

Abstract
Cornelia de Lange syndrome (CdLS) is a genetically heterogeneous disorder characterized by growth retardation, intellectual disability, upper limb abnormalities, hirsutism, and characteristic facial features. In this study we explored the occurrence of intragenic NIPBL copy number variations (CNVs) in a cohort of 510 NIPBL sequence-negative patients with suspected CdLS. Copy number analysis was performed by custom exon-targeted oligonucleotide array-comparative genomic hybridization and/or MLPA. Whole-genome SNP array was used to further characterize rearrangements extending beyond the NIPBL gene. We identified NIPBL CNVs in 13 patients (2.5%) including one intragenic duplication and a deletion in mosaic state. Breakpoint sequences in two patients provided further evidence of a microhomology-mediated replicative mechanism as a potential predominant contributor to CNVs in NIPBL. Patients for whom clinical information was available share classical CdLS features including craniofacial and limb defects. Our experience in studying the frequency of NIBPL CNVs in the largest series of patients to date widens the mutational spectrum of NIPBL and emphasizes the clinical utility of performing NIPBL deletion/duplication analysis in patients with CdLS.
Keywords: Array-CGH, copy number variation, Cornelia de Lange, MLPA, NIPBL
Introduction
Cornelia de Lange syndrome (CdLS [MIM 122470]) is a multisystem disorder characterized by characteristic facial features, growth retardation, intellectual disability, limb reduction defects, hirsutism, and moderate-to-severe neurodevelopmental delay (Kline et al. 1993). There is marked heterogeneity in the CdLS phenotype. At one end of the spectrum are individuals with classical CdLS features exhibiting profound growth and neurodevelopmental delay, sometimes accompanied by severe limb defects. Less severe growth retardation and developmental delay have been observed in mildly affected individuals. The prevalence of CdLS is estimated to be 1:10,000 live births, but the incidence may be underestimated given the existence of undiagnosed individuals with milder phenotypes.
Mutations in the NIPBL gene (MIM 608667) have been identified in ∼60% of classical CdLS patients (Krantz et al. 2004; Tonkin et al. 2004). NIPBL, located on chromosome 5p13, encodes for the human ortholog of Drosophila Nipped-B belonging to the family of chromosomal adherins, which are regulators of chromatin cohesion and enhancer–promoter communication in Drosophila (Rollins et al. 1999, 2004). Genotype–phenotype correlation studies have demonstrated that NIPBL mutation-positive patients tend to have a more severe phenotype than mutation-negative patients with truncating mutations, generally causing a more severe phenotype than missense mutations based on limb differences, growth, and cognitive function (Gillis et al. 2004). This suggests that NIPBL is a dosage-sensitive gene. Recently, somatic mosaicism of NIPBL mutations has been described as a relatively frequent occurrence (Huisman et al. 2013). Mutations in SMC1A (MIM 300040) and SMC3 (MIM 606062) account for ∼5% of patients with a milder variant of CdLS (Musio et al. 2006; Deardorff et al. 2007). More recently, de novo mutations in RAD21 (MIM 614701) and HDAC8 (MIM 300269) have been identified in individuals with growth retardation, minor skeletal anomalies, and cognitive and facial features consistent with those caused by mutations in NIPBL (Deardorff et al. 2012a,b2012b). The molecular etiology of the remaining CdLS cases remains unknown.
While the majority of mutations in Mendelian disorders are detected by sequence analysis, intragenic deletions and duplications are becoming an increasingly significant factor in elucidating the molecular etiology of many of these conditions (Aradhya et al. 2012). The identification of a wide spectrum of NIPBL mutations has made the molecular analysis of the NIPBL gene a routine component of the clinical and laboratory evaluation of patients with a suspected CdLS phenotype. Recent studies have shown that intragenic deletions in NIPBL are present in ∼2–5% of patient with CdLS (Bhuiyan et al. 2007; Pehlivan et al. 2012; Russo et al. 2012). Thus, the presence of NIPBL copy number changes has become an important factor to consider in CdLS molecular testing in suspected patients with negative NIPBL sequencing results. In this study we explored the occurrence of NIPBL copy number alterations in a cohort of 510 NIPBL sequence-negative patients referred to our laboratory for CdLS molecular diagnostic testing. We identified 13 cases with copy number alterations in the NIPBL gene, including one intragenic duplication and a deletion in the mosaic state. The size of this patient group is the largest among similar previously reported studies.
