

Abstract
Tandem repeats are common features of both prokaryote and eukaryote genomes, where they can be found not only in intergenic regions but also in both the noncoding and coding regions of a variety of different genes. The repeat expansion diseases are a group of human genetic disorders caused by long and highly polymorphic tandem repeats. These disorders provide many examples of the effects that such repeats can have on many biological processes. While repeats in the coding sequence can result in the generation of toxic or malfunctioning proteins, noncoding repeats can also have significant effects including the generation of chromosome fragility, the silencing of the genes in which they are located, the modulation of transcription and translation, and the sequestering of proteins involved in processes such as splicing and cell architecture.
Tandem repeats are a ubiquitous feature of the genomes of many organisms, and the human genome database is replete with examples of repeat tracts of different sequence and repeat number in different genomic locations (Riggins et al. 1992; Epplen et al. 1993; Subramanian et al. 2003; Subirana and Messeguer 2008). Estimates from the human genome sequencing project indicate that such repeats make up ∼3% of the sequenced human genome (Lander et al. 2001). However, the true number and length of such repeats may be much higher than the current database suggests since these sequences are prone to deletion when propagated in bacteria and yeast. Additionally, these repeats are often unstable or hypervariable in mammals as well. Thus, genomes are polymorphic with respect to many of these repeats, with some individuals or families having some tandem repeat tracts that are significantly longer than those seen in the general population. For example, most humans have 30 CGG•CGG repeats in the 5′ UTR of their FMR1 gene (Fu et al. 1991; Eichler et al. 1994; Strom et al. 2007). However, population studies in Caucasians, the only population for which significant data exist, indicate that ∼1 in 246–468 females have 55–200 repeats and ∼1 in 3717–8918 males have 200 to >1000 repeats at this locus (Crawford et al. 2001).
These larger repeat lengths are not necessarily biologically neutral. For example, FMR1 alleles with 55–200 CGG•CCG-repeats are associated with neurodegeneration (Hagerman and Hagerman 2004) and ovarian insufficiency (Murray 2000; Sherman 2000). Alleles with >200 repeats are associated with intellectual disability and autistic symptoms (Hagerman 2006). The FMR1 gene is not unique in having significant repeat length polymorphism that is associated with disease pathology. In fact, to date 20 disorders have been identified in humans that result from the presence of a large expansion-prone DNA tandem repeat. These diseases, known collectively as the repeat expansion diseases, are listed in Table 1. Two recent books discuss most of these diseases in some detail, and the reader is referred to these sources for more information as to their incidence, genetics, and pathology (Fry and Usdin 2006; Wells and Ashizawa 2006). Diseases are also known that involve an expanded amino acid tract generated by insertion or duplication of an imperfect trinucleotide repeat. These diseases differ in key ways from “classic” members of the repeat expansion diseases. This review will focus primarily on those “classic” or “nucleotide repeat expansion diseases.” More information about those diseases arising from expansions at the protein level can be found elsewhere (Albrecht and Mundlos 2005).
Table 1.
The repeat expansion diseases
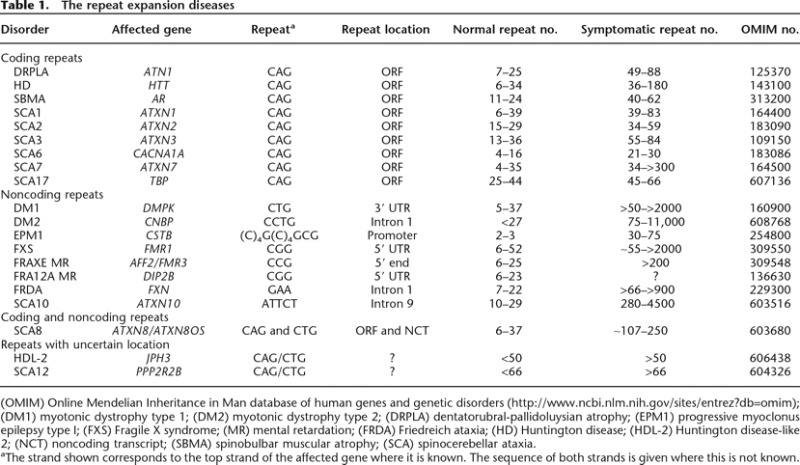
(OMIM) Online Mendelian Inheritance in Man database of human genes and genetic disorders (http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim); (DM1) myotonic dystrophy type 1; (DM2) myotonic dystrophy type 2; (DRPLA) dentatorubral-pallidoluysian atrophy; (EPM1) progressive myoclonus epilepsy type I; (FXS) Fragile X syndrome; (MR) mental retardation; (FRDA) Friedreich ataxia; (HD) Huntington disease; (HDL-2) Huntington disease-like 2; (NCT) noncoding transcript; (SBMA) spinobulbar muscular atrophy; (SCA) spinocerebellar ataxia.
aThe strand shown corresponds to the top strand of the affected gene where it is known. The sequence of both strands is given where this is not known.
Because of their association with human disease, the biological effects of the repeats responsible for these diseases have been studied intensively. As a result, a number of new paradigms have emerged for understanding how tandem repeats can affect genome structure and function. This review will focus on current ideas as to what effects these repeats have on the region of the genome in which they are located and what the ramifications may be for related repeats elsewhere.
The repeat expansion diseases can be divided into two categories: those like Huntington Disease (HD) or spinobulbar muscular atrophy (SBMA), where the repeat is located in an exon, and those like myotonic dystrophy (DM) or Fragile X syndrome (FXS), where the repeat is outside of the open reading frame (ORF) (Table 1). Both coding and noncoding repeats can have significant impact.
Open reading frame expansions
In all repeat expansion diseases identified thus far in which the repeat is in an exon, the repeat unit is a triplet with the sequence CAG•CTG. To date, at least 10 such diseases have been identified (Table 1). The result of the expansion is an increase in the length of a polyglutamine (polyQ) tract in the encoded protein. This is associated with the death of vulnerable neurons in the brain. The specific features of each disorder are thought to result from the particular combination of the properties of the expanded polyQ tract and the protein in which it is located (Orr and Zoghbi 2007). PolyQ is thought to cause conformational changes that confer toxic properties to the protein. This is accompanied by the appearance of polyQ-containing inclusions that may or may not be directly pathogenic. It has been suggested that failure to properly degrade the misfolded polyQ proteins via either the autophagic or the ubiquitin-proteasome pathways contributes to polyQ toxicity. Mitochondrial dysfunction, excitotoxicity, and disrupted intracellular trafficking have also been reported in cells expressing PolyQ proteins (for review, see Orr and Zoghbi 2007). Many of the polyQ proteins also interact with glutamine-rich transcription factors and cofactors like CREB-binding protein (CBP, also known as CREBBP) and SP1 (for review, see Orr and Zoghbi 2007). In some cases, factors like CBP have been found in inclusions, and CBP overexpression is known to rescue the polyQ toxicity in cells in tissue culture (Nucifora et al. 2001; Steffan et al. 2001). Sequestering of these proteins in aggregates may explain the widespread effect of polyQ on transcription. Reduced availability of transcription factors like SP1 could account for specific changes in gene expression, while inhibition of the histone acetyltransferase activities of proteins like CBP could account for the more global changes that are seen. Histone deacetylase inhibitors alleviate polyQ toxicity in various model systems (Steffan et al. 2001; Hughes 2002; Hockly et al. 2003; Bates et al. 2006), consistent with the hypothesis that polyQ toxicity results from the sequestering of proteins that affect gene expression by altering normal chromatin.
In addition to the toxic gain of function seen in expanded alleles, a reduction in the normal functioning of the affected protein can occur. For example, in SCA6, the small polyQ expansions seen in the affected protein, the alpha subunit of the P/Q-voltage gated calcium channel, affect channel function (Kordasiewicz and Gomez 2007). The relative contribution of impaired channel function and polyQ toxicity to SCA6 pathology is still the subject of some debate (Kordasiewicz and Gomez 2007). Small increases in the number of glutamines are also associated with a reduced transactivation ability of the androgen receptor (Mhatre et al. 1993; Chamberlain et al. 1994).
