

Abstract
One of the alluring aspects of examining chromatin modifications in the role of modulating transcription required for long-term memory processes is that these modifications may provide transient and potentially stable epigenetic marks in the service of activating and/or maintaining transcriptional processes. These, in turn, may ultimately participate in the molecular mechanisms required for neuronal changes subserving long-lasting changes in behavior. As an epigenetic mechanism of transcriptional control, chromatin modification has been shown to participate in maintaining cellular memory (e.g., cell fate) and may underlie the strengthening and maintenance of synaptic connections required for long-term changes in behavior. Epigenetics has become central to several fields of neurobiology, where researchers have found that regulation of chromatin modification has a significant role in epilepsy, drug addiction, depression, neurodegenerative diseases, and memory. In this review, we will discuss the role of chromatin modifying enzymes in memory processes, as well as how recent studies in yeast genetics and cancer biology may impact the way we think about how chromatin modification and chromatin remodeling regulate neuronal function.
The role of transcription in long-lasting forms of synaptic plasticity and memory has been actively investigated since initial experiments showing that transcription is required for long-term memory in goldfish nearly 40 years ago (Agranoff et al. 1967). Over the past four decades, technology has allowed researchers to study how individual transcription factors regulate gene expression required for long-term memory processes in finer detail, especially with advances in spatial and temporal control of genetic manipulations. As the molecular mechanisms controlling transcription required for long-term memory processes were being unraveled, researchers focusing solely on mechanisms of transcription were using bioinformatic approaches to show that promoter architecture relies on combinatorial elements for transcription-factor binding and subsequent expression of specific transcription profiles (e.g., Harbison et al. 2004).
Transcription is not occurring on naked DNA, but rather in the context of chromatin, providing an additional level of complexity yet to be fully understood. Chromatin is the protein complex that condenses and organizes genomic DNA. Thus, successful gene activation requires the orchestrated effort of not only transcription factors, but also very specific enzymatic protein complexes that modify chromatin structure. The regulation of chromatin structure is now the focus of intense investigation in several fields of neuroscience, including memory, neurodegenerative diseases, stem-cell research, and drug addiction: illustrating how important a fundamental process like the regulation of chromatin structure has become (for reviews, see Colvis et al. 2005; Levenson and Sweatt 2005; Tsankova et al. 2007). In this review, we will discuss the role of chromatin modifying enzymes in memory processes, as well as how recent studies in yeast genetics and cancer biology may impact the way we think about how chromatin modification and chromatin remodeling regulate neuronal function.
The term “epigenetics” is often used to describe general nongenetic functions, but a clear understanding of mechanisms involved in the regulation of chromatin structure and transcription requires very precise definitions. In one of an excellent set of reviews published in Cell (2007, volume 128), Allis et al. (2007) offered a modern molecular definition of epigenetics as “the sum of the alterations to the chromatin template that collectively establish and propagate different patterns of gene expression (transcription) and silencing from the same genome” (Allis et al. 2007). We will revisit this definition after describing the main mechanisms of altering chromatin structure. The basic repeating unit of chromatin is the nucleosome, which is comprised of a histone octamer that interacts with ∼147 bp of genomic DNA per nucleosome. The histone octamer contains pairs of histones H2A, H2B, H3, and H4 (see Fig. 1A). The amino-terminal tails of these core histone proteins are the sites of numerous post-translational modifications (e.g., acetylation, methylation, phosphorylation) (see Fig. 1A) carried out by an equally large number of histone modifying enzymes (acetyltransferases, deacetylases, methyltransferases, demethylases, kinases, etc.) (for review, see Kouzarides 2007).
Figure 1.
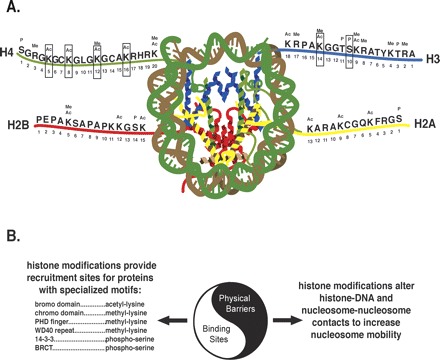
Modifications of residues on histone H4, H3, H2B, and H2A and their two main functions. (A) This schematic illustrates the basic repeating unit of chromatin, called the nucleosome, which is comprised of a histone octamer that interacts with ∼147 bp of genomic DNA per nucleosome. The histone octamer contains pairs of histone H4, H3, H2B, and H2A. The amino-terminal tails of these core histone proteins are the sites of numerous post-translational modifications (figure adapted from Luger et al. 1997 and Kouzarides 2007). Sites marked with a box are those that have been examined with regard to memory processes. (B) This schematic illustrates the two main functions these histone modifications are thought to perform. On the one hand, they are involved in relaxing physical restraints of compact chromatin structure. On the other hand, they provide docking and recruitment sites for additional factors that contain domains capable of binding specific histone modifications, such as the bromo and chromo domains, which bind acetylated lysines and methylated lysines, respectively.
The manipulation of chromatin via the addition of functional groups to histone tails is referred to as chromatin modification, which serves two main purposes (Fig. 1B). The first is to provide recruitment signals for non-histone proteins involved in transcriptional activation and silencing (Kouzarides 2007; Taverna et al. 2007). The second is to relax chromatin by disrupting contacts between nucleosomes and also interactions between histone tails and genomic DNA (Kouzarides 2007). The functional consequence of these modifications will be discussed in the next section. Importantly, chromatin modification should not be confused with chromatin (or nucleosome) remodeling, which refers to ATP-dependent enzymatic complexes (e.g., SWI/SNF, ISWI, INO80, NURD, etc.) that restructure, mobilize, and remove nucleosomes to regulate access to genomic DNA for transcriptional activation (Saha et al. 2006a, b). Chromatin structure may also be manipulated and regulated via histone variant incorporation (e.g., H3.3, macroH2A, H2AZ, H2AX) (Ausio 2006).