Material and Methods
Patient samples
The patient group consisted of 510 patients with clinical features consistent with CdLS in whom no NIPBL mutation was identified by sequence analysis in our laboratory. Genomic DNA was isolated from blood leukocytes on the AutoGenFlex STAR robotic workstation (Autogen, Holliston, MA) or using the MagNA Pure Compact DNA isolation system (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions.
Array-CGH
Deletion/duplication analysis of the NIPBL gene was performed using a high-resolution, exon-targeted 8X60K array-comparative genomic hybridization (CGH) platform (Agilent Technologies, Santa Clara, CA) designed to detect copy number changes in 53 genes including NIPBL. The array contained 2416 probes spanning the NIPBL gene and flanking 1-kb upstream and downstream regions with an average resolution of ∼1 probe/80 bp across the entire NIPBL locus. Genomic DNA samples of the patients and gender-matched controls were processed and cohybridized onto microarray slides according to the manufacturer's recommended procedures (Agilent Technologies). Microarray images were scanned at 2 μm resolution and the data were extracted using ImaGene (9.0) and analyzed using the Nexus software (6.0) (BioDiscovery, Hawthorne, CA). The genomic copy number was defined by analysis of the normalized log 2 (Cy5/Cy3) ratio average of the CGH signal. Regions that reached a log 2 threshold of at least −0.32 were considered losses consistent with deletion, and thresholds of at least 0.26 were considered gains consistent with duplication.
MLPA
Multiplex ligation-dependent probe amplification (MLPA) analysis was performed using the SALSA P141/P142 MLPA kit (MRC-Holland, Amsterdam, the Netherlands) in accordance with the manufacturer's instructions. Ligation products were polymerase chain reaction (PCR) amplified and resolved on an ABI-3730 genetic analyzer (Life Technologies, Carlsbad, CA). For quantitative analysis, peak heights of the patient and normal control were analyzed using GeneMarker Software (Soft Genetics Inc., State College, PA). Peak heights outside the range 0.7–1.3 times the control peak height were considered abnormal, with those below 0.7 representing deletions and those above 1.3 representing duplications.
Whole-genome microarray
Whole-genome array analysis was performed using Affymetrix CytoScan HD arrays (Affymetrix, Santa Clara, CA). The CytoScan HD array contains around 2.6 million probes including 7,50,000 single nucleotide polymorphisms (SNPs) and 1.9 million nonpolymorphic markers. Whole-genome array analysis was performed according to the manufacturer's recommended protocol. Images were acquired using the GeneChip Scanner 3000 7G and analyzed using Chromosome Analysis Suite (ChAS) 1.2.3 software (Affymetrix). Human genome build 19 was used for annotation.
Breakpoint junction sequence analyses
Break-point analysis of Patients 7 and 8 was performed by PCR primer walking using Taq polymerase and the Expand Long Template PCR System (Roche Applied Science). PCR primers were designed from the reference sequence, GenBank accession number NM_133433, across the deleted and duplicated regions derived from the array-CGH and MLPA results assuming the most likely rearrangements. PCR products were sequenced in both forward and reverse directions on an ABI 3730 DNA Analyzer (Life Technologies). Sequences were compared with the NIPBL reference sequence (NM_133433) using Mutation Surveyor software version 3.01 (Soft Genetics Inc.).
Results
In total, 13/510 patients (2.5%) were found to harbor NIPBL structural variations (Table 1).
Table 1.
