

Abstract
Influenza viruses have been responsible for the largest pandemics in the previous century. Although vaccination and prophylactic antiviral therapeutics are the primary defense against influenza virus, there is a pressing need to develop new antiviral agents to circumvent the limitations of current therapies. The endonuclease activity of the influenza virus PAN protein is essential for virus replication and is a promising target for novel anti-influenza drugs. To facilitate the discovery of endonuclease inhibitors, we have developed a high-throughput fluorescence polarization (FP) assay, utilizing a novel fluorescein-labeled compound (Kd = 0.378 μM) and a PAN construct, to identify small molecules that bind to the PAN endonuclease active site. Several known 4-substituted 2,4-dioxobutanoic acid inhibitors with high and low affinities have been evaluated in this FP-based competitive binding assay, and there was a general correlation between binding and the reported inhibition of endonuclease activity. Additionally, we have demonstrated the utility of this assay for identifying endonuclease inhibitors in a small diverse targeted fragment library. These fragment hits were used to build a follow up library that that led to new active compounds which demonstrate FP binding and anti-influenza activities in plaque inhibition assays. The assay offers significant advantages over previously reported assays, and is suitable for high-throughput and fragment-based screening studies. Additionally the demonstration of the applicability of a mechanism-based ‘targeted fragment’ library supports the general potential of this novel approach for other enzyme targets. These results serve as a sound foundation for the development of new therapeutic leads targeting influenza endonuclease.
INTRODUCTION
Influenza viruses cause some of the most common and aggressive infections of the upper respiratory tract and lungs (1). During the 2010–2011 season, 89% of total viruses detected in North America were influenza type A viruses (2). Infections with unusually virulent strains of the influenza A virus have led to many millions of deaths in a single season, notably during the 1918 influenza pandemic (3). Although vaccination can prevent influenza in 70%–90% of healthy adults (4), vaccines are only protective against a limited range of strains and are not effective against new, potentially pandemic strains. Also, even in the best case scenarios, the rate of protection is less than 40% in high-risk groups such as infants, the elderly, pregnant women, and individuals with weakened immune systems. In addition, the ~6 month lag time for the development and manufacture of new vaccines will always limit their applicability (5-7). The neuraminidase inhibitors oseltamivir (Tamiflu) and zanamivir (Relenza) are two very effective drugs developed to attenuate the incidence of influenza infection when used prophylactically and to reduce the severity of symptoms when given within 1–2 days of infection (8). However, the long-term effectiveness of these drugs is a concern due to the emergence of drug-resistant strains. Thus, there is an urgent need for new measures to prevent and treat influenza virus infection, especially in high-risk groups and during an influenza pandemic.
There have been significant efforts to identify novel targets in the influenza life cycle against which small molecule inhibitors can be developed (9 - 18). The influenza RNA-dependent RNA polymerase (RdRp) catalyzes both transcription and replication during infection and is highly conserved among influenza A, B, and C strains. Influenza RdRp is a heterotrimer composed of 3 subunits (PA, PB1, and PB2) that associates with the 5′- and 3′-ends of each viral RNA (vRNA) nucleoprotein genome segment where it is poised to perform both transcription and replication (19-21). Transcription begins with the binding of PB2 to capped host cell premRNAs, following which PA N-terminal domain (PAN) catalyzes endonuclease activity that results in cleavage of the host pre-mRNA. This “cap-snatching” mechanism generates short capped RNA oligoribonucleotides which are then used to prime viral mRNA transcription by PB1 (22, 23, 26 – 28).
Recent crystal structures have revealed that the endonuclease active site resides within the PA N-terminal domain (PAN) (22, 25, 26). In two separate studies strong structural evidence that PA contains the endonuclease site (22) was further refined by calorimetry studies, which demonstrated that Mn++ binds preferentially over Mg++ and that two Mn++ ions are bound to the construct (23). These studies when taken together lend strong support for a two-metal active-site model for PA endonuclease.
New work in the design of small molecule inhibitors that target this polymerase is an area of growing interest and these recent structural studies on RdRp sub-domains (22, 25, 26) will facilitate these efforts. The cap-snatching process is essential for influenza infection, and the structurally characterized endonuclease active site of PAN has been recognized as a promising target for the discovery of novel anti-influenza drugs. A series of 4-subsituted-2,4-dioxobutanoic acids that selectively target the endonuclease activity was previously discovered by researchers at Merck (29, 30). Bunyavirus is an RNA virus that also contains a similar endonuclease for cap-snatching during transcription, and a co-crystal structure with 2,4-dioxo-4-phenylbutanoic acid (DPBA, compound 1, Figure 1) revealed that DPBA interacts directly with the two active-site metal ions (31). Given the high active-site homology between the influenza endonuclease active-site these structural data also strongly suggest that DPBA binds to PA in an orientation similar to that of the bunyavirus endonuclease site (31). This structural confirmation of the presumed inhibitor-endonuclease complex supports the development of rapid and reproducible assays that can measure the binding of small molecules to the two-metal endonuclease active-site in PAN.
Figure 1.