Thus, epigenetic regulation of transcription may be thought of as the coordinated interplay of mechanisms (histone modifying enzymes, nucleosome remodeling complexes, histone variant incorporation, DNA methylation, etc.) that translate and integrate incoming signaling events by altering chromatin structure in a specific and precise manner, which in turn regulates gene expression profiles for defined cellular functions. To date, chromatin (or nucleosome) remodeling and histone variant incorporation have not been examined with regard to regulating transcription required during memory processes. Thus, we will focus this review on chromatin modifying enzymes and histone modifications that have been shown to be necessary for regulating transcription required during memory formation.
Histone acetyltransferases (HATs) and memory
Histone acetylation is a chromatin modification critically involved in gene regulation during many neural processes. The enzymes that regulate levels of histone acetylation are histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs acetylate histones, while HDACs deacetylate histones, and together with other modifications they regulate transcription profiles for specific cellular functions (for review, see Kouzarides 2007). One primary function of histone acetylation (as opposed to non-histone acetylation) carried out by HATs is to neutralize the charges on histones to relax chromatin structure, allowing for greater access to the DNA by transcription factors (Norton et al. 1989; Vettese-Dadey et al. 1996). Another main function is to mark chromatin for the recruitment of additional chromatin modifying and remodeling enzymes, and transcription-related proteins, to facilitate and regulate transcriptional processes (Felsenfeld and Groudine 2003). To date, only three HATs have been implicated in the molecular mechanisms underlying memory formation (Table 1). Although CREB-binding protein (CBP) is the most thoroughly studied HAT with regard to learning and memory (Oike et al. 1999; Bourtchouladze et al. 2003; Alarcon et al. 2004; Korzus et al. 2004; Wood et al. 2005, 2006), very recent studies have shown a role for E1A-binding protein (p300) (Oliveira et al. 2006, 2007) and p300/CBP-associated factor (PCAF) (Maurice et al. 2008) in memory processes.
Table 1.
Types of memory deficits observed in genetically modified mice and memory enhancements generated by HDAC inhibition
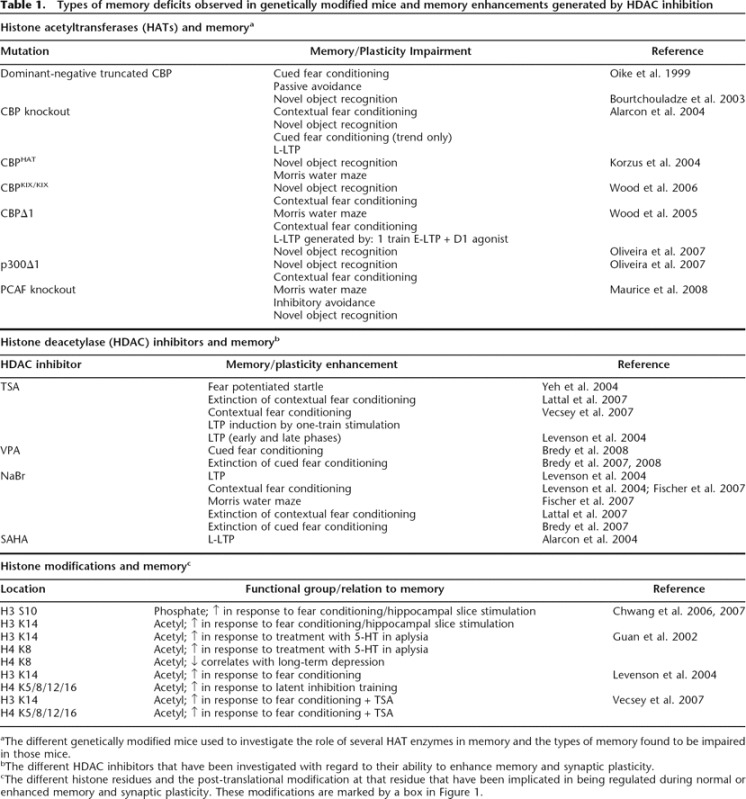
aThe different genetically modified mice used to investigate the role of several HAT enzymes in memory and the types of memory found to be impaired in those mice.
bThe different HDAC inhibitors that have been investigated with regard to their ability to enhance memory and synaptic plasticity.
cThe different histone residues and the post-translational modification at that residue that have been implicated in being regulated during normal or enhanced memory and synaptic plasticity. These modifications are marked by a box in Figure 1.
CBP is well known as a coactivator recruited by cyclicAMP responsive element-binding protein (CREB) via the interaction between the Ser 133 phosphorylated kinase-inducible domain (KID) of CREB and the KIX domain of CBP (Chrivia et al. 1993; for review, see Goodman and Smolik 2000). The first indication that CBP may have a role in memory came from human patients with Rubinstein-Taybi Syndrome (RTS) (Rubinstein and Taybi 1963; Padfield et al. 1968). RTS is caused by mutations in Cbp (Petrij et al. 1995) and is characterized by developmental abnormalities and mental retardation (Rubinstein and Taybi 1963; Padfield et al. 1968; Hennekam et al. 1992; Cantani and Gagliesi 1998).
A number of mouse models generated with various Cbp mutations have provided a great deal of insight into the role of CBP in learning and memory (Table 1). Supporting an important role for CBP in learning and memory, memory impairments have been observed in five different types of Cbp mutant mice: hetereozygous knock-out (Tanaka et al. 1997; Alarcon et al. 2004), dominant-negative (Oike et al. 1999; Bourtchouladze et al. 2003), spatially restricted transgenic dominant-negative (Wood et al. 2005), spatially and temporally restricted conditional transgenic dominant-negative (Korzus et al. 2004), and homozygous knock-in (Wood et al. 2006; Table 1).