Long polyalanine repeat tracts generated by imperfect trinucleotide repeats share many of the properties of polyglutamine repeats. They have been implicated in a variety of congenital abnormalities, including cleidocranial dysplasia, holoprosencephaly, oculopharyngeal muscular dystrophy, and synpolydactyly 1 (for review, see Albrecht and Mundlos 2005). Thus, long polyalanine tracts can also be deleterious. In HD and SCA3, a small amount of frameshifting occurs during translation of the repeat (Davies and Rubinsztein 2006). This results in the low levels of proteins with polyalanine and polyserine tracts. Their contribution to disease pathology is currently unknown. Reduced protein translation is also sometimes seen (Brockschmidt et al. 2007).
It has been estimated that as many as 1 in 20 human proteins have tandem repeat polymorphisms (O'Dushlaine et al. 2005). Of these, approximately one fifth involve tandem repeat units that are not multiples of three, and thus these polymorphisms involve frameshifts (O'Dushlaine et al. 2005). Many examples of tandem repeat-mediated frameshifts are seen in pathogenic microorganisms, where they are a frequent source of phase variation in bacteria like Neisseria and Mycoplasma (van Belkum et al. 1999). Frameshifting may be mediated at the RNA level, perhaps by affecting ribosomal scanning. This phenomenon will be discussed in more detail below in the context of RNA-mediated effects of noncoding repeats.
Noncoding expansions
Many of the repeat expansion diseases involve a repeat that is in a noncoding region of the gene. In these disorders, self-evidently, the relationship between repeat expansion and disease pathology is not simply a matter of a change in the properties of the protein product of the affected gene. These diseases include progressive myoclonus epilepsy type 1 (EPM1); neurodegenerative disorders such as Friedreich ataxia (FRDA), spinocerebellar ataxia type 8 (SCA8) and 10 (SCA10), and fragile X-associated tremor and ataxia syndrome (FXTAS); the mental retardation (MR) syndromes, fragile X syndrome (FXS) and FRAXE and FRA12A MR, as well as myotonic dystrophy type 1 (DM1) and 2 (DM2).
EPM1 is caused by a dodecamer repeat in the promoter of the cystatin B gene. It is characterized by severe stimulus-sensitive myoclonus, generalized tonic-clonic seizures (Lalioti et al. 1997; Virtaneva et al. 1997). FRDA is a relentlessly progressive ataxia with associated hypertrophic cardiomyopathy resulting from the presence of >66 GAA•TTC repeats in intron 1 of the frataxin (FXN) gene (Pandolfo 2002). SCA8 and SCA10 are characterized by cerebellar dysfunction and seizures, with variable expression of polyneuropathy, as well as cognitive and neuropsychiatric impairment. The CAG•CTG repeat responsible for SCA8 lies in the region of overlap of the 3′ end of the ORF of the ATXN8 gene and ATXN8OS, a gene that produces a noncoding transcript from the opposite strand (Moseley et al. 2006). SCA10 results from expansion of the repeat ATTCT•AGAAT in intron 9 of the ATXN10 gene (Matsuura et al. 2000).
FXTAS (Hagerman and Hagerman 2004) and premature ovarian insufficiency (Murray 2000; Sherman 2000) result from the presence of 55–200 CGG•CCG repeats in the 5′ UTR of the FMR1 gene. A quite different disorder, FXS, occurs when the repeat number in the FMR1 gene exceeds 200 (Hagerman 2006). Expansion in the region of overlap between the divergently transcribed genes AFF2 (formerly FMR2) and FMR3 results in a mild form of intellectual disability known as FRAXE MR (Gecz 2000a, b). FRA12A MR results from the presence of a large CGG•CCG-repeat tract in the 5′ UTR of the DIP2B gene (Winnepenninckx et al. 2007).
DM1, which results from a large CTG•CAG repeat in the 3′ UTR of the DMPK gene, is a dominantly inherited multisystemic disorder (for recent reviews, see Ranum and Cooper 2006; Wheeler and Thornton 2007). A juvenile-onset form and an adult-onset form are characterized by skeletal muscle myotonia, together with progressive muscle weakness and wasting, cardiac conduction defects, cataracts, insulin insensitivity, and testicular atrophy. Extremely large repeat numbers are associated with a congenital form of DM1 (CDM), which presents initially with delayed motor development, mental retardation, and hypotonia, with symptoms typical of adult-onset DM1 developing later. DM2 is a disorder with symptoms very similar to DM1 but that results from a large CTGG•CCAG-repeat tract in an intron of the unrelated CCHC-type zinc finger, nucleic acid binding protein (CNBP) gene (formerly known as ZNF9) (for recent reviews, see Ranum and Cooper 2006; Wheeler and Thornton 2007).
The noncoding repeats responsible for these diseases may affect cell and chromatin structure, as well as transcription, splicing, and translation in a variety of different ways.
Tandem repeats as origins of replication
The ATTCT•AGAATA repeats responsible for SCA10 are prone to unwinding in vitro and readily form unpaired regions in supercoiled plasmids (Potaman et al. 2003). This behavior is consistent with a role in the initiation of replication. Indeed, SCA10 alleles show elevated origin activity compared with unaffected alleles (Liu et al. 2007). CGG•CCG repeats have very different properties in vitro (Fry and Loeb 1994; Chen et al. 1995; Nadel et al. 1995; Usdin and Woodford 1995; Mariappan et al. 1996). Nonetheless, origins of replication have been mapped to the region containing the repeat in both the FMR1 (Brylawski et al. 2007; Gray et al. 2007) and the AFF2 genes (Chastain II et al. 2006). The ability to act as origins of replication may play a role in the tendency of these repeats to expand (Chastain II et al. 2006; Brylawski et al. 2007; Gray et al. 2007).
Paradoxically, the individual strands of the CGG•CCG and CTG•CAG repeats form folded structures like hairpins and tetraplexes that block DNA synthesis (Kang et al. 1995; Usdin and Woodford 1995; Weitzmann et al. 1996, 1998; Samadashwily et al. 1997; Ohshima et al. 1998; Usdin 1998). This may contribute to the association of repeats like these with chromosome fragility as discussed below.
Tandem repeats and chromosome fragility
Some large repeat tracts cause the formation of a cytogenetic abnormality known as a fragile site. Mammalian fragile sites are gaps or constrictions in chromosomes that are visible by microscopy when cells are grown in the presence of particular inducers. They are frequent sites of chromosome breakage and translocation (for review, see Arlt et al. 2006). Since microscopy is a relatively low-resolution technique, it may be that fragile sites occur with shorter repeat lengths but are not visible with this technique. Breakage-prone sites can be seen to occur spontaneously with even relatively short repeat tracts in yeast using genetic assays (Freudenreich et al. 1998; Balakumaran et al. 2000).
A number of inducers of mammalian fragile sites have been identified. These include folate stress, aphidicolin, bromodeoxyuridine, and distamycin. To date, all seven folate-sensitive fragile sites whose sequence is known contain long CGG•CCG-repeat tracts. This includes the disease-associated tracts seen in FXS, FRAXE MR, and FRA12A MR. The 33-bp AT-rich repeats responsible for the distamycin-inducible fragile site at FRA16B (Yu et al. 1997) and the ∼42-bp AT-rich repeats responsible for the bromodeoxyuridine-inducible site at FRA10B (Hewett et al. 1998) are not associated with any known disease. However, they do illustrate the potential for other tandem repeat sequences to cause chromosome fragility. Long CTG•CAG repeats are fragile in yeast (Freudenreich et al. 1998). However, no agent has yet been identified that induces fragility of these repeats in human cells, and whether, in fact, they are fragile in mammalian cells remains to be seen.