Three known RdRp influenza endonuclease activity inhibitors (1–3) and the structure of the fluorescent ligand (4) that was developed for our FP assay.
Several biochemical and cell-based assays have been developed to evaluate the activity of influenza RdRp (18, 32 - 34). Although there has been some notable success in increasing the throughput of RdRp polymerase assays (18), only two assays can readily differentiate between the endonuclease and polymerase activities of RdRp by the measurement of cap-dependent polymerase activity (19, 23, 29, 33), or by the direct measurement of nucleic acid hydrolysis by electrophoresis (22, 35). The cap-dependent endonuclease activity assay is radiometric, low-throughput, labor-intensive and impractical for high throughput approaches, while the electrophoretic method is primarily useful for the low-throughput qualitative assessment of endonuclease activity. These issues limit the applicability of these assays to lead discovery in this area. On the other hand, fluorescence polarization (FP) has been widely used in assays that are compatible with high-throughput screening (HTS) and fragment-based screens; this has allowed FP screening to be used to successfully identify small molecule binders for numerous protein targets (36 - 49). FP is used to monitor the change in molecular motion of a labeled species upon binding to a target, and is calculated as the ratio of the difference between the vertical and horizontal components of the emitted light over the sum of each component. Polarization is a dimensionless value (mP) and is therefore independent of the emitted light or the concentration of the fluorophore (50). These features make FP assays highly suitable for screening compound libraries in a high-throughput format to identify small molecule inhibitors. In this report, we describe the design, optimization and validation of a robust FP assay for binding to the influenza endonuclease active-site using the isolated PAN domain of RdRp and a novel fluorescently labeled 4-subsituted-2,4-dioxobutanoic acid probe (4). This FP assay has been adapted to a format that would be suitable for HTS, and was successfully used to demonstrate the specificity of known endonuclease inhibitors and to identify novel inhibitors of this promising influenza target.
RESULTS AND DISCUSSION
Fluorescent Ligand Design
Compound 2 (Figure 1) inhibits the influenza endonuclease-dependent polymerase activity with an IC50 of 0.43 μM, making it one of the most potent small molecule inhibitors of this type that has been reported in the literature (30). No structural data are available for the complex between 2 and PAN, but 2 is closely related to 1 (DPBA) which has been demonstrated to specifically inhibit the isolated endonuclease domain (22) and to coordinate the two metal ions in the related active site of the bunyavirus endonuclease (31). Our FP ligand design started from compound 2 because this compound has the best in vitro activity among all reported influenza endonuclease inhibitors (30), and derivatives of 2 would be expected to show tight binding. Previous SAR studies with influenza endonuclease inhibitory analogs of 2 have shown that hydrophobic groups were well tolerated when the piperazine nitrogen is substituted with hydrophobic groups (24), so we chose to add the fluorescent label (fluorescein) at this position. A fluorescent ligand with a low Kd is desirable to minimize the amount of protein and tracer needed to obtain a strong FP signal while maintaining the required dynamic range. A reasonably tight binding fluorescent ligand would also be able to measure a wide range of inhibitor potencies and provides the necessary sensitivity to identify more weakly binding inhibitors. We were pleased to find through direct binding experiments that 4 binds to PAN with a Kd of 0.378 μM and a dynamic range of 124.3 ± 0.9 mP (Figure 2).
Figure 2.