NIPBL copy number variations identified by targeted CGH/whole-genome arrays.
| Patient | Gender | Genotype (NCBI build 37) | Included region | CNV type | Minimum size (kb) |
|---|---|---|---|---|---|
| 1 | Female | chr5:g.(37005025_37005435)_(37007490_37007565)del | Ex 17–18 | Deletion | 2.0 |
| 2 | Male | chr5:g.(36952230_36952266)_(36956007_36956380)del | Ex 2–3 | Deletion | 3.7 |
| 3 | Male | chr5:g.(36993420_36993499)_(36997655_36997816)del | Ex 11 | Deletion | 4.1 |
| 4 | Female | chr5:g.(37061350_37062360)_(37066620_?)del | Ex 46–47 | Deletion | 4.3 |
| 5 | Male | chr5:g.(37061350_37062360)_(37066620_?)del | Ex 46–47 | Deletion | 4.3 |
| arr 5p13.2(37,065,152-37,089,599)x1** | 24.4** | ||||
| 6 | Female | chr5:g.(37044305_37044340)_(37049000_37049158)del | Ex 35–39 | Deletion | 4.7 |
| 7 | Female | chr5:g.(36995835_36995861)_(37004025_37004935)del* | Ex 12–14* | Deletion | 5.0 |
| 8 | Female | chr5:g.(37015062_37015875)_(37021035_37021455)dup | Ex 23–27 | Duplication | 5.2 |
| 9 | Female | chr5:g.(36968915_36969370)_(37004450_37005025)del | Ex 7–16 | Deletion | 35.1 |
| 10 | Female | chr5:g.(36964200_36965948)_(37010235_37010791)del | Ex 7–21 | Deletion | 44.3 |
| 11 | Male | chr5:g.(36997300_36997336)_(37066620_?)del | Ex 12–47 | Deletion | 69.3 |
| arr 5p13.2(36,994,939-37,188,352)x1** | 193.4** | ||||
| 12 | Male | chr5:g.(36900635_36903625)_(36998050_36998150)del | Ex 2–11 | Deletion | 94.4 |
| 13 | Male | chr5:g.(?_36877675)_(37066620_?)del | Whole gene | Deletion | 189.0 |
| arr 5p13.1p13.2(35,232,614-40,365,530)x1** | 5.1 (Mb)** |
Deletion in mosaic state.
Nomenclature/size based on SNP array results. NIPBL RefSeq NM_133433.
NIPBL deletions
Patients 1–3 and 6 had relatively small single-and multiexon intragenic deletions ranging from ∼2 to ∼5 kb in size. Patients 9, 10, and 12 were found to have larger multiexon intragenic NIPBL deletions ranging from ∼35 to ∼94 kb in size. Patients 4, 5, and 11 had deletions involving the last coding exon of NIPBL and extending downstream of the gene. To further characterize the extent of the deletions in Patients 5 and 11, whole-genome microarray analysis was performed (Fig. 1). In Patient 5, a 24.4 kb deletion was identified at cytogenetic band position 5p13.2 that included the terminal region of NIPBL and extended past the gene into the intergenic region (arr 5p13.2(37,065,152-37,089,599)x1). A 193.4 kb deletion (arr 5p13.2(36,994,939-37,188,352)x1) was observed in Patient 11 and included exons 12–47 of NIBPL to exons 22–52 of C5orf42 (NM_023073.3), transcribed in reverse orientation to NIPBL. Patient 13 had a deletion encompassing the entire NIPBL gene. Whole-genome microarray analysis revealed that the deletion spanned ∼5.1 Mb and involved NIPBL and 22 other genes at the cytogenetic band position 5p13.1p13.2 (arr 5p13.1p13.2 (35,232,614-40,365,530)x1). No additional DNA was available to further characterize the extent of the deletion detected in Patient 4.
Figure 1.

Characterization of deletions involving NIPBL by whole-genome microarray analysis in Patients 5, 11, and 13. In Patient 5 (Pt_5), a 24.4 kb deletion extending from the distal end of NIPBL into the intergenic region was observed. In Patient 11 (Pt_11), a 193.4 kb deletion was detected and extended to C5orf42. In Patient 13 (Pt_13), the deletion spanned 5.1 Mb and included NIPBL and 22 other genes. The copy number state segments (red for deletion) and copy number state data are shown for each patient. Genes involved are indicated. Not drawn to scale.
