative method for retrieving SNP information in XML format. Our system implements a Remote Procedure Call (RPC) interface, that allows other programs to send ad hoc HTTP requests and receive in response an XML document rather then an HTML page [16]. This feature is meant to increase the value and usefulness of the SNPper database, by allowing alternative systems using different user interfaces to be built on top of the same underlying data
Comparison with other SNP resources
Several other public SNP databases exist, each with its own strengths and limitations. In this section we briefly review the most important ones, pointing out their characteristic features and comparing them with what is offered by SNPper.
The HGVBASE database [17] is focused on very accurate manual curation and annotation of the available SNP data [18]. Therefore, it is smaller than dbSNP in size, but its data is extremely useful for researchers interested in exploring the genetic component of human phenotypic variation. It provides extensive annotations for its SNPs, and several search options including the ability to look for SNPs in a user-supplied genomic sequence that is automatically aligned to the human genome.
ALFRED [19] and JSNPs [20] are two smaller SNP databases that aim at providing accurate frequency information. JSNPs focuses on the Japanese population, while ALFRED provides data on a large number of very diverse populations from the whole world in order to enhance their differences in frequency and to facilitate the study of haplotypes [21]. Both sites offer a relatively small number of SNPs, but the frequency data they provide for them is extremely important. JSNPs offers a useful graphical chromosome browser and the ability to display sequencer traces for its SNPS. On the other hand, data export features are somewhat limited in both systems.
The GeneSNPs [22] site integrates gene, sequence and SNP data into highly annotated gene models. It provides extensive visualization and data export features, including a way of displaying SNPs within the genomic sequence of the gene they belong to similar to the one available in SNPper. Compared to SNPper, its main limitation is that it only contains SNPs on a small number of genes related with susceptibility to environmental exposure.
EnsMart [23] is a very comprehensive data mining tool to extract data from the Ensembl database. Ensembl is by far the most exhaustive and sophisticated database of all the ones described here. It provides approximately the same set of data items on SNPs as SNPper, with the main difference being that it uses its own database of genes rather than being linked to Goldenpath. Although its interface is very powerful and effective, it is not specifically tailored for the needs of SNP-based research; for example, it does not provide ways to generate sets of SNPs having a desired average spacing, as is instead possible with SNPper.
Features that are unique to SNPper are mainly related to the management of SNPsets and to its interoperability features. SNPper is the only resource giving users the ability to manipulate, refine, save and export SNPsets as a whole, and to allow creating sets of SNPs through complex queries (such as the one using GeneOntology classes described in section 4.a). Also, to our knowledge, SNPper is the only SNP resource that provides full access to its database as machine-readable XML files through a Remote Procedure Call interface, and that allows the user to upload a set of private SNPs to its database and to analyze or display them through its standard interface.
Conclusions
The main purpose of SNPper is to integrate and make available as a single, consistent resource a large amount of annotation data about Single Nucleotide Polymorphisms in the human genome. This was accomplished by combining the information available in several public online resources, and creating a local database that is accessible through a web-based front-end. The resulting system supports different forms of SNP-based research, providing the required information for each case: from SNP localization (in genes or in arbitrary genomic regions) to SNP roles in genes (e.g. to restrict the analysis to coding SNPs), to population-specific frequency information.
SNPper is an important and widely used SNP resource, receiving several thousand hits per day and counting over 2,000 registered users. It also constitutes an integral component of an entirely automated data pipeline for SNP discovery and analysis developed by the Innate Immunity PGA project. We take its success as proof that there exist a great need for tools that integrate biomedical information from different sources, offering flexible data visualization and export functions, and promoting interoperability with other systems.
Future work on SNPper will proceed in two main directions. On the application side, we will provide a wider range of analysis tools aimed at evaluating and characterizing SNPs in terms of their potential effects on genes and genomic regulatory elements. These tools will be tightly integrated with SNPper, allowing for automated, large-scale SNP analysis.