Direct binding experiment to determine the Kd and dynamic range of 4. (◊) 4 alone (0% binding) (●) PAN protein + 4 after a 15-min incubation (Kd = 0.378 μM). (■) PAN protein + 4 after a 24-h incubation (Kd = 0.353 μM).
To confirm that the complex between 4 and PAN was the result of a specific binding interaction, the direct binding experiment was repeated with immunoglobulin G (IgG) and bovine serum albumin (BSA) (2-fold serial dilutions from 100 μM) in the presence of 4 (60 nM). Non-specific binding to IgG and BSA was not observed as indicated by the absence of a binding curve (Supplementary Figure 1). The difference in mP values between buffer and protein is due to the tryptophan residues in the protein.
Assay Development with potential for HTS
In order to assess the stability of the FP assay, we first examined three variables: incubation time, total fluorescence, and DMSO concentration, before embarking on the competitive binding studies. The Kd and ΔmP values observed for measurements at 15-min and 24-h incubations at room temperature were practically identical (Figure 2), an incubation time of 15 min was therefore chosen for further optimization steps to facilitate rapid screening of compounds.
To demonstrate that FP is independent of total fluorescence, increasing concentrations of 4 (0.080–60 nM) were titrated against a constant PAN concentration of 0.8 μM. The data are summarized in Supplementary Figure 2 and show a linear increase in total fluorescence with increasing concentrations of 4, whereas the FP remains constant. Therefore, small molecule inhibitors that are intrinsically fluorescent are likely to have negligible effects on the outcome of the assay (50).
DMSO is a commonly used solvent in HTS, and its influence on the FP assay was investigated by titrating it into a mixture of 4 (60 nM) and PAN (0.8 μM). A stable total fluorescence and FP was observed at 10% DMSO (Supplementary Figure 3). 10% DMSO was therefore used for all subsequent FP assays to maximize the solubilities of the diverse library compounds that will be used for screening.
The Z′ factor is a statistical benchmark to assess the suitability of an assay for HTS (51) and is a measure of the reproducibility in the difference in signal between free and bound tracer controls across a large number of assay wells. An assay with ideal reproducibility displays a Z′ factor of 1, whereas a Z′ factor greater than 0.5 is considered acceptable for a good high-throughput assay (51). The Z′ factor calculated for our FP assay is 0.91 (Supplementary Figure 4), confirming that these assay conditions are suitable for HTS.
Competition FP Assay
On the basis of the Kd value and dynamic range determined in the direct binding FP experiments, we established a competition FP assay to screen compounds for their ability to displace 4 from PAN. The affinity of competitors for PAN was quantified as an equilibrium dissociation constant (Ki), which was calculated from the inhibition curve (52). Compound 2 was able to compete with 4 with a Ki value of 0.09 μM and therefore was used as a positive control in subsequent competitive binding experiments. Two other known PAN inhibitors, 1 (DPBA) and 3 were measured for competitive binding and showed Ki values of 0.48 μM and 0.85 μM respectively (Figure 3 and Table 1). This FP assay was also able to accurately identify a compound known to be inactive against influenza endonuclease (19, Figure 3 and Table 1), which is important since this represents an essential feature of any useful binding assay.
Figure 3.

Competitive binding experiment measuring the displacement of 4 from PAN. (▼) DMSO negative control (0% inhibition) (◊) 250 μM compound 2 (100% inhibition) (●) Compound 2 (Ki = 0.09 μM) (■) DPBA (Ki = 0.48 μM) (▲) Compound 3 (Ki = 0.85 μM) (♦) Compound 19 (no inhibition).
Table 1.
Comparison of results of our FP competitive binding assay to reported anti-influenza activities of known endonuclease inhibitors. Reported FP values are the average of triplicate measurements ± standard error of the mean (SEM). ND = no data.
| Compound | Structure | Reported IC50 (MM) | FP EC50 (MM) | FP Ki (MM) | BEIa | LEIb | |
|---|---|---|---|---|---|---|---|
| 1 (DPBA) |