The first Cbp mutant mice contained a mutant allele of Cbp, resulting in a deletion of amino acids 29–265, effectively reducing CBP expression to 50% of wild-type levels (Tanaka et al. 1997). The homozygous mutants died between 8 and 10 d post-fertilization, demonstrating a critical role for CBP in development (Tanaka et al. 1997). The heterozygous mutant mice exhibited physical abnormalities similar to those seen in RTS patients. Oike et al. (1999) generated Cbp mutant mice expressing one wild-type allele of Cbp and one truncated allele of Cbp, which lacked the C terminus of CBP, including the HAT domain. Embryonic lethality was observed in the homozygous mice. The heterozygous dominant-negative mice showed significant developmental abnormalities as observed in RTS patients as well as memory impairments. However, to study the role of CBP in memory independently of its role in development, it became essential to generate animals with more specific spatial and temporal regulation of mutations in Cbp.
Several genetically modified Cbp mutant mice have been generated that do not exhibit developmental abnormalities. These include conditional transgenic mice expressing a dominant-negative Cbp allele with a point mutation in the HAT domain, which is spatially restricted by the CaMKII promoter and temporally restricted by the tetracycline system (Korzus et al. 2004); mice expressing a dominant-negative C-terminal truncation mutant under control of the CaMKII promoter to restrict transgene expression spatially to forebrain neurons in transgenic mice (Wood et al. 2005); and homozygous knock-in mice expressing Cbp alleles carrying a triple point mutation in the KIX domain of CBP (Wood et al. 2006). All of these Cbp mutant mice exhibit normal development and significant impairments in specific forms of synaptic plasticity and long-term memory.
The specific details of the memory deficits differ in all of the Cbp mutant mice generated thus far, but this is not surprising in light of the differences in the types of mutations and expression patterns (summarized in Table 1). CBP dominant-negative mice exhibit impaired cued fear conditioning, yet normal memory for contextual fear and spatial learning in the water maze (Oike et al. 1999). In contrast, transgenic mice expressing a similar dominant-negative CBP mutant transgene from the CaMKII promoter exhibit deficits in memory for contextual fear and spatial learning and memory in the Morris water maze, yet normal cued fear conditioning (Wood et al. 2005). CBP heterozygous knockout mice exhibit impaired memory for contextual fear, but normal memory for cued fear and spatial learning and memory in the water maze (Alarcon et al. 2004). In homozygous knock-in mice expressing mutant CBP carrying a triple point mutation in the KIX domain, contextual fear conditioning is impaired, but cued fear conditioning is spared (Wood et al. 2006). Out of all the currently studied genetically modified Cbp mutant mice, the most sophisticated are the conditional transgenic mice generated by Korzus et al. (2004). These mice express a Cbp transgene expressing a mutant CBP protein with a point mutation in the HAT domain, which is spatially restricted by the CaMKII promoter and temporally restricted by the tetracycline system. These mice exhibit normal memory for contextual and cued fear conditioning, but impaired spatial learning and memory in the water maze. Interestingly, all of the different types of genetically modified Cbp mutant mice studied to date exhibit deficits in long-term memory for novel object recognition (Bourtchouladze et al. 2003; Alarcon et al. 2004; Korzus et al. 2004; Wood et al. 2006; Oliveira et al. 2007). This evidence suggests that brain regions required for long-term memory of novel object recognition (including the perirhinal cortex, hippocampus, amygdala, and insular cortex) may be particularly sensitive to alterations in CBP activity and histone acetylation (Ennaceur and Aggleton 1997; Murray and Mishkin1998; Brown and Aggleton 2001; Mumby et al. 2002; Stupien et al. 2003; Broadbent et al. 2004; Winters et al. 2004; Bermudez-Rattoni et al. 2005).
Results from hippocampal long-term potentiation experiments demonstrate that CBP is necessary for several forms of hippocampal L-LTP, but not E-LTP (Alarcon et al. 2004; Wood et al. 2005) and behavioral studies show that CBP is required for hippocampus-dependent memory as well (Alarcon et al. 2004; Wood et al. 2005, 2006). These findings suggest that the hippocampus is particularly sensitive to alterations in CBP activity and histone acetylation. Additional studies are necessary now, using more focally restricted manipulations of CBP, to understand the role of CBP and histone acetylation in different brain regions and memory systems.
Examination of the different genetically modified Cbp mutant mice has also revealed which domains of CBP are critical for learning and memory. CBP has two functional domains that are particularly critical for transcription, the HAT domain and the CREB binding domain (the KIX domain). The conditional transgenic dominant-negative mice generated by Korzus et al. (2004) express a transgene in which CBP carries a point mutation in the HAT domain of CBP, which disrupts CBP HAT activity. These mice were used to demonstrate the role of the CBP HAT domain in long-term memory formation. The CBP C-terminal truncation mutants lacking the HAT domain expressed in dominant-negative mice generated by Oike et al. (1999) and transgenic dominant-negative mice generated by Wood et al. (2005) also indicate a role for the HAT domain in memory processes. Homozygous knock-in mice expressing mutant CBP with a triple point mutation in the KIX domain exhibit long-term memory impairments and decreased expression of specific CRE-containing genes (Wood et al. 2006). These results demonstrate a role for the KIX domain in transcriptional processes required for memory formation. Future studies examining the role of the bromo domain of CBP, the domain used by CBP to interact with acetylated lysine residues, will be very interesting with regard to how chromatin modifications (including acetylation) recruit chromatin modifying enzymes directly to modulate transcription. This idea will be discussed in greater detail below.