On the infrastructure side, we will increase the amount of data offered by SNPper by integrating more external databases. Additional information, such as genotypes, allelic frequencies, and the ancestral allele of SNPs, will be added to the database as it becomes available. We will also strive to automate as much as possible the construction and updating of the database, since keeping the local database up to date and synchronized with the multiple data sources it is built on currently requires frequent manual updates and ad-hoc changes to the import procedures, whenever the data formats of the external resources change. Finally, we are planning to extend SNPper to include information about SNPs in other organisms besides human, starting with the mouse.
Availability and requirements
The SNPper database is implemented using the MySQL relational database management system [24], while the application, including the web interface, was entirely developed in Common Lisp. The system runs on two GNU/Linux machines (one hosting the application and one for the database), and can be accessed at http://snpper.chip.org/. Its use is free for academic and research purposes.
Authors' contributions
AR designed and implemented the SNPper system. ISK provided guidance and supervision for the whole project.
Acknowledgments
Acknowledgements
This work was supported by grant U01-HL66795 from the National Heart, Lung and Blood Institute of NIH. We are grateful to the staff of the Children's Hospital Informatics Program and of the Innate Immunity PGA project for their feedback and constant encouragement.
Contributor Information
Alberto Riva, Email: alberto.riva@tch.harvard.edu.
Isaac S Kohane, Email: isaac_kohane@harvard.edu.
References
- Brookes A. The essence of SNPs. Gene. 1999:177–186. doi: 10.1016/S0378-1119(99)00219-X. [DOI] [PubMed] [Google Scholar]
- Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000;58:250–264. doi: 10.1034/j.1399-0004.2000.580402.x. [DOI] [PubMed] [Google Scholar]
- Stein LD. Integrating biological databases. Nat Rev Genet. 2003;4:337–345. doi: 10.1038/nrg1065. [DOI] [PubMed] [Google Scholar]
- SNPper http://snpper.chip.org/
- Lazarus R, Vercelli D, Palmer LJ, Klimecki WJ, Silverman EK, Richter B, Riva A, Ramoni M, Martinez FD, Weiss ST, Kwiatkowski DJ. Single nucleotide polymorphisms in innate immunity genes: abundant variation and potential role in complex human disease. Immunol Rev. 2002;190:9–25. doi: 10.1034/j.1600-065X.2002.19002.x. [DOI] [PubMed] [Google Scholar]
- Riva A, Kohane IS. SNPper: retrieval and analysis of human SNPs. Bioinformatics. 2002;18:1681–1685. doi: 10.1093/bioinformatics/18.12.1681. [DOI] [PubMed] [Google Scholar]
- dbSNP http://ncbi.nih.gov/SNP
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSC http://snp.cshl.org/
- Thorisson GA, Stein LD. The SNP Consortium website: past, present and future. Nucleic Acids Res. 2003;31:124–127. doi: 10.1093/nar/gkg052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenpath http://genome.ucsc.edu/
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LocusLink http://www.ncbi.nih.gov/LocusLink/
- GeneOntology http://www.geneontology.org/
- SwissProt http://www.expasy.org/sprot/
- Riva A, Kohane IS. Accessing genomic data through XML-based remote procedure calls. Proc AMIA Symp. 2002:662–666. [PMC free article] [PubMed] [Google Scholar]
- HGVbase http://hgvbase.cgb.ki.se/
- Fredman D, Siegfried M, Yuan YP, Bork P, Lehväslaiho H, Brookes AJ. HGVbase: A human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Research. 2002;30:387–391. doi: 10.1093/nar/30.1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfred http://alfred.med.yale.edu/
- JSNPs http://snp.ims.u-tokyo.ac.jp/
- Cheung KH Osier MV, Kidd JR, Pakstis AJ, Miller PL, Kidd KK. ALFRED: an allele frequency database for diverse populations and DNA polymorphisms. Nucleic Acids Res. 2000;1:361–363. doi: 10.1093/nar/28.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneSNPs http://www.genome.utah.edu/genesnps/
- EnsMart http://www.ensembl.org/EnsMart/
- mySQL http://www.mysql.com/