Agents that induce the different classes of fragile sites all interfere with DNA replication. In addition, many fragile sites are inherently difficult to replicate (Kang et al. 1995; Usdin and Woodford 1995; Samadashwily et al. 1997; Ohshima et al. 1998) or are associated with regions of late replication. This has led to the idea that fragile sites result from some sort of replication block or from incompletely replicated regions of the chromosome (Le Beau et al. 1998).
Most recent studies in human cells have been carried out on the common aphidicolin-inducible fragile sites, whose sequence basis is unknown (Rassool et al. 1996). It has been shown that reduced levels of ataxia telangiectasia-related (ATR), CHEK1, BRCA1, and SMC1 proteins lead to elevated levels of expression of these sites (Casper et al. 2002, 2004; Arlt et al. 2004; Musio et al. 2005; Durkin et al. 2006). Since these proteins are key components of the cellular response to stalled replication forks, it supports the idea that fragile sites block or retard the progress of the DNA replication machinery. Common fragile sites may represent double-strand breaks formed because of stalled replication forks that were not rescued via the ATR/CHEK1/BRCA1/SMC1 pathway. Neoplastic transformation may result from those cells that escape the ATR checkpoint and enter mitosis with an incompletely resolved or improperly repaired fragile site (Musio et al. 2005). While similar experiments have not yet been reported for other classes of fragile sites, there is reason to think that they arise by a similar mechanism. In this regard, it may be relevant that ATR is required for the stable maintenance of long CGG•CCG repeats in mice (Entezam and Usdin 2008).
It is unlikely that chromosome fragility per se plays any role in FXS, FRAXE MR, or FRA12A MR. However, some of the deletions common in this region may be related to this phenomenon, and at least one balanced translocation involving a breakpoint in AFF2 is known (Honda et al. 2007). In other locations, such repeat tracts may cause the loss or disruption of vital genes or the generation of an oncogene. For example, Jacobsen syndrome (JBS) (OMIM #147791), a disorder that includes trigonocephaly, cardiac anomalies, and thrombocytopenia, results from deletions involving the long arm of chromosome 11. Deletion breakpoints map to FRA11B, a folate-sensitive fragile site, as well as to other more distal CGG•CCG-repeat tracts (Jones et al. 2000). Fragile sites in primates have been shown to constitute breakpoints that have contributed to chromosome evolution (Ruiz-Herrera et al. 2005, 2006). In fact, it has been suggested that mammalian chromosomal evolution has been driven by such sites and that “the human genome can be considered a mosaic comprising regions of fragility that are prone to reorganization” (Ruiz-Herrera et al. 2005).
Tandem repeats as intrinsic promoter components
The bidirectional promoter for the AFF2 and FMR3 genes implicated in FRAXE MR is comprised largely of CGG•CCG repeats, with the transcription start site for each gene being located within 30 bp of one end of the repeat tract (Gecz 2000a). Thus, the repeats present in the normal allele may play a positive role in regulating transcription from this promoter. Antisense transcripts are produced from the FMR1 gene (Ladd et al. 2007). The major transcription start sites on normal alleles are within 350 bp of the CGG-repeat tract. Thus, it is possible that the repeats are an integral part of this promoter as well. Since normal FMR1 and AFF2/FMR3 alleles have 5–54 repeats and 3–42 repeats respectively, it suggests that even relatively short CGG•CCG-repeat tracts may be able to contribute significantly to promoter activity. A variety of CGG•CCG-binding proteins have been described that have the potential to affect transcription, including CGGBP (Deissler et al. 1997), the multifunctional Pur proteins (Gallia et al. 2000), and the Egr family of transcription factors (Christy and Nathans 1989).
In EPM1, repeat numbers in the disease range lead to decreased cystatin B mRNA production. As few as 12 repeats can cause a significant reduction in promoter activity in a reporter assay (Alakurtti et al. 2000). Since a random sequence of the same length has the same effect, the consequences of repeat expansion in EPM1 have been attributed to a nonspecific effect of the repeats on the relative position of important regulatory elements (Lalioti et al. 1999). A similar mechanism is thought to explain the effect of the polymorphic GAA•TTC repeat in the Mycoplasma gallisepticum pMGA promoter on the phase variation of the adhesin proteins (Glew et al. 2000). It is also possible to envision a scenario in which the repeats in the promoter may bind an activator or repressor, thereby positively or negatively affecting gene activity.
Tandem repeats as transcription enhancers
Increased numbers of CGG•CCG repeats are associated with increased FMR1 transcription up to ∼200 repeats (Tassone et al. 2000, 2007). The antisense transcript shows a similar repeat-related increase (Ladd et al. 2007). A more open chromatin architecture may be responsible, perhaps related to the tendency of CGG•CCG repeats to exclude nucleosomes (Wang et al. 1996). Alternatively, the repeat may bind more CGG•CCG-binding proteins that facilitate transcription initiation or elongation. The increased transcription is associated with the increased usage of upstream transcription start sites for both the sense and antisense transcripts (Beilina et al. 2004; Ladd et al. 2007). In the case of the antisense transcript, this upstream start site is located ∼10 kb away. This demonstrates that the effect of these repeats on transcription initiation can be exerted over a large distance. A positive effect of the repeats on transcription has also been seen in transient transfection experiments in which transcription is driven by the cytomegalovirus intermediate-early promoter (Chen et al. 2003). Thus, the effect of the repeats is not promoter specific.
Tandem repeats and gene silencing
In contrast to the elevated levels of transcription seen when the CGG•CCG-repeat number in the FMR1 gene is 55–200, the gene becomes silenced when the repeat number exceeds 200 (Pieretti et al. 1991). This silencing is responsible for FXS. Repeat-mediated gene silencing is also responsible for both FRAXE MR (Gu et al. 1996) and FRAX12A MR (Winnepenninckx et al. 2007). In addition to being hypermethylated, the 5′ end of the FMR1 gene is associated with marks of transcriptionally silent chromatin. H3 histones dimethylated at lysine 9 and hypomethylated at lysine 4 accumulate on expanded alleles (Coffee et al. 1999, 2002; Tabolacci et al. 2005). H4K16 is also hypoacetylated (Biacsi et al. 2008). Similar epigenetic changes are also seen in CDM and FRDA (Otten and Tapscott 1995; Thornton et al. 1997; Filippova et al. 2001; Cho et al. 2005; Herman et al. 2006; Greene et al. 2007).
Of the three repeats associated with heterochromatin formation, only CGG•CCG repeats can be methylated. Therefore, DNA methylation cannot be the trigger for all of these chromatin changes as initially envisioned for FXS (Smith et al. 1994; Chen et al. 1995). RNAs ∼21 nucleotides in length that are homologous to the region containing the repeats in the DMPK locus are seen (Cho et al. 2005). These small RNAs are characteristic of the products of Dicer (DICER1) digestion. DICER1 is a key component of the RNA interference (RNAi) pathway. RNAi leads to post-transcriptional gene silencing by targeting homologous mRNAs for degradation or by inhibiting their translation (for review, see Peters and Meister 2007). In some organisms, the RNAi machinery can also be involved in the formation of transcriptionally silent chromatin (Mette et al. 2000).
The presence of these small RNAs would be consistent with a role of an RNAi-like mechanism in heterochromatin formation at the DMPK locus, although a role for DICER1 has yet to be demonstrated. One potential source of dsRNA for this process could be the antisense transcript that is initiated downstream of the repeat (Cho et al. 2005). Interaction of the sense and antisense transcript would then generate a dsRNA substrate for DICER1. A similar sense-antisense pair is possible for the FMR1 gene (Ladd et al. 2007), and the FRDA repeat is upstream of a short interspersed nuclear element that would be transcribed in the antisense direction (Greene et al. 2007). However, another source of dsRNA is present in both CDM and diseases involving long CGG•CCG repeats: Both CGG-RNA and CUG-RNA form stable hairpins involving a mixture of Watson-Crick (WC) and non-WC base pairs (Napierala and Krzyzosiak 1997; Handa et al. 2003; Zumwalt et al. 2007). Despite the presence of the non-WC base pairs, these RNAs are also DICER1 substrates (Handa et al. 2003; Krol et al. 2007). Thus, it may be that the repeats directly contribute to gene silencing. In this regard, it is interesting to remember that every large CGG•CCG repeat that has been described to date is associated with DNA methylation. While antisense transcripts are relatively common, the methylation of all of these repeats may indicate that it is the repeat itself that is the trigger for gene silencing.