In addition, a deletion of exons 12–14 in an apparent mosaic state was found in Patient 7. The array-CGH data showed low-level reduction in the Cy5/Cy3 fluorescence log 2 ratio of oligonucleotide probes interrogating exons 11–16 (Fig. 2A). The decreased log 2 (Cy5/Cy3) fluorescence ratios did not reach the lower defined threshold value of −0.32 and therefore this aberration was not called by the analysis software. MLPA analysis showed a ∼30% decrease in signal intensity for probes specific for NIPBL exons 12, 13, and 14, supporting the finding of mosaicism (Fig. 2B). Various combinations of PCR primer pairs were designed to amplify the putative deletion breakpoint junction based on the array-CGH and MLPA results. A unique 5 kb PCR product was obtained in the patient and not in the control when using primers 5′ and 3′ of introns 11 and 14, respectively. Sequence analysis of the PCR product revealed a 4968 base-pair deletion of chr5: 36,997,269-37,002,237 that included NIPBL exons 12–14 with a 4-bp microhomology (AGGA) at the breakpoint junction (Fig. 2C). No other tissue was available for the study.
Figure 2.
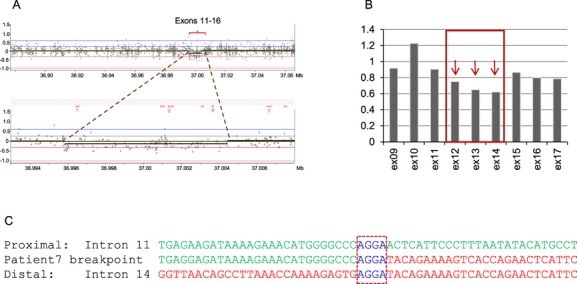
Break-point analysis of the mosaic deletion of NIPBL exons 12–14 in Patient 7. (A) Array-CGH profile (top) with the magnified region of interest (bottom) showing low-level reduction in the Cy5/Cy3 fluorescence log 2 ratio of oligonucleotide probes spanning exons 11–16; (B) MLPA histogram: NIPBL exons arrowed in red (12–14) showing reduced height ratio in comparison with control probes (∼0.7 vs. 1); (C) nucleotide sequence of the break points. Proximal reference sequence and patient break-point sequence that match with the proximal reference sequence are shown in green, whereas the distal reference sequence and patient break-point sequence that match with the distal reference sequence are shown in red. Dash boxed sequence corresponds to a region of microhomology and reveals the break-point junction.
NIPBL duplications
One novel multiexon NIPBL duplication was also identified in this cohort: a ∼5 kb duplication of NIPBL exons 23–27 in Patient 8. The breakpoint junction of the duplication detected by array-CGH and MLPA (Fig. 3A and B) was successfully amplified by long-range PCR using primers positioned at the very end of the duplication breakpoints, as determined by array-CGH, under the assumption that the repeated copies were arranged in tandem (Fig. 3C). Sequence analysis demonstrated that exons 23–27 were duplicated in tandem and revealed a 7-bp insertion (ATATAAT) and a 1-bp microhomology (T) at the breakpoint junction (Fig. 3D). Follow-up MLPA analysis of the patient's parents confirmed that the duplication was de novo in the patient.
Figure 3.

Breakpoint analysis of the duplication of NIPBL exons 23–27 in Patient 8. (A) Array-CGH results revealed a duplication of NIPBL exons 23–27; (B) MLPA histogram: NIPBL exons arrowed in red (23–27) showing increased height ratio (∼1.4 vs. 1); (C) schematic diagram of the duplicated region with dashed square boxes representing the exons duplicated in tandem. Arrow heads indicate the location of PCR primers used to amplify the breakpoint junction of the duplication; (D) Nucleotide sequence of the breakpoint revealing insertion (purple) of seven nucleotides (ATATAAT) and 1bp-microhomology (dash box) at the breakpoint junction; (E) Photo of Patient 8 taken at two weeks of age.