In early studies, p300 and CBP were thought to have mostly redundant functions because they are highly homologous and share several conserved domains. In vitro experiments demonstrated that they interact with the same transcription factors (for review, see Goodman and Smolik 2000). However, recent in vivo studies suggest that they have nonredundant functions during embryogenesis and hematopoiesis (Tanaka et al. 1997; Yao et al. 1998; Kasper et al. 2002, 2006; for review, see Kalkhoven 2004). Using genetically modified p300 and CBP mutant mice, a study comparing heterozygous p300 mice (in which one allele of p300 carries a triple point mutation in the KIX domain) to heterozygous CBP mice (in which one allele of Cbp carries an analogous triple point mutation in the KIX domain) demonstrated a differential role for p300 and CBP in motor-skill learning (Oliveira et al. 2006). A more recent study using conditional transgenic mice expressing an inhibitory truncated form of p300, lacking the HAT domain, that is spatially regulated by the CaMKII promoter and temporally regulated by the tetracycline system, showed that p300 is required for long-term recognition and contextual fear memory (Oliveira et al. 2007). These memory deficits are very similar to what has been observed in several different types of genetically modified CBP mutant mice, suggesting functional overlap in vivo with regard to some, but not all types of memory processes.
To date, there are only a handful of studies examining the expression of CBP and p300 in the brain. Several studies have shown CBP (Stromberg et al. 1999; Chung et al. 2002; Fiore and Gannon 2003; Wood et al. 2006) and p300 (Fiore and Gannon 2003; Oliveira et al. 2007) to be expressed ubiquitously in forebrain neurons, including the cortex and hippocampus. Thus, differences between CBP and p300 with regard to memory are not simply explained by differential expression patterns. It is more likely that even though they share similar functions/interactions/activities, they are also known to be distinct at the level of enzymatic activity, both with regard to their different interaction partners (Perissi et al. 1999) as well as their different histone lysine residue targets (McManus and Hendzel 2003).
To investigate the role of PCAF in learning, memory, and stress, Maurice et al. (2008) examined traditional PCAF knockout mice. At 2 mo of age, PCAF knockout mice exhibited impairments in short-term memory, but not long-term memory. At 6 and 12 mo of age, PCAF knockout mice exhibit long-term memory impairments as measured by water maze and passive avoidance performance. PCAF knockout mice also exhibit altered response to stress, which was associated with increased plasma corticosterone levels. Thus, as with any germ line knockout mouse, there are several factors that may affect the development and/or performance of these knockout mice, which may confound interpretation of data in tasks used to indirectly measure memory. Future studies using more precise manipulations of PCAF will be essential for understanding the role of this HAT in learning and memory.
It is important to note that although the HATs described above (CBP, p300, and PCAF) are well known for acetylating histones, they can also acetylate non-histone proteins, which then regulate transcription and memory. One example of a non-histone HAT target is CREB, with three of the five lysines within CREB’s activation domain being targets of CBP. Acetylation of CREB by CBP leads to enhancement of CREB-mediated gene expression (Lu et al. 2003). CBP can also acetylate NFκB, which leads to enhanced long-term memory, but not short-term memory (Yeh et al. 2004). Together with PCAF, CBP regulates both the transcriptional activation and deactivation of INFβ by acetylating specific residues within the INFβ enhanceosome (Munshi et al. 1998). p53 is another protein regulated by CBP/p300-dependent acetylation (Gu and Roeder 1997). p53 acetylation results in stronger binding of p53 to DNA, which correlates with increased transcriptional activity. Clearly, the role of CBP as an acetyltransferase is not limited to histones, and as with any HAT, the non-histone acetylation substrates should be kept in mind.
Histone deacetylases (HDACs) and memory
In general, HAT activity and histone acetylation have been associated with transcriptional activation, whereas HDAC activity and histone deacetylation have been linked to transcriptional repression (Jenuwein and Allis 2001; Narlikar et al. 2002). These two types of histone modifying enzymes function together, balancing one another, to modulate gene expression (Mahlknecht and Hoelzer 2000; Guan et al. 2002; Soejima et al. 2004; Clayton et al. 2006). Because mutations in CBP and decreases in histone acetylation result in memory impairments, it was quickly realized that HDAC inhibitors may be able to induce a histone hyperacetylated state that would compensate for deficits in CBP activity in CBP mutant mice and even enhance synaptic plasticity and memory in wild-type mice (summarized in Table 1).
Indeed, studies have shown that memory deficits due to mutations of CBP can be ameliorated pharmacologically using HDAC inhibitors. Alarcon et al. (2004) demonstrated that the HDAC inhibitor SAHA could ameliorate contextual fear-conditioning memory impairments in CBP heterozygous knock-out mice. Similarly, Korzus et al. (2004) showed that the HDAC inhibitor TSA could ameliorate deficits in novel object recognition memory in conditional transgenic CBP mutant mice. These studies demonstrate that HDAC inhibitors may be considered an important therapeutic for treating cognitive disorders associated with impairments in CBP activity, as in Rubinstein-Taybi Syndrome.
These studies agree with previous findings in the Huntington’s disease (HD) literature, in which pathology is associated with impaired CBP activity and decreased histone acetylation. HDAC inhibition has been shown to increase histone acetylation, arrest ongoing progressive neuronal degeneration, and reduce lethality in a Drosophila model of HD (Steffan et al. 2001), as well as ameliorate motor deficits in a mouse model of HD (Hockly et al. 2003). These studies suggest that it is the effect of HDAC inhibition on histone acetylation that is important. However, as discussed above with regard to non-histone substrates of HATs, it may also involve non-histone substrates of HDACs. Huntington’s disease is also characterized by altered microtubule-dependent transport of organelles, including vesicles containing brain-derived neurotrophic factor (BDNF) (Gauthier et al. 2004). Recently, HDAC inhibition was shown to increase vesicular transport of BDNF via increased acetylation of α-tubulin (Dompierre et al. 2007). The authors demonstrate that HDAC inhibition, via TSA, specifically increases lys40 acetylation of α-tubulin in an HDAC6-dependent manner, and that HDAC inhibition may thus ameliorate vesicular transport defects in HD. Taken together, these studies suggest that HDAC inhibition may affect both histone and non-histone acetylation mechanisms with regard to ameliorating certain pathology in HD. HDAC inhibitors are currently being tested in clinical trials with regard to neurological disorders, some of which have been published (Mercuri et al. 2004; Brahe et al. 2005; Hogarth et al. 2007;).