There is another human genetic disorder that lends support to the generality of tandem repeat-mediated epigenetic changes in humans. Facioscapulohumeral muscular dystrophy (FSHD1) (OMIM #158900) is caused by contraction of the subtelomeric D4Z4 repeat array on chromosome 4. This array consists of 1–150 large G + C-rich repeats (Wijmenga et al. 1993). Contraction of the array to <11 repeats results in hypomethylation of the region in affected individuals (van Overveld et al. 2003). This relative demethylation is thought to lead to the activation of an otherwise silent gene, whose identity is, as yet, unknown, that has negative effects on muscular development.
A repeat-mediated effect on chromatin structure in these noncoding disorders suggests an unexpected parallel with disorders resulting from expansions in the coding sequence. While the mechanisms involved are distinctly different, the ability of both polyQ and these long noncoding repeats to alter normal chromatin suggests that both groups of diseases are chromatinopathies.
Tandem repeats as blocks to transcription elongation
The GAA•TTC repeat that causes FRDA forms triplexes as well as related structures known as “sticky DNA” (Bidichandani et al. 1998; Sakamoto et al. 1999; Grabczyk and Usdin 2000a, b; Vetcher et al. 2002; Potaman et al. 2004). Experiments in vitro and in bacteria show that during transcription the repeats trap RNA polymerase on the template thus blocking transcription elongation. The pattern of transcription termination and the effects of blocking oligonucleotides are consistent with the formation of some sort of three-stranded structure. This effect on transcription elongation could contribute to the transcription deficit responsible for FRDA. In principle, a similar phenomenon could occur at other polypurine:polypyridimine sequences in the genome, since they too have the potential to form triplexes.
Transcribed repeats as protein traps
Since the symptoms of DM1 and DM2 are similar despite the different genes affected, the mechanism of disease pathology is probably not related specifically to DMPK or CNBP (Ranum and Cooper 2006; Wheeler and Thornton 2007). The expanded repeats in both diseases generate numerous cytoplasmic foci or inclusions in affected cells. These foci include, in addition to the repeat-containing transcript, members of the muscleblind (MBNL) family of splicing proteins (Mankodi et al. 2005). MBNL proteins interact antagonistically with members of the CUGBP1 and CUGBP2-like (CELF) family of splicing factors. The interaction of these different proteins is important for developmentally appropriate RNA splicing. MBNL proteins favor the adult splice isoforms and CUGBP1 favors the retention of fetal exons (Ho et al. 2004). Consistent with the idea that DM pathology results from sequestering of MBNL proteins by the DM RNA, DM1 and DM2 cells have aberrant splicing of cardiac troponin T, chloride channel 1, and insulin receptor transcripts. Furthermore, Mbnl1 nullizygous mice develop symptoms similar to that of DM and have aberrant splicing of the same transcripts (Kanadia et al. 2003). Similar effects are also seen in transgenic mice expressing large numbers of CUG repeats in the transcriptional unit of an unrelated gene (Mankodi et al. 2000), lending support to the idea that it is the CUG-RNA that is responsible for this effect.
FMR1 mRNA-containing intranuclear neuronal inclusions are seen in the brains of individuals with FXTAS (Greco et al. 2002) and knock-in mice (Willemsen et al. 2003; Entezam et al. 2007). An RNA-mediated mechanism of pathology is supported by the observations that RNA with large numbers of CGG repeats is toxic to human cells (Arocena et al. 2005; Handa et al. 2005) and causes neurodegeneration in flies (Jin et al. 2003). Some of the antisense transcripts seen in carriers of alleles with 55–200 repeats also contain the repeat region (Ladd et al. 2007), and CCG repeats also cause neurodegeneration in flies (Sofola et al. 2007a). The antisense transcript may thus also contribute to FXTAS pathology. The FXTAS inclusions contain many proteins (Iwahashi et al. 2006). This makes it difficult to identify a single responsible protein, if there is one. Lamin A/C is one of the proteins found in these inclusions, and cells from FXTAS brains show aberrant lamin localization and abnormal nuclear morphology (Arocena et al. 2005). This has led to the suggestion that FXTAS is a laminopathy. On the other hand, overexpression of Pur-α, HNRNPA2B1, or CUGBP1 suppresses the phenotype of the CGG transgenic fly (Jin et al. 2007; Sofola et al. 2007b). These proteins all interact directly or indirectly with the CGG-RNA, supporting the idea that neurodegeneration may result from the sequestering of such binding proteins. The presence of MBNL1 in the inclusions is interesting given the role of this protein in DM1 and DM2. MBNL1 binds to CCG-RNA but not CGG-RNA (Kino et al. 2004) lending support to the idea that the repeats in the antisense transcript may be significant in FXTAS pathology. This transcript also contains an ORF that encodes polyproline, but whether this peptide is made and whether protein-mediated effects contribute to the pathology remains to be seen.
In addition to a polyQ tract in the ATXN8 gene of individuals with SCA8 (Ikeda et al. 2008), repeat expansion at this locus also results in a long CUG tract in the antisense transcript produced from the ATXN8OS gene. Long CUG repeats cause neurodegeneration in a fly model (Mutsuddi et al. 2004). The symptoms of SCA8 may thus result from the combined effect of protein and RNA-mediated pathology. Interestingly, overexpression of even normal numbers of CUG repeats in this model causes pathology (Mutsuddi et al. 2004), thus demonstrating that even very short repeats may be deleterious if their transcript is abundant.
Tandem repeats as translational regulators
Paradoxical effects on translation have been demonstrated for the CGG•CCG repeats in the 5′ UTR of the FMR1 gene. Short repeat tracts are associated with increased levels of expression of a reporter construct (Chen et al. 2003). This effect may be related to enhanced recruitment of CGG-RNA binding proteins that facilitate translation. In contrast, large numbers of repeats produce a transcript that is translated poorly (Feng et al. 1995). This results in decreased FMR1 protein in humans (Kenneson et al. 2001) and knock-in mice with similar numbers of repeats (Entezam et al. 2007). This decrease may be responsible for some of the FXS-like symptoms seen in carriers of alleles with 55–200 repeats. This negative effect on translation is thought to be due to stalling of the 40S ribosomal subunit in the vicinity of the repeat (Feng et al. 1995). Secondary structures, particularly when they occur in the 5′ UTR, are associated with problems with ribosome scanning. It is thus reasonable to think that the translation deficit is related to the ability of CGG-RNA to form stable RNA hairpins (Handa et al. 2003; Zumwalt et al. 2007). A similar negative effect of CUG repeats on translation has also been reported (Raca et al. 2000). This would be consistent with the ability of CUG-RNA to form stable hairpins (Napierala and Krzyzosiak 1997). CAG-RNA hairpins are also stable (Krol et al. 2007) and may be responsible for the frameshifting and decreased translation seen in repeat expansion diseases where the repeat is in the coding sequence (Davies and Rubinsztein 2006; Brockschmidt et al. 2007). Other repeats involving palindromes or quasipalindromic sequences that can form stable RNA hairpins would be expected to behave in a similar way.
As mentioned previously, some of the disease-associated hairpins are substrates for the ribonuclease DICER1. DICER1 knockdown leads to a 40% increase in the amount of the CUG repeat-containing transcript produced from the DM1 locus (Krol et al. 2007). Even relatively short repeats can have this effect. For example, the levels of transcripts from a HD allele with 44 CAG repeats and transcripts from a SCA1 allele with 53 CAG repeats are twice as high in cells in which DICER1 is depleted, compared with cells with normal levels of DICER1 (Krol et al. 2007). The effect, if any, on disease pathology in these disorders is unclear. However, it does illustrate how even relatively short, transcribed tandem repeats can affect mRNA levels. Furthermore, this effect can be exerted in trans (Krol et al. 2007), raising the possibility that the repeats may also reduce the transcript levels of unrelated genes as well. Once again, other repeats with the potential to form RNA hairpins may have similar effects.