|
R = H | 21.3c | 2.07 ± 0.03 | 0.48 ± 0.01 | 33.0 | 0.62 |
| 5 | R = Ph | 3.7c | 3.56 ± 0.07 | 0.96 ± 0.02 | 22.5 | 0.42 | |
| 6 | R = piperazine | ND | 1.5 ± 0.8 | 0.3 ± 0.3 | 23.7 | 0.45 | |
| 7 | R = methoxy | ND | 6.7 ± 0.1 | 1.97 ± 0.03 | 25.7 | 0.49 | |
| 8 | R = oxyphenyl | ND | 1.45 ± 0.07 | 0.28 ± 0.02 | 23.1 | 0.43 | |
| 9 | R = benzyloxy | ND | 6.8 ± 0.2 | 1.99 ± 0.06 | 19.1 | 0.30 | |
| 2 (L-742,001) |
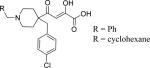
|
R = Ph | 0.430c | 0.86 ± 0.04 | 0.09 ± 0.01 | 17.0 | 0.34 |
| 10 | R = cyclohexane | 0.33c | 0.8 ± 0.2 | 0.084 ± 0.001 | 16.9 | 0.34 | |
| 3 |

|
15.0d | 3.22 ± 0.04 | 0.85 ± 0.02 | 34.3 | 0.65 | |
| 11 |

|
ND | 6.8 ± 0.5 | 2.0 ± 0.2 | 27.8 | 0.52 | |
| 12 |

|
ND | 8.17 ± 0.02 | 2.440 ± 0.006 | 24.0 | 0.46 | |
| 13 |

|
ND | >250 | ||||
| 14 |

|
ND | >250 | ||||
| 15 |

|
ND | >250 | ||||
| 16 |
|
R = CH3 | >500c | >250 | |||
| 17 | R = tert-butyl | ND | >250 | ||||
| 18 |

|
>500c | >250 | ||||
| 19 |

|
>500c | >250 | ||||
BEI = Binding efficiency index based on FP data (pKi,/MW), where MW is molecular weight in kDa (55).
LE = Ligand efficiency based on FP data (ΔG/N), where ΔG = -RTlnKi and N is the number of heavy atoms (56).
Published results in an influenza virus in vitro transcription assay (29).
Published results in an influenza virus in vitro transcription assay (53).
Assay Validation
A series of 4-subsituted 2,4-dioxobutanoic acids has been previously shown to selectively target the cap-dependent endonuclease of the influenza transcriptase complex (29) over a broad range of inhibitory concentrations (IC50) in the radiometric cap-dependent transcription assay. We synthesized seven compounds from this series and an additional N-hydroxyimide derivative (3) that was previously shown to have a similar mode of action to 2 (53), and quantitatively compared our FP assay with the reported cap-dependent RNA polymerase assay (Table 1) (54). Analogues 2 and 10 were the most potent in both assays, with negligible differences in each assay. Overall, the IC50 values from the reported cap-dependent transcriptase inhibitory data generally correlate with the values from our FP binding assay (Figure 3 and Table 1). This correlation confirms that our FP assay provides reliable information on the binding of potential inhibitors to the relevant active-site of PAN that generally parallels the inhibition of the enzymatic activity of RdRp endonuclease activity.
Our FP data also provide useful structure activity relationship (SAR) information on this compound series. The reproducibility of inactive compounds 18 and 19 confirms the importance of the oxygen at C2 in facilitating the two-metal interaction with PAN. Also, the terminal carboxylic acid moiety at C1 is necessary for binding to PAN as indicated by the inactivity of esters 16 and 17. Replacement of the phenyl ring with a pyridyl group (13), as well as fusing C3 to the aromatic ring with an ether (14) or alkyl group (15) also renders compounds inactive. As for the remaining analogues, it appears that ortho- and para-substituents are tolerated (6–9, 11 and 12) and lipophilic moieties are preferred (6 and 8). In addition to SAR information, other factors such as binding efficiency index (BEI) (55) and ligand efficiency (LE) (56) are important to consider when selecting active fragments for subsequent modification (Tables 1, 2, and 3). SAR, BEI, and LE, combined with potency, drug-likeness, and cytotoxicity (CC50, Tables 2 and 3) are useful criteria for analog design and the selection of compounds for advancement to in vitro, and eventually, to in vivo antiviral efficacy studies.
Table 2.
Results of the FP competitive binding assay for a set of representative fragments. (See Supporting Information, Table S1 for the full set of inactive fragments). Reported FP values are the average of triplicate measurements ± SEM. NT = not tested.
| Compound | Scaffold | R1 | R2 | R3 | R4 | FP EC50 (μM) | FP Ki (μM) | BEIa | LEIb | CC50 (μM)c |
|---|---|---|---|---|---|---|---|---|---|---|
| 20 |
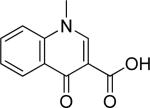
|
>250 | NT | |||||||
| 21 |
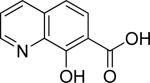
|
>250 | NT | |||||||
| 22 |