Phenotypes of CdLS patients with genomic rearrangements in NIPBL
Clinical information was collected for 10 of the 13 patients with NIPBL deletions/duplications and is presented in Table S1. Information on growth, development, craniofacial, limb abnormalities, as well as other systemic/organ involvement was requested. Age of patients ranged from 1 day to 19 years old. Complete clinical information was not available for all parameters requested and due to the young ages (1 day to 3 months) of the majority of patients, information on intellectual deficiencies and developmental delay were not fully ascertained. All reported patients had facial features consistent with CdLS, regardless of the size or location of their deletion/duplication. All patients on whom information was available had a thin upper lip, long smooth philtrum, upturned nares, and all but one had synophyrs and long thick eyelashes. All reported patients had characteristic limb abnormalities ranging from mild (fifth finger clinodactyly and 2–3 toe syndactyly) to severe (monodactyly and missing forearms). Other recurrent systems affected included cardiovascular defects, hearing loss, genitourinary anomalies, and gastroesophageal reflux, all of which are features seen in patients with CdLS.
Discussion
We identified 13 copy number variations (CNVs) in a cohort of 510 CdLS cases. Copy number analysis was performed utilizing high-density array-CGH targeted to the NIPBL gene, whole-genome SNP array, and MLPA. Deletions were found to be more common, with 12 deletions and 1 duplication identified. The CNVs ranged in size from 2 kb to 5.1 Mb with the minimum affecting one exon to the maximum affecting NIPBL and other adjacent genes at 5p13 (Fig. 4).
Figure 4.
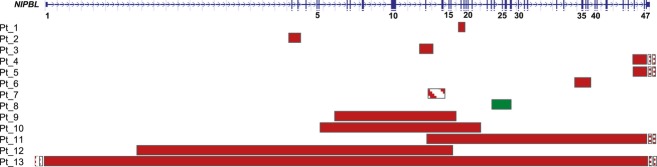
Schematic diagram of the NIPBL gene displaying exonic deletions and duplications in 13 patients. On top: graphic view of 47 exons (vertical blue bars) of NIPBL: solid horizontal bars represent NIPBL genomic regions deleted (red) or duplicated (green) and approximate sizes. The deletion in mosaic state in Patient 7 is indicated by a striped red bar. Narrow dotted bars indicate rearrangements extending beyond the gene in Patients 4, 5, 11, and 13. The graphical data for each patient were obtained by inputting the most distal and proximal oligonucleotide genomic probe coordinates into the custom track at the University of California, Santa Cruz website: http://genome.ucsc.edu/cgi-bin/hgGateway.
The CNVs identified in this study appear to not have been previously described in the literature supporting the broad allelic heterogeneity of NIPBL mutations in CdLS. An exception could be possibly represented by the 4.1 kb deletion of exon 11 observed in Patient 3 as a deletion similar in size and location has been reported previously (Pehlivan et al. 2012), and the apparently similar deletions of exons 46 and 47 identified in Patients 4 and 5. Interestingly, comparison of the sequences flanking the break points, based on array-CGH coordinates in our Patient 3 and sequence information available for the previously reported patient, revealed the presence of Alu sequences with ∼80% sequence similarity (AluY and AluJo) in the proximity of the borders of both deletions. Moreover, an enrichment of repetitive elements (AluJb and AluSq2) and MER2 DNA elements was also noted in the region of intron 45 (chr5: 37061350–37062360) containing the proximal breakpoint of the exons 46–47 deletions found in Patients 4 and 5. As previously suggested (Stankiewicz et al. 2003; Lupski 2007), it is possible that repetitive elements may play a role in predisposing some of these NIPBL regions to structural instability, although whether these motifs have any mechanistic role in the formation of some NIPBL deletions has yet to be determined.
Partial NIPBL gene deletions extending beyond the 5′ end of the gene were observed in Patients 4, 5, and 11. Whole-genome microarray analysis performed in Patients 5 and 11 (no additional DNA was available for Patient 4) revealed that the deletion in Patient 11 included the terminal part of the C5orf42 gene. Point mutations of the C5orf42 gene have recently been associated with autosomal recessive Joubert syndrome (Srour et al. 2012). As Joubert syndrome is a recessively inherited multisystemic disorder, we feel that its involvement in this deletion is unlikely to contribute to this patient's phenotype.