Concluding remarks
The repeat expansion diseases illustrate the myriad ways that tandem repeats can affect gene structure and function: Exonic repeats can have relatively large effects on protein synthesis and function even with small changes in repeat number, while the very much larger part of the genome that is comprised of noncoding sequences creates a large window of opportunity for repeat-mediated effects to occur that do not involve changes in protein sequence.
While genome sequencing efforts are likely to underestimate the prevalence of long repeats because of difficulties with stable cloning of these sequences, a number of relatively long repeat sequences have been identified in a number of sequenced organisms. For example, 402 GAA•TTC-repeat tracts with >99 repeats have been identified in the genome of the malaria vector, Anopheles gambiae, and >44 such repeat tracts have been found on the human X chromosome alone (Subirana and Messeguer 2008). Thus, it is reasonable to project that other examples of strong repeat-mediated effects like those discussed here may emerge with time.
There are many examples of polymorphic tandem repeat tracts with smaller repeat numbers that are overrepresented in patient populations, suggesting a link with disease pathology. This includes those with short repeat units, sometimes known as variable number tandem repeats (VNTRs), and those with much larger repeat units, sometimes known as copy number variants (CNVs). Examples of promoter VNTRs include the VNTR in the insulin gene where alleles with fewer repeats are associated with increased insulin gene transcription (Lucassen et al. 1995). Smaller alleles have been proposed to contribute to susceptibility to diabetes, an elevated body mass index, and metabolic syndrome (O'Dell et al. 1999). An increase in the number of (TAAAA) repeats in the promoter of the sex hormone-binding globulin gene (SHBG) has been linked to cardiac artery disease and polycystic ovary syndrome (Hogeveen et al. 2001; Ferk et al. 2007; Alevizaki et al. 2008). CNVs have also been implicated in a number of disorders including rheumatoid arthritis, bipolar disorder, and early-onset Parkinson disease (for review, see Estivill and Armengol 2007).
While more data is needed to firmly establish the role of these VNTRs and CNVs in disease pathology, there is good reason to think that some could be important modifiers of disease severity or contribute to disease symptoms in polygenic disorders.
Acknowledgments
This work was made possible by funding from the Intramural program of NIDDK (NIH).
Footnotes
Article is online at http://www.genome.org/cgi/doi/10.1101/gr.070409.107.
References
- Alakurtti K., Virtaneva K., Joensuu T., Palvimo J.J., Lehesjoki A.E. Characterization of the cystatin B gene promoter harboring the dodecamer repeat expanded in progressive myoclonus epilepsy, EPM1. Gene. 2000;242:65–73. doi: 10.1016/s0378-1119(99)00550-8. [DOI] [PubMed] [Google Scholar]
- Albrecht A., Mundlos S. The other trinucleotide repeat: Polyalanine expansion disorders. Curr. Opin. Genet. Dev. 2005;15:285–293. doi: 10.1016/j.gde.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Alevizaki M., Saltiki K., Xita N., Cimponeriu A., Stamatelopoulos K., Mantzou E., Doukas C., Georgiou I. The importance of the (TAAAA)n alleles at the SHBG gene promoter for the severity of coronary artery disease in postmenopausal women. Menopause. 2008;15:461–468. doi: 10.1097/gme.0b013e31814faf41. [DOI] [PubMed] [Google Scholar]
- Arlt M.F., Xu B., Durkin S.G., Casper A.M., Kastan M.B., Glover T.W. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol. Cell. Biol. 2004;24:6701–6709. doi: 10.1128/MCB.24.15.6701-6709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt M.F., Durkin S.G., Ragland R.L., Glover T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst.) 2006;5:1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Arocena D.G., Iwahashi C.K., Won N., Beilina A., Ludwig A.L., Tassone F., Schwartz P.H., Hagerman P.J. Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet. 2005;14:3661–3671. doi: 10.1093/hmg/ddi394. [DOI] [PubMed] [Google Scholar]
- Balakumaran B.S., Freudenreich C.H., Zakian V.A. CGG/CCG repeats exhibit orientation-dependent instability and orientation-independent fragility in Saccharomyces cerevisiae. Hum. Mol. Genet. 2000;9:93–100. doi: 10.1093/hmg/9.1.93. [DOI] [PubMed] [Google Scholar]
- Bates E.A., Victor M., Jones A.K., Shi Y., Hart A.C. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 2006;26:2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A., Tassone F., Schwartz P.H., Sahota P., Hagerman P.J. Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum. Mol. Genet. 2004;13:543–549. doi: 10.1093/hmg/ddh053. [DOI] [PubMed] [Google Scholar]
- Biacsi R., Kumari D., Usdin K. SIRT1 inhibition alleviates gene silencing in Fragile X mental retardation syndrome. PLoS Genet. 4:e1000017. doi: 10.1371/journal.pgen.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidichandani S.I., Ashizawa T., Patel P.I. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am. J. Hum. Genet. 1998;62:111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockschmidt F.F., Nothen M.M., Hillmer A.M. The two most common alleles of the coding GGN repeat in the androgen receptor gene cause differences in protein function. J. Mol. Endocrinol. 2007;39:1–8. doi: 10.1677/JME-06-0072. [DOI] [PubMed] [Google Scholar]
- Brylawski B.P., Chastain P.D., Cohen S.M., Cordeiro-Stone M., Kaufman D.G. Mapping of an origin of DNA replication in the promoter of fragile X gene FMR1. Exp. Mol. Pathol. 2007;82:190–196. doi: 10.1016/j.yexmp.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper A.M., Nghiem P., Arlt M.F., Glover T.W. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- Casper A.M., Durkin S.G., Arlt M.F., Glover T.W. Chromosomal instability at common fragile sites in Seckel syndrome. Am. J. Hum. Genet. 2004;75:654–660. doi: 10.1086/422701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain N.L., Driver E.D., Miesfeld R.L. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22:3181–3186. doi: 10.1093/nar/22.15.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastain P.D., Cohen S.M., Brylawski B.P., Cordeiro-Stone M., Kaufman D.G. A late origin of DNA replication in the trinucleotide repeat region of the human FMR2 gene. Cell Cycle. 2006;5:869–872. doi: 10.4161/cc.5.8.2648. [DOI] [PubMed] [Google Scholar]
- Chen X., Mariappan S.V., Catasti P., Ratliff R., Moyzis R.K., Laayoun A., Smith S.S., Bradbury E.M., Gupta G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: Structure and biological implications. Proc. Natl. Acad. Sci. 1995;92:5199–5203. doi: 10.1073/pnas.92.11.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.S., Tassone F., Sahota P., Hagerman P.J. The (CGG)n repeat element within the 5′ untranslated region of the FMR1 message provides both positive and negative cis effects on in vivo translation of a downstream reporter. Hum. Mol. Genet. 2003;12:3067–3074. doi: 10.1093/hmg/ddg331. [DOI] [PubMed] [Google Scholar]
- Cho D.H., Thienes C.P., Mahoney S.E., Analau E., Filippova G.N., Tapscott S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell. 2005;20:483–489. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Christy B., Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proc. Natl. Acad. Sci. 1989;86:8737–8741. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B., Zhang F., Warren S.T., Reines D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999;22:98–101. doi: 10.1038/8807. [DOI] [PubMed] [Google Scholar]