|
>250 | NT | |||||||
| 23 |
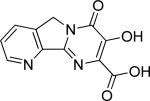
|
67.4 ± 2.4 | 21.6 ± 1.1 | 19.0 | 0.36 | NT | ||||
| 24 |

|

|
H | OH | OH | 58.9 ± 0.5 | 18.7 ± 0.2 | 16.3 | 0.31 | NT |
| 25 |
|
H | OH | OH | 18.09 ±0.04 | 5.01 ± 0.02 | 22.4 | 0.43 | NT | |
| 26 | H | OH | OMe | 71.3 ± 0.6 | 20.3 ± 0.2 | 18.8 | 0.39 | NT | ||
| 27 |
|
H | OH | OH | 5.92 ± 0.05 | 1.84 ± 0.01 | 24.6 | 0.47 | NT | |
| 28 | H | OH | OMe | 156.1 ± 1.9 | 50.42 ± 0.59 | 17.4 | 0.33 | NT | ||
| 29 | CH3 | OH | OH | 151.8 ± 1.5 | 49.0 ± 0.7 | 17.4 | 0.34 | NT | ||
| 30 |
|
H | OH | OH | 119.7 ± 1.5 | 38.6 ± 0.6 | 17.9 | 0.34 | NT | |
| 31 |

|
H | OH | OH | 3.8 ± 0.3 | 1.1 ± 0.1 | 25.4 | 0.48 | >60 | |
| 32 | H | OH | OMe | 25.46 ± 0.15 | 8.00 ± 0.09 | 20.6 | 0.39 | NT |
Table 3.
Results of FP competitive binding and compound cytotoxicity for a set of carboxamides based on the most potent fragment (31). Reported values are the average of triplicate measurements ± SEM. NT = not tested.
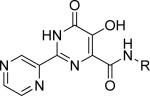
| ||||||
|---|---|---|---|---|---|---|
| Compound | R | FP EC50 (μM) | FP Ki (μM) | BEIa | LEIb | CC50 (μM)c |
| 33 |
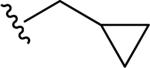
|
3.66 ± 0.08 | 0.99 ± 0.03 | 20.9 | 0.40 | >60 |
| 34 |
|
88.12 ± 0.05 | 28.57 ± 0.01 | 15.0 | 0.30 | NT |
| 35 |

|
216.7 ± 1.5 | 70.5 ± 0.7 | 13.0 | 0.27 | NT |
| 36 |
|
>250 | NT | |||
| 37 |

|
11.48 ± 0.01 | 3.556 ± 0.004 | 17.3 | 0.33 | NT |
| 38 |

|
5.41 ± 0.06 | 1.65 ± 0.02 | 17.9 | 0.33 | >60 |
| 39 |

|
22.6 ± 0.2 | 7.1 ± 0.1 | 15.3 | 0.28 | >60 |
| 40 |
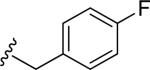
|
3.7 ± 0.5 | 1.2 ± 0.2 | 17.3 | 0.33 | >60 |
| 41 |
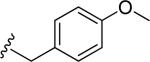
|
35.3 ± 0.8 | 11.2 ± 0.3 | 14.0 | 0.26 | >60 |
| 42 |