With regard to enhancing memory in normal mice, HDAC inhibitors have been found to enhance long-term memory for contextual fear conditioning (Levenson et al. 2004; Fischer et al. 2007; Vecsey et al. 2007), spatial learning in the water maze (Fischer et al. 2007), fear potentiated startle (Yeh et al. 2004), extinction of fear memory (Bredy et al. 2007; Lattal et al. 2007; Bredy and Barad 2008), and synaptic plasticity (Alarcon et al. 2004; Levenson et al. 2004; Vecsey et al. 2007). One major open question that arises from these studies is what is the mechanism by which HDAC inhibitors enhance memory and synaptic plasticity? This question is answered in part by two very recent studies. In the first, Fischer et al. (2007) found that HDAC inhibition induced histone hyperacetylation correlating with dendritic sprouting and increased number of synapses, suggesting a cellular mechanism by which HDAC inhibitors may modulate memory. In the second, Vecsey et al. (2007), using homozygous CREB knockout mice as well as homozygous knock-in mice expressing CBP carrying a triple point mutation in the CREB-binding (KIX) domain of CBP, have shown that CREB and the interaction between CREB and CBP is necessary for HDAC inhibition to enhance long-term memory and hippocampal long-term potentiation. In this study, different types of genetically modified Cbp mutant mice were essential to demonstrate that at least one wild-type allele of Cbp is required to mediate the effects of HDAC inhibition. Thus, simply inducing a histone hyperacetylated state is not sufficient, most likely because of the role CBP plays in recruiting basal transcription machinery. Understanding the molecular and cellular mechanisms underlying the activity of HDAC inhibitors in neurons will be pivotal to the future design and use of the next generation of HDAC inhibitors with increased specificity, ability to cross the blood-brain barrier, and cell permeability.
Dynamic interactions between histone modifying enzymes and nucleosome remodeling complexes: Essential for neuronal function?
The interplay between HATs and HDACs involved in regulating transcription required for synaptic plasticity was elegantly demonstrated in a study in Aplysia. Guan et al. (2002) used a bifurcated single sensory neuron-two motor neuron culture preparation (Martin et al. 1997; Casadio et al. 1999) to measure synapse-specific long-term depression and facilitation (forms of synaptic plasticity in Aplysia). Long-term depression was induced using FMRFa, a neuropeptide related to enkephalins, which was shown to require the repressor CREB2. Long-term facilitation was induced using 5-HT (serotonin), which is a form of facilitation known to require the activator CREB1 (Bartsch et al. 1998). The bifurcated prep allowed the authors to induce long-term depression at one synapse and long-term facilitation at another to determine the effect of one form of plasticity on the other, as well as examine the underlying mechanisms of signal integration.
Guan et al. (2002) found that long-term depression could over-ride long-term facilitation. To understand how, they examined the molecular events taking place at the C/EBP promoter in dissected ganglia. They found that under 5-HT-induced long-term facilitation, histone acetylation at histones H4 Lys 8 and H3 Lys 14 significantly increased, and that this increase in acetylation correlated with the recruitment of CBP at the C/EBP promoter. 5-HT is thought to lead to PKA-dependent phosphorylation of CREB1, which serves as an inducible recruitment signal for CBP. In contrast, FMRFa induced long-term depression correlated with recruitment of the repressor CREB2 and HDAC5. Thus, the dynamic interplay between HAT and HDACs appears to be critical for the integration of neuronal signaling events underlying synaptic plasticity. However, the molecular mechanism involved in the regulation of transcription during synaptic plasticity and memory formation are likely to be much more complicated, as suggested by the interdependence of chromatin modifying enzymes (e.g., HATs, HDACs) and chromatin remodeling enzymes (e.g., SWI/SNF, BAF).
The coordinated recruitment of chromatin modifying enzymes and chromatin remodeling enzymes has been described in detail with respect to viruses and yeast, where the resulting complex is called the enhanceosome (Merika and Thanos 2001). A number of studies (Munshi et al. 1998, 2001; Agalioti et al. 2000, 2002; Lomvardas and Thanos 2002) have described the temporal and spatial relationships between different transcription factors, chromatin modifiers, and chromatin remodelers. The order of recruitment earlier in transcription can alter later recruitment, which allows the enhanceosome to accommodate variations in nucleosomal architecture. Acetylation by CBP can facilitate nucleosome remodeling by the mammalian SWI/SNF (or BAF complex), which in turn is necessary for the binding of other transcription factors and can accelerate their recruitment. The placement of nucleosomes can alter the necessity for certain parts of the enhanceosome. For example, if a nucleosome is blocking a core promoter, then remodeling machinery is necessary for transcription to occur. However, if the nucleosome is elsewhere, then this machinery is not as critical. The enhanceosome, including CBP, transcription factors, and chromatin remodeling machines thus works in an integrated fashion (see Agalioti et al. 2000, Fig. 6 for schematic model; for review, see Merika and Thanos 2001).
The order of CBP recruitment is crucial for its functionality. Agalioti et al. (2000) demonstrated the ordered recruitment leading to IFN-β transcription in response to viral infection. First, there is initial acetylation of the nucleosome by GCN5, a HAT, which allows for recruitment of the CBP–PolII holoenzyme complex. CBP further acetylates the nucleosome and recruits SWI/SNF, which is a nucleosome remodeling complex. Nucleosome remodeling facilitates transcription by moving the nucleosome out of the way of transcriptional machinery. Because SWI/SNF has no sequence specificity (Vignali et al. 2000), recruitment by CBP and the involvement of the already assembled enhanceosome lends the necessary specificity. Therefore, CBP cannot be recruited without the proper composition of the enhanceosome, but CBP itself is necessary for completion of the enhanceosome.