- Coffee B., Zhang F., Ceman S., Warren S.T., Reines D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile X syndrome. Am. J. Hum. Genet. 2002;71:923–932. doi: 10.1086/342931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford D.C., Acuna J.M., Sherman S.L. FMR1 and the fragile X syndrome: Human genome epidemiology review. Genet. Med. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.E., Rubinsztein D.C. Polyalanine and polyserine frameshift products in Huntington's disease. J. Med. Genet. 2006;43:893–896. doi: 10.1136/jmg.2006.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deissler H., Wilm M., Genc B., Schmitz B., Ternes T., Naumann F., Mann M., Doerfler W. Rapid protein sequencing by tandem mass spectrometry and cDNA cloning of p20-CGGBP. A novel protein that binds to the unstable triplet repeat 5′-d(CGG)n-3′ in the human FMR1 gene. J. Biol. Chem. 1997;272:16761–16768. doi: 10.1074/jbc.272.27.16761. [DOI] [PubMed] [Google Scholar]
- Durkin S.G., Arlt M.F., Howlett N.G., Glover T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- Eichler E.E., Holden J.J., Popovich B.W., Reiss A.L., Snow K., Thibodeau S.N., Richards C.S., Ward P.A., Nelson D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- Entezam A., Usdin K. ATR protects the genome against CGG•CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008;36:1050–1056. doi: 10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G.E., Grabczyk E., Nussbaum R.L., Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epplen C., Melmer G., Siedlaczck I., Schwaiger F.W., Maueler W., Epplen J.T. On the essence of “meaningless” simple repetitive DNA in eukaryote genomes. EXS. 1993;67:29–45. doi: 10.1007/978-3-0348-8583-6_3. [DOI] [PubMed] [Google Scholar]
- Estivill X., Armengol L. Copy number variants and common disorders: Filling the gaps and exploring complexity in genome-wide association studies. PLoS Genet. 2007;3:e190. doi: 10.1371/journal.pgen.0030190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Zhang F., Lokey L.K., Chastain J.L., Lakkis L., Eberhart D., Warren S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- Ferk P., Teran N., Gersak K. The (TAAAA)n microsatellite polymorphism in the SHBG gene influences serum SHBG levels in women with polycystic ovary syndrome. Hum. Reprod. 2007;22:1031–1036. doi: 10.1093/humrep/del457. [DOI] [PubMed] [Google Scholar]
- Filippova G.N., Thienes C.P., Penn B.H., Cho D.H., Hu Y.J., Moore J.M., Klesert T.R., Lobanenkov V.V., Tapscott S.J. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 2001;28:335–343. doi: 10.1038/ng570. [DOI] [PubMed] [Google Scholar]
- Freudenreich C.H., Kantrow S.M., Zakian V.A. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- Fry M., Loeb L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. 1994;91:4950–4954. doi: 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M., Usdin K. Human Nucleotide Expansion Diseases. Springer; Heidelberg, Germany: 2006. [Google Scholar]
- Fu Y.H., Kuhl D.P., Pizzuti A., Pieretti M., Sutcliffe J.S., Richards S., Verkerk A.J., Holden J.J., Fenwick R.G., Warren S.T., et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Gallia G.L., Johnson E.M., Khalili K. Purα: A multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res. 2000;28:3197–3205. doi: 10.1093/nar/28.17.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gecz J. FMR3 is a novel gene associated with FRAXE CpG island and transcriptionally silent in FRAXE full mutations. J. Med. Genet. 2000a;37:782–784. doi: 10.1136/jmg.37.10.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gecz J. The FMR2 gene, FRAXE and non-specific X-linked mental retardation: Clinical and molecular aspects. Ann. Hum. Genet. 2000b;64:95–106. doi: 10.1017/S0003480000007983. [DOI] [PubMed] [Google Scholar]
- Glew M.D., Browning G.F., Markham P.F., Walker I.D. pMGA phenotypic variation in Mycoplasma gallisepticum occurs in vivo and is mediated by trinucleotide repeat length variation. Infect. Immun. 2000;68:6027–6033. doi: 10.1128/iai.68.10.6027-6033.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E., Usdin K. Alleviating transcript insufficiency caused by Friedreich's ataxia triplet repeats. Nucleic Acids Res. 2000a;28:4930–4937. doi: 10.1093/nar/28.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E., Usdin K. The GAA•TTC triplet repeat expanded in Friedreich's ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000b;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray S.J., Gerhardt J., Doerfler W., Small L.E., Fanning E. An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol. Cell. Biol. 2007;27:426–437. doi: 10.1128/MCB.01382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco C.M., Hagerman R.J., Tassone F., Chudley A.E., Del Bigio M.R., Jacquemont S., Leehey M., Hagerman P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- Greene E., Mahishi L., Entezam A., Kumari D., Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35:3383–3390. doi: 10.1093/nar/gkm271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Shen Y., Gibbs R.A., Nelson D.L. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat. Genet. 1996;13:109–113. doi: 10.1038/ng0596-109. [DOI] [PubMed] [Google Scholar]
- Hagerman R.J. Lessons from fragile X regarding neurobiology, autism, and neurodegeneration. J. Dev. Behav. Pediatr. 2006;27:63–74. doi: 10.1097/00004703-200602000-00012. [DOI] [PubMed] [Google Scholar]
- Hagerman P.J., Hagerman R.J. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment. Retard. Dev. Disabil. Res. Rev. 2004;10:25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- Handa V., Saha T., Usdin K. The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by DICER1. Nucleic Acids Res. 2003;31:6243–6248. doi: 10.1093/nar/gkg818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa V., Goldwater D., Stiles D., Cam M., Poy G., Kumari D., Usdin K. Long CGG-repeat tracts are toxic to human cells: Implications for carriers of Fragile X premutation alleles. FEBS Lett. 2005;579:2702–2708. doi: 10.1016/j.febslet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Herman D., Jenssen K., Burnett R., Soragni E., Perlman S.L., Gottesfeld J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- Hewett D.R., Handt O., Hobson L., Mangelsdorf M., Eyre H.J., Baker E., Sutherland G.R., Schuffenhauer S., Mao J.I., Richards R.I. FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol. Cell. 1998;1:773–781. doi: 10.1016/s1097-2765(00)80077-5. [DOI] [PubMed] [Google Scholar]
- Ho T.H., Charlet B.N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E., Richon V.M., Woodman B., Smith D.L., Zhou X., Rosa E., Sathasivam K., Ghazi-Noori S., Mahal A., Lowden P.A., et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogeveen K.N., Talikka M., Hammond G.L. Human sex hormone-binding globulin promoter activity is influenced by a (TAAAA)n repeat element within an Alu sequence. J. Biol. Chem. 2001;276:36383–36390. doi: 10.1074/jbc.M104681200. [DOI] [PubMed] [Google Scholar]
- Honda S., Hayashi S., Kato M., Niida Y., Hayasaka K., Okuyama T., Imoto I., Mizutani S., Inazawa J. Clinical and molecular cytogenetic characterization of two patients with non-mutational aberrations of the FMR2 gene. Am. J. Med. Genet. A. 2007;143:687–693. doi: 10.1002/ajmg.a.31638. [DOI] [PubMed] [Google Scholar]
- Hughes R.E. Polyglutamine disease: Acetyltransferases awry. Curr. Biol. 2002;12:R141–R143. doi: 10.1016/S0960-9822(02)00709-1. [DOI] [PubMed] [Google Scholar]
- Ikeda Y., Daughters R.S., Ranum L.P. Bidirectional expression of the SCA8 expansion mutation: One mutation, two genes. Cerebellum. 2008 doi: 10.1080/14734220701413781. [DOI] [PubMed] [Google Scholar]