|
3.5 ± 0.3 | 0.72 ± 0.05 | 18.2 | 0.34 | >60 |
| 43 |

|
>250 | >60 | |||
| 44 |
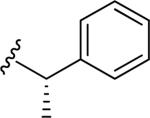
|
7.67 ± 0.03 | 2.28 ± 0.01 | 16.7 | 0.31 | >60 |
| 45 |

|
1.5 ± 0.2 | 0.29 ± 0.06 | 19.4 | 0.36 | >60 |
| 46 |

|
5.52 ± 0.09 | 1.61 ± 0.03 | 16.5 | 0.31 | >60 |
| 47 |

|
4.851 ± 0.007 | 1.392 ± 0.002 | 17.0 | 0.32 | 7.1 |
| 48 |
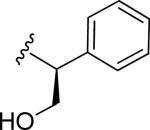
|
3.5 ± 0.6 | 1.0 ± 0.3 | 17.0 | 0.32 | >60 |
| 49 |

|
24.3 ± 0.4 | 7.6 ± 0.3 | 14.1 | 0.26 | 10.2 |
Fragment-Based FP Screen
The 4-substituted 2,4-dioxobutanoic acids developed by Merck did not lead to a report of a clinical compound. However, recent progress in the development of inhibitors that target the mechanistically similar sites in the RdRp of hepatitis C virus (HCV) (57), and the integrase of the human immunodeficiency virus (HIV) (58), suggests that clinically useful compounds may be found for influenza RdRp, if a more drug-like scaffold could be identified. This approach has been used by the Webb group to design a two-metal polymerase targeted library (59), using a 2-substituted-4,5-dihydroxypyrimidine scaffold that matches the pharmacophore identified by the Parkes group (53). The mechanistic basis of this two-metal targeted library (59) has recently been given additional structural validation by the publication of structures of prototype foamy virus integrase co-crystalized with DNA and the structurally similar MK0518 (60). The co-crystal structures clarify the specific binding mode to the two-metal active site for 2-substituted-4,5-dihydroxypyrimidine derivatives (60) that is consistent with the previously proposed pharmacophore (59). Using this 2-substituted-4,5-dihydroxypyrimidine scaffold type, which is common to some HCV RdRp and HIV integrase inhibitors (57, 58), we synthesized a series of acid and ester fragments that has been used previously for targeted library synthesis (see Table 2) (59). We also supplemented this small group of fragments with structurally distinct fragments that also have the potential to interact with a two-metal site, or that have are similar to substructures that are present in known HIV integrase inhibitors (Supporting Information Table S1) (60, 61). From this library of 42 compounds, we were able to identify several active fragments by screening in a dose response mode with concentrations up to 250 μM (see the Supporting Information Extended Methods, competitive binding FP assays section). Table 2 summarizes these results for the active fragments (defined as those compounds with an EC50 <250 μM) and the closely related inactive compounds. This table also includes BEI and LE data for each fragment based on the observed FP binding (55, 56). Among the fragments investigated only those based on the 2-substituted-4,5-dihydroxypyrimindine scaffold showed significant activity (see Table 2). These active compounds show a preference for the unsubstituted aromatic moieties at the R1 position (Table 2), among the fragments screened (see Supporting Information, Table S1 for the full set of inactive fragments). The 2,5-pyrazine derivative 31 is the most potent and also shows good binding efficiency (BEI of 25.4 and LE of 0.48) without exhibiting significant cytotoxicity in MDCK cells (CC50 >60 μM).
FP Screen of Carboxamide Derivatives of 31
The Webb group has previously reported the synthesis of a two-metal polymerase targeted library containing a 2-substituted-4,5-dihydroxypyrimidine scaffold, and this has led to the identification of a compound with in vitro activity against the Sendai virus (59). Using the chemistry developed in this work, we synthesized a small set of carboxamides based on the most active fragment (31) and evaluated their binding as measured by FP as well as their cytotoxicity in MDCK cells. As shown in Table 3, benzyl amides (38–42) show good activity, but the increase in chain length to two carbons (43) is not desirable. Interestingly, α-substitution at the benzylic position results in increased potency (42). To further explore the potency of this racemic compound, we synthesized the enantiomerically pure carboxamides (44 and 45) from chiral amines which revealed a binding preference for the (R) enantiomer 45. Next, we synthesized a set of analogues to elucidate the SAR around 45 (Table 3). Once again, substitution at the para-position results in a decrease in potency (46). Both the replacement of phenyl group in 45 with a cyclohexyl moiety (47) and the substitution of the α-methyl with a hydroxyl group (48) are well tolerated, but the constrained amide analogue (49) is less active than 45. None of these compounds showed significant cytotoxicity in MDCK cells, the cell line that is used for influenza plaque inhibition to assess the potential for in vitro anti-influenza activity of compounds.
Antiviral Activity of FP Hits Measured by Plaque Inhibition
In order to evaluate the in vitro antiviral activity of endonuclease inhibitors identified by our FP assay, we measured their ability to inhibit influenza A/PR/8/34 viral plaque formation in MDCK cells (Figure 4). As shown in Figure 4 the compounds 42, 44 and 45 show similar activities and the same rank order for activity as seen with the FP assay, although the ratios of activity are not identical between these two assay types. Compound 31 shows significantly less activity (IC50 >50 μM), which could in part be due to a reduction of the cell permeability of this particular ionizable carboxylic acid, though we have not explored the permeability of our fragment library. As expected, the FP inactive control compounds (16 and 43) were also inactive in inhibiting viral plaque formation. As positive controls, 2 and oseltamivir carboxylate, were also measured for antiviral activity by viral plaque inhibition, and the activities obtained for these compounds compare well with those reported previously (62, 63).
Figure 4.