A large deletion involving the entire NIPBL gene was identified in Patient 13. This deletion spans ∼5 Mb in the 5p13.1-13.2 region and encompasses NIPBL and 22 other genes as indicated by whole-genome SNP array analysis. Physical examination of Patient 13 showed the presence of the classical craniofacial features of CdLS, bilateral upper limb reduction, genital abnormalities, bilateral hydronephrosis, Dandy Walker malformation, and developmental delay. The complexity of this patient's phenotype can be attributed to the combined haploinsufficiency of dosage-sensitive genes located within the deletion; however, the fact that the patient has classic CdLS features suggests that NIPBL is the major dosage-sensitive gene. Large deletions involving the NIPBL gene have been reported previously, and while phenotypic heterogeneity exists between patients, all display minimally diagnostic features of CdLS (Russo et al. 2012; Gervasini et al. 2013b).
Somatic mosaicism has previously been described in patients with CdLS and is not an uncommon occurrence in the mutational landscape of the NIPBL gene (Huisman et al. 2013). The array-CGH and MLPA data of mild-to-medium probe signal reductions infer the mechanism of somatic mosaicism for a deletion involving exons 12–14 in Patient 7. Breakpoint sequence analysis confirmed the presence of the deletion and revealed a 4-bp microhomology at the break-point junction consistent with a possible replicative mechanism such as FoSTes/microhomology-mediated break-induced replication as previously suggested (Pehlivan et al. 2012). A case of somatic mosaicism for a frameshift mutation in NIPBL has been reported previously in a patient showing a phenotype milder than that predicted by a truncating mutation (Castronovo et al. 2010). More recently, high level of mosaicism for a large deletion encompassing exons 2–32 of the NIPBL gene was identified in a patient with severe CdLS phenotype (Gervasini et al. 2013a). Our patient demonstrates typical CdLS facial features, but no severe limb reduction defects or other major abnormalities. The clinical phenotype of Patient 7 was comparable with other NIPBL deletion patients and was reported to be “classic CdLS” by the referring physician. A caveat is that the clinical examination of Patient 7 was done at 3 months of age; therefore, the cognitive and developmental information was limited and additional follow-up of the patient was not performed. Other tissues were not available for study and therefore we cannot exclude that the levels of somatic mosaicism might be higher in other tissues, thus leading to further functional impairments.
Intragenic duplications in the NIPBL gene appear to be rarer than deletions, with only one other patient harboring a single-exon duplication being reported recently (Russo et al. 2012). In this study, we identified a previously unreported de novo duplication of NIPBL exons 23–27 in Patient 8. Breakpoint analysis revealed the presence of a 7-bp insertion flanked by 1-bp microhomology, again suggesting a microhomology-mediated replicative mechanism as a potential predominant contributor to this rearrangement. No clinical information was available for the patient reported by Russo et al. (2012) with a duplication of exon 32 to compare with our duplication patient. 5q13 duplication syndrome [MIM#613174] is generally considered a distinct phenotype than the one observed in CdLS patients, and recently Novara et al. (2013) reported a patient with a 5p13 duplication including the NIPBL gene only and observed overlapping features with 5p13 microduplication syndrome. The phenotype of Patient 8 did not diverge dramatically from the classic CdLS spectrum. The patient's characteristic CdLS features included synophyrs, hirsutism, low posterior hairline, flat nasal bridge and upturned nose, thin upper lip, and downturn corners of the mouth (Fig. 3E). While 5p13 microduplication patients present with long fingers, large hands and feet, Patient 8 had small fingers and hands, and complete 2–3 toe syndactyly. In addition, Patient 8 had complete, unbalanced AV canal with hypoplastic left heart, and whereas congenital heart malformations occur in 25% of patients with CdLS, none have been reported to date in patients with 5p13 microduplication syndrome. The exons 23–27 duplication in Patient 8 is predicted to result in an out-of-frame protein and is a plausible cause of the patient's CdLS phenotype.