- Iwahashi C.K., Yasui D.H., An H.J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J., Hagerman P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- Jin P., Zarnescu D.C., Zhang F., Pearson C.E., Lucchesi J.C., Moses K., Warren S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- Jin P., Duan R., Qurashi A., Qin Y., Tian D., Rosser T.C., Liu H., Feng Y., Warren S.T. Pur α binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C., Mullenbach R., Grossfeld P., Auer R., Favier R., Chien K., James M., Tunnacliffe A., Cotter F. Co-localisation of CCG repeats and chromosome deletion breakpoints in Jacobsen syndrome: Evidence for a common mechanism of chromosome breakage. Hum. Mol. Genet. 2000;9:1201–1208. doi: 10.1093/hmg/9.8.1201. [DOI] [PubMed] [Google Scholar]
- Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kang S., Ohshima K., Shimizu M., Amirhaeri S., Wells R.D. Pausing of DNA synthesis in vitro at specific loci in CTG and CGG triplet repeats from human hereditary disease genes. J. Biol. Chem. 1995;270:27014–27021. doi: 10.1074/jbc.270.45.27014. [DOI] [PubMed] [Google Scholar]
- Kenneson A., Zhang F., Hagedorn C.H., Warren S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Kino Y., Mori D., Oma Y., Takeshita Y., Sasagawa N., Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- Kordasiewicz H.B., Gomez C.M. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics. 2007;4:285–294. doi: 10.1016/j.nurt.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Krol J., Fiszer A., Mykowska A., Sobczak K., de Mezer M., Krzyzosiak W.J. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell. 2007;25:575–586. doi: 10.1016/j.molcel.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Ladd P.D., Smith L.E., Rabaia N.A., Moore J.M., Georges S.A., Hansen R.S., Hagerman R.J., Tassone F., Tapscott S.J., Filippova G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- Lalioti M.D., Scott H.S., Buresi C., Rossier C., Bottani A., Morris M.A., Malafosse A., Antonarakis S.E. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature. 1997;386:847–851. doi: 10.1038/386847a0. [DOI] [PubMed] [Google Scholar]
- Lalioti M.D., Scott H.S., Antonarakis S.E. Altered spacing of promoter elements due to the dodecamer repeat expansion contributes to reduced expression of the cystatin B gene in EPM1. Hum. Mol. Genet. 1999;8:1791–1798. doi: 10.1093/hmg/8.9.1791. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Le Beau M.M., Rassool F.V., Neilly M.E., Espinosa R., Glover T.W., Smith D.I., McKeithan T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998;7:755–761. doi: 10.1093/hmg/7.4.755. [DOI] [PubMed] [Google Scholar]
- Liu G., Bissler J.J., Sinden R.R., Leffak M. Unstable spinocerebellar ataxia type 10 (ATTCT)•(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 2007;27:7828–7838. doi: 10.1128/MCB.01276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucassen A.M., Screaton G.R., Julier C., Elliott T.J., Lathrop M., Bell J.I. Regulation of insulin gene expression by the IDDM associated, insulin locus haplotype. Hum. Mol. Genet. 1995;4:501–506. doi: 10.1093/hmg/4.4.501. [DOI] [PubMed] [Google Scholar]
- Mankodi A., Logigian E., Callahan L., McClain C., White R., Henderson D., Krym M., Thornton C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- Mankodi A., Lin X., Blaxall B.C., Swanson M.S., Thornton C.A. Nuclear RNA foci in the heart in myotonic dystrophy. Circ. Res. 2005;97:1152–1155. doi: 10.1161/01.RES.0000193598.89753.e3. [DOI] [PubMed] [Google Scholar]
- Mariappan S.V., Catasti P., Chen X., Ratliff R., Moyzis R.K., Bradbury E.M., Gupta G. Solution structures of the individual single strands of the fragile X DNA triplets (GCC)n•(GGC)n. Nucleic Acids Res. 1996;24:784–792. doi: 10.1093/nar/24.4.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marz P., Probst A., Lang S., Schwager M., Rose-John S., Otten U., Ozbek S. Ataxin-10, the spinocerebellar ataxia type 10 neurodegenerative disorder protein, is essential for survival of cerebellar neurons. J. Biol. Chem. 2004;279:35542–35550. doi: 10.1074/jbc.M405865200. [DOI] [PubMed] [Google Scholar]
- Matsuura T., Yamagata T., Burgess D.L., Rasmussen A., Grewal R.P., Watase K., Khajavi M., McCall A.E., Davis C.F., Zu L., et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- Mette M.F., Aufsatz W., van der Winden J., Matzke M.A., Matzke A.J. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 2000;19:5194–5201. doi: 10.1093/emboj/19.19.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre A.N., Trifiro M.A., Kaufman M., Kazemi-Esfarjani P., Figlewicz D., Rouleau G., Pinsky L. Reduced transcriptional regulatory competence of the androgen receptor in X-linked spinal and bulbar muscular atrophy. Nat. Genet. 1993;5:184–188. doi: 10.1038/ng1093-184. [DOI] [PubMed] [Google Scholar]
- Moseley M.L., Zu T., Ikeda Y., Gao W., Mosemiller A.K., Daughters R.S., Chen G., Weatherspoon M.R., Clark H.B., Ebner T.J., et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- Murray A. Premature ovarian failure and the FMR1 gene. Semin. Reprod. Med. 2000;18:59–66. doi: 10.1055/s-2000-13476. [DOI] [PubMed] [Google Scholar]
- Musio A., Montagna C., Mariani T., Tilenni M., Focarelli M.L., Brait L., Indino E., Benedetti P.A., Chessa L., Albertini A., et al. SMC1 involvement in fragile site expression. Hum. Mol. Genet. 2005;14:525–533. doi: 10.1093/hmg/ddi049. [DOI] [PubMed] [Google Scholar]
- Mutsuddi M., Marshall C.M., Benzow K.A., Koob M.D., Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr. Biol. 2004;14:302–308. doi: 10.1016/j.cub.2004.01.034. [DOI] [PubMed] [Google Scholar]
- Nadel Y., Weisman-Shomer P., Fry M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 1995;270:28970–28977. doi: 10.1074/jbc.270.48.28970. [DOI] [PubMed] [Google Scholar]
- Napierala M., Krzyzosiak W.J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 1997;272:31079–31085. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- Nucifora F.C., Sasaki M., Peters M.F., Huang H., Cooper J.K., Yamada M., Takahashi H., Tsuji S., Troncoso J., Dawson V.L., et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- O'Dell S.D., Bujac S.R., Miller G.J., Day I.N. Associations of IGF2 ApaI RFLP and INS VNTR class I allele size with obesity. Eur. J. Hum. Genet. 1999;7:821–827. doi: 10.1038/sj.ejhg.5200381. [DOI] [PubMed] [Google Scholar]
- O'Dushlaine C.T., Edwards R.J., Park S.D., Shields D.C. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 2005;6:R69. doi: 10.1186/gb-2005-6-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K., Montermini L., Wells R.D., Pandolfo M. Inhibitory effects of expanded GAA•TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J. Biol. Chem. 1998;273:14588–14595. doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Otten A.D., Tapscott S.J. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc. Natl. Acad. Sci. 1995;92:5465–5469. doi: 10.1073/pnas.92.12.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M. The molecular basis of Friedreich ataxia. Adv. Exp. Med. Biol. 2002;516:99–118. doi: 10.1007/978-1-4615-0117-6_5. [DOI] [PubMed] [Google Scholar]