It is likely that, although these large enhanceosome complexes have been studied mainly in yeast, they are also necessary for regulation of the transcriptional regulation of learning and memory. This aspect of transcriptional regulation has so far been overlooked in studies of learning and memory. Its critical involvement in other models, however, suggests that it may be a central piece of the puzzle, an idea supported by the existence of neuron-specific chromatin remodeling complexes (nBAF complexes) described by Crabtree and colleagues (for review, see Kouzarides 2007; Wu et al. 2007).
Stable and transient histone modifications
One of the alluring aspects of examining chromatin modifications in the role of modulating transcription required for long-term memory processes is that these modifications may provide transient as well as stable epigenetic marks in the service of activating and/or maintaining transcriptional processes. These in turn may ultimately participate in the molecular mechanisms required for neuronal changes subserving long-lasting changes in behavior. As an epigenetic mechanism of transcription, chromatin modification in combination with DNA methylation has been shown to maintain cellular memory (e.g., cell fate) (for review, see Turner 2002; Hirose 2007) and may underlie the strengthening and maintenance of synaptic connections required for long-term changes in behavior (e.g., maternal behavior) (Meaney et al. 2007) and memory (Miller and Sweatt 2007; Miller et al. 2007). While a role for rapid DNA methylation in regulating transcription required for memory has been demonstrated (Miller and Sweatt 2007; Miller et al. 2007), no stable DNA or histone modifications have been demonstrated to be involved in memory processes. Perhaps this is because at each level, molecular, cellular, systems, etc., there are emergent properties specific to each level, and thus, correlating stable epigenetic marks to remote long-term memories will be impossible. On the other hand, it may be that investigation of other histone modifications will reveal long-lasting modification changes. Currently, the best-studied modifications are those with reliable site-specific antibodies raised against them. As those tools improve, so may our understanding of transient versus stable modifications. Regardless, all current studies demonstrate that transient histone modifications are observed during memory consolidation and there are several very interesting possibilities for these transient modifications. For example, they may play an integral part of epigenetically “bookmarking” genes that have recently been transcriptionally activated. This mechanism has been described in the yeast literature as molecular short-term memory (for review, see Turner 2003) and may be one type of mechanism by which timing of activity-dependent events may be processed.
Investigating how chromatin modification, chromatin remodeling, and DNA methylation regulate transcription required for long-term memory addresses a crucial, and still unanswered question in neurobiology of how long-lasting changes in behavior may occur. As we understand more about different chromatin modifying and remodeling enzymes, types of histone modifications, DNA methylation, and the incredible combinatorial complexity of epigenetics, the epigenome will hopefully reveal fundamental insight into the molecular mechanism of memory processes as well as avenues to rational therapeutic design for human cognitive diseases.
Acknowledgments
We thank K.M. Lattal, C. Miller, and T. Miyashita for helpful discussions and critical reading of the manuscript. We apologize to our colleagues whose work was not included in this review due to space limitations. This work was supported by a pre-doctoral Training Program in Cellular and Molecular Neuroscience fellowship (R.M.B.; PI: Arthur D. Lander, T32 NS007444-7) and by the Whitehall Foundation (M.A.W.).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.917508.
References
- Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- Agalioti T., Chen G., Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- Agranoff B.W., Davis R.E., Casola L., Lim R. Actinomycin D blocks formation of memory of shock-avoidance in goldfish. Science. 1967;158:1600–1601. doi: 10.1126/science.158.3808.1600. [DOI] [PubMed] [Google Scholar]
- Alarcon J.M., Malleret G., Touzani K., Vronskaya S., Ishii S., Kandel E.R., Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: A model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Allis C.D., Jenuwein T., Reinberg D. Epigenetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2007. [Google Scholar]
- Ausio J. Histone variants—The structure behind the function. Brief. Funct. Genomic Proteomic. 2006;5:228–243. doi: 10.1093/bfgp/ell020. [DOI] [PubMed] [Google Scholar]
- Bartsch D., Casadio A., Karl K.A., Serodio P., Kandel E.R. CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 1998;95:211–223. doi: 10.1016/s0092-8674(00)81752-3. [DOI] [PubMed] [Google Scholar]
- Bermudez-Rattoni F., Okuda S., Roozendaal B., McGaugh J.L. Insular cortex is involved in consolidation of object recognition memory. Learn. Mem. 2005;12:447–449. doi: 10.1101/lm.97605. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R., Lidge R., Catapano R., Stanley J., Gossweiler S., Romashko D., Scott R., Tully T. A mouse model of Rubinstein-Taybi syndrome: Defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl. Acad. Sci. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahe C., Vitali T., Tiziano F.D., Angelozzi C., Pinto A.M., Borgo F., Moscato U., Bertini E., Mercuri E., Neri G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur. J. Hum. Genet. 2005;13:256–259. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- Bredy T.W., Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn. Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy T.W., Wu H., Crego C., Zellhoefer J., Sun Y.E., Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy T.W., Barad M. The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem. 2008;15:39–45. doi: 10.1101/lm.801108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent N.J., Squire L.R., Clark R.E. Spatial memory, recognition memory, and the hippocampus. Proc. Natl. Acad. Sci. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.W., Aggleton J.P. Recognition memory: What are the roles of the perirhinal cortex and hippocampus? Nat. Rev. Neurosci. 2001;2:51–61. doi: 10.1038/35049064. [DOI] [PubMed] [Google Scholar]