As CdLS is a well-described multiple malformation syndrome, we compared several CdLS facial features in our patient group with previously reported patients with NIPBL point mutations (Table S1). All patients for whom information was available shared the characteristic facial features of CdLS, including thin upper lip, long smooth philtrum, upturned nares, and all but one had synophyrs and long thick eyelashes. These facial features are the clinical hallmark of CdLS with synophyrs in 98%, long thick eyelashes in 99%, thin upper lip in 94%, and upturned nares in 85% of affected individuals (Kline et al. 2007). Cleft palate, which was identified in 2/7 (29%) of patients in our study, is seen in ∼20% of patients with CdLS (Kline et al. 2007). No significant differences were identified in the presence of these features in our patients with NIPBL CNVs and in patients with NIPBL point mutations (Borck et al. 2007; Schoumans et al. 2007).
Previous studies analyzing genotype–phenotype correlation in mutation-positive cases have suggested that patients with missense mutations are associated with milder phenotypes than those with truncating mutations (Gillis et al. 2004). Thus, we have further explored the possible genotype–phenotype correlation in our patient cohort. The majority of our patient cohort fell within the moderate-to-mild range (Table S2). Some correlation can be seen with regards to the size of deletion and severity of growth retardation. Patients with larger multiexonic deletions, like Patient 11 (69 kb deletion), had severe growth retardation, and patients with smaller single-exon deletions, like Patient 3 (4.1 kb deletion), had mild growth retardation. The severity pattern of limb reduction defects of our patient cohort is also similar and more in line with missense mutation patients (Table S2). One explanation of this is that several of our intragenic deletion cases (Patient 1, Patient 3, and Patient 7), which correlated with mild limb reduction defects, are predicted to result in in-frame deletions that may still lead to the formation of a NIPBL protein with some residual function. In addition, Patient 2, who also had mild limb reduction defects, has a deletion of exons 2–3. As exon 2 contains the primary start codon, it is possible that a downstream start codon at c.334 in exon 4 may possibly be utilized to initiate protein translation. While the deletion in Patient 6 is predicted to result in an out-of-frame deletion, the milder limb defects observed in this patient could be related to the smaller size of this patient's deletion and potentially to some functional aspect of the protein being preserved due to its more distal location within the gene. The large size of the deletions in Patients 9, 10, and 13 correlate with greater upper limb involvement and is consistent with the proposal that phenotypic severity is proportional to number of exons involved (Pehlivan et al. 2012). Other modifying factors, either at the NIPBL locus or at other genomic sites, may also play a role in the severity of limb reduction defects. Importantly, as all but three of our patients were 3 months old or younger, the lack of information regarding cognitive function is likely due to the inability to make such evaluations at the time of assessment.
In this study we further documented the heterogeneity of NIPBL genomic rearrangements in a cohort of 510 sequence-negative CdLS cases and identified NIPBL copy number aberrations in 13 (2.5%) unrelated patients. Our detection rate is lower than previously described studies, which identified NIPBL structural rearrangements in ∼5% of mutation-negative patients (Pehlivan et al. 2012; Russo et al. 2012). This is potentially explained by the wide phenotypic variability of patients sent to our laboratory for NIPBL analysis for diagnostic testing purposes. Our detection rate likely represents the true mutation detection rate of NIPBL copy number changes in the clinical setting.
In summary, we have shown that intragenic NIPBL deletion/duplication events are not uncommon in CdLS patients and result in a similar phenotype to patients with NIPBL point mutations. Our data contribute additional information regarding the NIPBL mutation spectrum in CdLS and emphasize the utility of NIPBL deletion and duplication analysis in the molecular diagnosis of CdLS, especially in the absence of identifiable NIPBL point mutations.
Conflict of Interest
None declared.
Funding Information
No funding information provided.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Clinical features of 10 patients with copy number variations in NIPBL. The final column summarizes the facial features of CdLS patients identified with point mutations in NIBPL in comparison with the facial features of our patient cohort (second to last column).
Limb reduction defect and growth retardation correlation of NIPBL copy number changes found in CdLS patients in our study.