- Peters L., Meister G. Argonaute proteins: Mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- Potaman V.N., Bissler J.J., Hashem V.I., Oussatcheva E.A., Lu L., Shlyakhtenko L.S., Lyubchenko Y.L., Matsuura T., Ashizawa T., Leffak M., et al. Unpaired structures in SCA10 (ATTCT)n•(AGAAT)n repeats. J. Mol. Biol. 2003;326:1095–1111. doi: 10.1016/s0022-2836(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Potaman V.N., Oussatcheva E.A., Lyubchenko Y.L., Shlyakhtenko L.S., Bidichandani S.I., Ashizawa T., Sinden R.R. Length-dependent structure formation in Friedreich ataxia (GAA)n•(TTC)n repeats at neutral pH. Nucleic Acids Res. 2004;32:1224–1231. doi: 10.1093/nar/gkh274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raca G., Siyanova E.Y., McMurray C.T., Mirkin S.M. Expansion of the (CTG)n repeat in the 5′-UTR of a reporter gene impedes translation. Nucleic Acids Res. 2000;28:3943–3949. doi: 10.1093/nar/28.20.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranum L.P., Cooper T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Rassool F.V., Le Beau M.M., Shen M.L., Neilly M.E., Espinosa R., Ong S.T., Boldog F., Drabkin H., McCarroll R., McKeithan T.W. Direct cloning of DNA sequences from the common fragile site region at chromosome band 3p14.2. Genomics. 1996;35:109–117. doi: 10.1006/geno.1996.0329. [DOI] [PubMed] [Google Scholar]
- Riggins G.J., Lokey L.K., Chastain J.L., Leiner H.A., Sherman S.L., Wilkinson K.D., Warren S.T. Human genes containing polymorphic trinucleotide repeats. Nat. Genet. 1992;2:186–191. doi: 10.1038/ng1192-186. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrera A., Garcia F., Giulotto E., Attolini C., Egozcue J., Ponsa M., Garcia M. Evolutionary breakpoints are co-localized with fragile sites and intrachromosomal telomeric sequences in primates. Cytogenet. Genome Res. 2005;108:234–247. doi: 10.1159/000080822. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrera A., Castresana J., Robinson T.J. Is mammalian chromosomal evolution driven by regions of genome fragility? Genome Biol. 2006;7:R115. doi: 10.1186/gb-2006-7-12-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto N., Chastain P.D., Parniewski P., Ohshima K., Pandolfo M., Griffith J.D., Wells R.D. Sticky DNA: Self-association properties of long GAA•TTC repeats in R•R•Y triplex structures from Friedreich's ataxia. Mol. Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- Samadashwily G.M., Raca G., Mirkin S.M. Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- Sherman S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000;97:189–194. doi: 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Smith S.S., Laayoun A., Lingeman R.G., Baker D.J., Riley J. Hypermethylation of telomere-like foldbacks at codon 12 of the human c-Ha-ras gene and the trinucleotide repeat of the FMR-1 gene of fragile X. J. Mol. Biol. 1994;243:143–151. doi: 10.1006/jmbi.1994.1640. [DOI] [PubMed] [Google Scholar]
- Sofola O.A., Jin P., Botas J., Nelson D.L. Argonaute-2-dependent rescue of a Drosophila model of FXTAS by FRAXE premutation repeat. Hum. Mol. Genet. 2007a;16:2326–2332. doi: 10.1093/hmg/ddm186. [DOI] [PubMed] [Google Scholar]
- Sofola O.A., Jin P., Qin Y., Duan R., Liu H., de Haro M., Nelson D.L., Botas J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007b;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Strom C.M., Crossley B., Redman J.B., Buller A., Quan F., Peng M., McGinnis M., Fenwick R.G., Sun W. Molecular testing for Fragile X Syndrome: Lessons learned from 119,232 tests performed in a clinical laboratory. Genet. Med. 2007;9:46–51. doi: 10.1097/gim.0b013e31802d833c. [DOI] [PubMed] [Google Scholar]
- Subirana J.A., Messeguer X. Structural families of genomic microsatellites. Gene. 2008;408:124–132. doi: 10.1016/j.gene.2007.10.025. [DOI] [PubMed] [Google Scholar]
- Subramanian S., Madgula V.M., George R., Mishra R.K., Pandit M.W., Kumar C.S., Singh L. Triplet repeats in human genome: Distribution and their association with genes and other genomic regions. Bioinformatics. 2003;19:549–552. doi: 10.1093/bioinformatics/btg029. [DOI] [PubMed] [Google Scholar]
- Tabolacci E., Pietrobono R., Moscato U., Oostra B.A., Chiurazzi P., Neri G. Differential epigenetic modifications in the FMR1 gene of the fragile X syndrome after reactivating pharmacological treatments. Eur. J. Hum. Genet. 2005;13:641–648. doi: 10.1038/sj.ejhg.5201393. [DOI] [PubMed] [Google Scholar]
- Tassone F., Hagerman R.J., Taylor A.K., Gane L.W., Godfrey T.E., Hagerman P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F., Beilina A., Carosi C., Albertosi S., Bagni C., Li L., Glover K., Bentley D., Hagerman P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C.A., Wymer J.P., Simmons Z., McClain C., Moxley R.T. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997;16:407–409. doi: 10.1038/ng0897-407. [DOI] [PubMed] [Google Scholar]
- Usdin K. NGG-triplet repeats form similar intrastrand structures: Implications for the triplet expansion diseases. Nucleic Acids Res. 1998;26:4078–4085. doi: 10.1093/nar/26.17.4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin K., Woodford K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A., van Leeuwen W., Scherer S., Verbrugh H. Occurrence and structure-function relationship of pentameric short sequence repeats in microbial genomes. Res. Microbiol. 1999;150:617–626. doi: 10.1016/s0923-2508(99)00129-1. [DOI] [PubMed] [Google Scholar]
- van Overveld P.G., Lemmers R.J., Sandkuijl L.A., Enthoven L., Winokur S.T., Bakels F., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- Vetcher A.A., Napierala M., Wells R.D. Sticky DNA: Effect of the polypurine•polypyrimidine sequence. J. Biol. Chem. 2002;277:39228–39234. doi: 10.1074/jbc.M205210200. [DOI] [PubMed] [Google Scholar]
- Virtaneva K., D'Amato E., Miao J., Koskiniemi M., Norio R., Avanzini G., Franceschetti S., Michelucci R., Tassinari C.A., Omer S., et al. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat. Genet. 1997;15:393–396. doi: 10.1038/ng0497-393. [DOI] [PubMed] [Google Scholar]
- Wang Y.H., Gellibolian R., Shimizu M., Wells R.D., Griffith J. Long CCG triplet repeat blocks exclude nucleosomes: A possible mechanism for the nature of fragile sites in chromosomes. J. Mol. Biol. 1996;263:511–516. doi: 10.1006/jmbi.1996.0593. [DOI] [PubMed] [Google Scholar]
- Weitzmann M.N., Woodford K.J., Usdin K. The development and use of a DNA polymerase arrest assay for the evaluation of parameters affecting intrastrand tetraplex formation. J. Biol. Chem. 1996;271:20958–20964. doi: 10.1074/jbc.271.34.20958. [DOI] [PubMed] [Google Scholar]
- Weitzmann M.N., Woodford K.J., Usdin K. The mouse Ms6-hm hypervariable microsatellite forms a hairpin and two unusual tetraplexes. J. Biol. Chem. 1998;273:30742–30749. doi: 10.1074/jbc.273.46.30742. [DOI] [PubMed] [Google Scholar]
- Wells R.D., Ashizawa T. Genetic Instabilities and Neurological Diseases. Elsevier; Oxford, UK: 2006. [Google Scholar]
- Wheeler T.M., Thornton C.A. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- Wijmenga C., Frants R.R., Hewitt J.E., van Deutekom J.C., van Geel M., Wright T.J., Padberg G.W., Hofker M.H., van Ommen G.J. Molecular genetics of facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1993;3:487–491. doi: 10.1016/0960-8966(93)90102-p. [DOI] [PubMed] [Google Scholar]
- Willemsen R., Hoogeveen-Westerveld M., Reis S., Holstege J., Severijnen L.A., Nieuwenhuizen I.M., Schrier M., van Unen L., Tassone F., Hoogeveen A.T., et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 2003;12:949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Winnepenninckx B., Debacker K., Ramsay J., Smeets D., Smits A., FitzPatrick D.R., Kooy R.F. CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 2007;80:221–231. doi: 10.1086/510800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S., Mangelsdorf M., Hewett D., Hobson L., Baker E., Eyre H.J., Lapsys N., Le Paslier D., Doggett N.A., Sutherland G.R., et al. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell. 1997;88:367–374. doi: 10.1016/s0092-8674(00)81875-9. [DOI] [PubMed] [Google Scholar]
- Zumwalt M., Ludwig A., Hagerman P.J., Dieckmann T. Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol. 2007;4:93–100. doi: 10.4161/rna.4.2.5039. [DOI] [PubMed] [Google Scholar]