- Cantani A., Gagliesi D. Rubinstein-Taybi syndrome. Review of 732 cases and analysis of the typical traits. Eur. Rev. Med. Pharmacol. Sci. 1998;2:81–87. [PubMed] [Google Scholar]
- Casadio A., Martin K.C., Giustetto M., Zhu H., Chen M., Bartsch D., Bailey C.H., Kandel E.R. A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell. 1999;99:221–237. doi: 10.1016/s0092-8674(00)81653-0. [DOI] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok R.P., Lamb N., Hagiwara M., Montminy M.R., Goodman R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Chung Y.H., Kim E.J., Shin C.M., Joo K.M., Kim M.J., Woo H.W., Cha C.I. Age-related changes in CREB binding protein immunoreactivity in the cerebral cortex and hippocampus of rats. Brain Res. 2002;956:312–318. doi: 10.1016/s0006-8993(02)03562-x. [DOI] [PubMed] [Google Scholar]
- Chwang W.B., O’Riordan K.J., Levenson J.M., Sweatt J.D. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang W.B., Arthur J.S., Schumacher A., Sweatt J.D. The nuclear kinase mitogen- and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J. Neurosci. 2007;14:268–276. doi: 10.1523/JNEUROSCI.2522-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A.L., Hazzalin C.A., Mahadevan L.C. Enhanced histone acetylation and transcription: A dynamic perspective. Mol. Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Colvis C.M., Pollock J.D., Goodman R.H., Impey S., Dunn J., Mandel G., Champagne F.A., Mayford M., Korzus E., Kumar A., et al. Epigenetic mechanisms and gene networks in the nervous system. J. Neurosci. 2005;25:10379–10389. doi: 10.1523/JNEUROSCI.4119-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre J.P., Godin J.D., Charrin B.C., Cordelieres F.P., King S.J., Humbert S., Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A., Aggleton J.P. The effects of neurotoxic lesions of the perirhinal cortex combined to fornix transection on object recognition memory in the rat. Behav. Brain Res. 1997;88:181–193. doi: 10.1016/s0166-4328(97)02297-3. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G., Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Fiore P., Gannon R.L. Expression of the transcriptional coactivators CBP and p300 in the hamster suprachiasmatic nucleus: Possible molecular components of the mammalian circadian clock. Brain Res. Mol. Brain Res. 2003;111:1–7. doi: 10.1016/s0169-328x(02)00663-0. [DOI] [PubMed] [Google Scholar]
- Fischer A., Sananbenesi F., Wang X., Dobbin M., Tsai L.H. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Goodman R.H., Smolik S. CBP/p300 in cell growth, transformation, and development. Genes & Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Gu W., Roeder R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Guan Z., Giustetto M., Lomvardas S., Kim J.H., Miniaci M.C., Schwartz J.H., Thanos D., Kandel E.R. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111:483–493. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- Harbison C.T., Gordon D.B., Lee T.I., Rinaldi N.J., Macisaac K.D., Danford T.W., Hannett N.M., Tagne J.B., Reynolds D.B., Yoo J., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekam R.C., Baselier A.C., Beyaert E., Bos A., Blok J.B., Jansma H.B., Thorbecke-Nilsen V.V., Veerman H. Psychological and speech studies in Rubinstein-Taybi syndrome. Am. J. Ment. Retard. 1992;96:645–660. [PubMed] [Google Scholar]
- Hirose S. Crucial roles for chromatin dynamics in cellular memory. J. Biochem. 2007;141:615–619. doi: 10.1093/jb/mvm092. [DOI] [PubMed] [Google Scholar]
- Hockly E., Richon V.M., Woodman B., Smith D.L., Zhou X., Rosa E., Sathasivam K., Ghazi-Noori S., Mahal A., Lowden P.A., et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth P., Lovrecic L., Krainc D. Sodium phenylbutyrate in Huntington's disease: A dose-finding study. Mov. Disord. 2007;22:1962–1964. doi: 10.1002/mds.21632. [DOI] [PubMed] [Google Scholar]
- Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Kasper L.H., Boussouar F., Ney P.A., Jackson C.W., Rehg J., van Deursen J.M., Brindle P.K. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature. 2002;419:738–743. doi: 10.1038/nature01062. [DOI] [PubMed] [Google Scholar]
- Kasper L.H., Fukuyama T., Biesen M.A., Boussouar F., Tong C., de Pauw A., Murray P.J., van Deursen J.M., Brindle P.K. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol. Cell. Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E., Rosenfeld M.G., Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lattal K.M., Barrett R.M., Wood M.A. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav. Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J.M., Sweatt J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson J.M., O'Riordan K.J., Brown K.D., Trinh M.A., Molfese D.L., Sweatt J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lomvardas S., Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 2002;110:261–271. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- Lu Q., Hutchins A.E., Doyle C.M., Lundblad J.R., Kwok R.P. Acetylation of cAMP-responsive element-binding protein (CREB) by CREB-binding protein enhances CREB-dependent transcription. J. Biol. Chem. 2003;278:15727–15734. doi: 10.1074/jbc.M300546200. [DOI] [PubMed] [Google Scholar]
- Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mahlknecht U., Hoelzer D. Histone acetylation modifiers in the pathogenesis of malignant disease. Mol. Med. 2000;6:623–644. [PMC free article] [PubMed] [Google Scholar]
- Martin K.C., Casadio A., Zhu H., Yaping E., Rose J.C., Chen M., Bailey C.H., Kandel E.R. Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: A function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Maurice T., Duclot F., Meunier J., Naert G., Givalois L., Meffre J., Celerier A., Jacquet C., Copois V., Mechti N., et al. Altered memory capacities and response to stress in p300/CBP-associated factor (PCAF) histone acetylase knockout mice. Neuropsychopharmacology. 2008;33:1584–1602. doi: 10.1038/sj.npp.1301551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus K.J., Hendzel M.J. Quantitative analysis of CBP- and P300-induced histone acetylations in vivo using native chromatin. Mol. Cell. Biol. 2003;23:7611–7627. doi: 10.1128/MCB.23.21.7611-7627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney M.J., Szyf M., Seckl J.R. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol. Med. 2007;13:269–277. doi: 10.1016/j.molmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Mercuri E., Bertini E., Messina S., Pelliccioni M., D'Amico A., Colitto F., Mirabella M., Tiziano F.D., Vitali T., Angelozzi C., et al. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul. Disord. 2004;14:130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Merika M., Thanos D. Enhanceosomes. Curr. Opin. Genet. Dev. 2001;11:205–208. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- Miller C.A., Sweatt J.D. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Miller C.A., Campbell S.L., Sweatt J.D. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol. Learn. Mem. 2007;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby D.G., Gaskin S., Glenn M.J., Schramek T.E., Lehmann H. Hippocampal damage and exploratory preferences in rats: Memory for objects, places, and contexts. Learn. Mem. 2002;9:49–57. doi: 10.1101/lm.41302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi N., Merika M., Yie J., Senger K., Chen G., Thanos D. Acetylation of HMG I(Y) by CBP turns off IFNβ expression by disrupting the enhanceosome. Mol. Cell. 1998;2:457–467. doi: 10.1016/s1097-2765(00)80145-8. [DOI] [PubMed] [Google Scholar]
- Munshi N., Agalioti T., Lomvardas S., Merika M., Chen G., Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293:1133–1136. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- Murray E.A., Mishkin M. Object recognition and location memory in monkeys with excitotoxic lesions of the amygdala and hippocampus. J. Neurosci. 1998;18:6568–6582. doi: 10.1523/JNEUROSCI.18-16-06568.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar G.J., Fan H.Y., Kingston R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Norton V.G., Imai B.S., Yau P., Bradbury E.M. Histone acetylation reduces nucleosome core particle linking number change. Cell. 1989;57:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- Oike Y., Hata A., Mamiya T., Kaname T., Noda Y., Suzuki M., Yasue H., Nabeshima T., Araki K., Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: Implications for a dominant-negative mechanism. Hum. Mol. Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Oliveira A.M., Abel T., Brindle P.K., Wood M.A. Differential role for CBP and p300 CREB-binding domain in motor skill learning. Behav. Neurosci. 2006;120:724–729. doi: 10.1037/0735-7044.120.3.724. [DOI] [PubMed] [Google Scholar]
- Oliveira A.M., Wood M.A., McDonough C.B., Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn. Mem. 2007;14:564–572. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padfield C.J., Partington M.W., Simpson N.E. The Rubinstein-Taybi syndrome. Arch. Dis. Child. 1968;43:94–101. doi: 10.1136/adc.43.227.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V., Dasen J.S., Kurokawa R., Wang Z., Korzus E., Rose D.W., Glass C.K., Rosenfeld M.G. Factor-specific modulation of CREB-binding protein acetyltransferase activity. Proc. Natl. Acad. Sci. 1999;96:3652–3657. doi: 10.1073/pnas.96.7.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F., Giles R.H., Dauwerse H.G., Saris J.J., Hennekam R.C., Masuno M., Tommerup N., van Ommen G.J., Goodman R.H., Peters D.J., et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Rubinstein J.H., Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am. J. Dis. Child. 1963;105:588–608. doi: 10.1001/archpedi.1963.02080040590010. [DOI] [PubMed] [Google Scholar]
- Saha A., Wittmeyer J., Cairns B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006a;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- Saha A., Wittmeyer J., Cairns B.R. Mechanisms for nucleosome movement by ATP-dependent chromatin remodeling complexes. Results Probl. Cell Differ. 2006b;41:127–148. doi: 10.1007/400_005. [DOI] [PubMed] [Google Scholar]
- Soejima H., Joh K., Mukai T. Gene silencing in DNA damage repair. Cell. Mol. Life Sci. 2004;61:2168–2172. doi: 10.1007/s00018-004-4178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Stromberg H., Svensson S.P., Hermanson O. Distribution of CREB-binding protein immunoreactivity in the adult rat brain. Brain Res. 1999;818:510–514. doi: 10.1016/s0006-8993(98)01219-0. [DOI] [PubMed] [Google Scholar]
- Stupien G., Florian C., Roullet P. Involvement of the hippocampal CA3-region in acquisition and in memory consolidation of spatial but not in object information in mice. Neurobiol. Learn. Mem. 2003;80:32–41. doi: 10.1016/s1074-7427(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Naruse I., Maekawa T., Masuya H., Shiroishi T., Ishii S. Abnormal skeletal patterning in embryos lacking a single Cbp allele: A partial similarity with Rubinstein-Taybi syndrome. Proc. Natl. Acad. Sci. 1997;94:10215–10220. doi: 10.1073/pnas.94.19.10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S.D., Li H., Ruthenburg A.J., Allis C.D., Patel D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N., Renthal W., Kumar A., Nestler E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Turner B.M. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- Turner B.M. Memorable transcription. Nat. Cell Biol. 2003;5:390–393. doi: 10.1038/ncb0503-390. [DOI] [PubMed] [Google Scholar]
- Vecsey C.G., Hawk J.D., Lattal K.M., Stein J.M., Fabian S.A., Attner M.A., Cabrera S.M., McDonough C.B., Brindle P.K., Abel T., et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettese-Dadey M., Grant P.A., Hebbes T.R., Crane-Robinson C., Allis C.D., Workman J.L. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- Vignali M., Hassan A.H., Neely K.E., Workman J.L. ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters B.D., Forwood S.E., Cowell R.A., Saksida L.M., Bussey T.J. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: Heterogeneity of function within the temporal lobe. J. Neurosci. 2004;24:5901–5908. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M.A., Kaplan M.P., Park A., Blanchard E.J., Oliveira A.M., Lombardi T.L., Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn. Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M.A., Attner M.A., Oliveira A.M., Brindle P.K., Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn. Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.I., Lessard J., Olave I.A., Qiu Z., Ghosh A., Graef I.A., Crabtree G.R. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron. 2007;56:94–108. doi: 10.1016/j.neuron.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Yao T.P., Oh S.P., Fuchs M., Zhou N.D., Ch'ng L.E., Newsome D., Bronson R.T., Li E., Livingston D.M., Eckner R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- Yeh S.H., Lin C.H., Gean P.W. Acetylation of nuclear factor-κB in rat amygdala improves long-term but not short-term retention of fear memory. Mol. Pharmacol. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]