References
- Aradhya S, Lewis R, Bonaga T, Nwokekeh N, Stafford A, Boggs B. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet. Med. 2012;14:594–603. doi: 10.1038/gim.2011.65. [DOI] [PubMed] [Google Scholar]
- Bhuiyan ZA, Stewart H, Redeker EJ, Mannens MM, Hennekam RC. Large genomic rearrangements in NIPBL are infrequent in Cornelia de Lange syndrome. Eur. J. Hum. Genet. 2007;15:505–508. doi: 10.1038/sj.ejhg.5201776. [DOI] [PubMed] [Google Scholar]
- Borck G, Redon R, Sanlaville D, Rio M, Prieur M, Lyonnet S. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J. Med. Genet. 2007;41:e128. doi: 10.1136/jmg.2004.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castronovo P, Delahaye-Duriez A, Gervasini C, Azzollini J, Minier F, Russo S. Somatic mosaicism in Cornelia de Lange syndrome: a further contributor to the wide clinical expressivity? Clin. Genet. 2010;78:560–564. doi: 10.1111/j.1399-0004.2010.01408.x. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012a;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012b;90:1014–1027. doi: 10.1016/j.ajhg.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasini C, Parenti I, Picinelli C, Azzollini J, Masciadri M, Cereda A. Molecular characterization of a mosaic NIPBL deletion in a Cornelia de Lange patient with severe phenotype. Eur. J. Med. Genet. 2013a;56:138–143. doi: 10.1016/j.ejmg.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Gervasini C, Picinelli C, Azzollini J, Rusconi D, Masciadri M, Cereda A. Genomic imbalances in patients with a clinical presentation in the spectrum of Cornelia de Lange syndrome. BMC Med. Genet. 2013b;14:41. doi: 10.1186/1471-2350-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J. Med. Genet. 2013;50:339–344. doi: 10.1136/jmedgenet-2012-101477. [DOI] [PubMed] [Google Scholar]
- Kline AD, Stanley C, Belevich J, Brodsky K, Barr M, Jackson LG. Developmental data on individuals with the Brachmann-de Lange syndrome. Am. J. Med. Genet. 1993;47:1053–1058. doi: 10.1002/ajmg.1320470724. [DOI] [PubMed] [Google Scholar]
- Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR. Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A. 2007;143A:1287–1296. doi: 10.1002/ajmg.a.31757. [DOI] [PubMed] [Google Scholar]
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat. Genet. 2007;39(Suppl. 7):S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- Novara F, Alfei E, D'Arrigo S, Pantaleoni C, Beri S, Achille V. 5p13 microduplication syndrome: a new case and better clinical definition of the syndrome. Eur. J. Med. Genet. 2013;56:54–58. doi: 10.1016/j.ejmg.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Pehlivan D, Hullings M, Carvalho CM, Gonzaga-Jauregui CG, Loy E, Jackson LG. NIPBL rearrangements in Cornelia de Lange syndrome: evidence for replicative mechanism and genotype-phenotype correlation. Genet. Med. 2012;14:313–322. doi: 10.1038/gim.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Morcillo P, Dorsett D. Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics. 1999;152:577–593. doi: 10.1093/genetics/152.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Korom M, Aulner N, Martens A, Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell. Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo S, Masciadri M, Gervasini C, Azzollini J, Cereda A, Zampino G. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur. J. Hum. Genet. 2012;20:734–741. doi: 10.1038/ejhg.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumans J, Wincent J, Barbaro M, Djureinovic T, Maguire P, Forsberg L. Comprehensive mutational analysis of a cohort of Swedish Cornelia de Lange syndrome patients. Eur. J. Hum. Genet. 2007;15:143–149. doi: 10.1038/sj.ejhg.5201737. [DOI] [PubMed] [Google Scholar]
- Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet. 2012;90:693–700. doi: 10.1016/j.ajhg.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Inoue K, Bi W, Walz K, Park SS, Kurotaki N. Genomic disorders: genome architecture results in susceptibility to DNA rearrangements causing common human traits. Cold Spring Harb. Symp. Quant. Biol. 2003;68:445–454. doi: 10.1101/sqb.2003.68.445. [DOI] [PubMed] [Google Scholar]
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical features of 10 patients with copy number variations in NIPBL. The final column summarizes the facial features of CdLS patients identified with point mutations in NIBPL in comparison with the facial features of our patient cohort (second to last column).
Limb reduction defect and growth retardation correlation of NIPBL copy number changes found in CdLS patients in our study.