Viral plaque inhibition of candidates identified by FP. (▼) DMSO negative control (0% inhibition), (♦) oseltamivir carboxylate positive control (100% inhibition), (○) Compound 16 (IC50 >100 μM), (□) Compound 43 (IC50 >100 μM), (∆) Compound 31 (IC50 >50 μM), (◊) No infection, (●) Compound 42 (IC50 = 14.28 ± 0.07 μM), (▽) Compound 44 (IC50 = 18.61 ± 0.09 μM), (■) Compound 45 (IC50 = 12.68 ± 0.08μM), (▲) Compound 2 (IC50 = 3.13 ± 0.08 μM).
In conclusion we have developed the first reported binding assay that can be used to screen for molecules that bind to the PAN endonuclease subunit of influenza polymerase. The assay correlates with endonuclease enzymatic activity for a series of inhibitors and should be widely applicable for use in a low, medium or high-throughput formats that include relatively high concentrations of potential inhibitors (up to 250 μM). During the course of assay miniaturization, various factors that would have an effect on screening efficiency and applicability, including incubation time, temperature, DMSO tolerance, and total fluorescence were each subjected to optimization. Miniaturized FP assays not only enable screening of larger libraries, but are also cost effective and can provide validation of the direct target of active compounds. We have demonstrated the use of this assay for the initial identification of active inhibitors and their subsequent development, which has led to novel antiviral compounds with an associated SAR. Blocking the active site of the PA subunit of the influenza polymerase is a potentially powerful therapeutic strategy for inhibiting viral transcription. Although small molecules have previously been developed to inhibit this cellular event (24, 30), none have thus far reached the clinic. Our development of a facile screening method and initial identification of active drug-like compounds will facilitate the discovery and development of new diverse inhibitors with more drug-like properties and subsequently new lead compounds that may, in turn, ultimately advance through clinical trials to become the next generation of drugs for pandemic and seasonal influenza. Additionally the demonstration of the applicability of a mechanism-based ‘targeted-fragment’ library supports the general potential of this novel approach for other enzyme targets.
METHODS
Details describing the high throughput optimization of the FP assay (fluorescent ligand concentration, equilibration time and signal stability, FP independence on total fluorescence intensity, DMSO tolerance, accuracy and precision, estimation of Kd for binding of 4 to PAN, and competitive FP assays) are outlined in the Supporting Information. Also included in the Supporting Information are details describing the expression and purification of PAN, avian H1N1 influenza A virus (A/PuertoRico/8/34) culture, influenza virus plaque inhibition assays in MDCK cells, and quantification of compound cytotoxicity in MDCK cells using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Supplementary Material
Acknowledgements
We thank R. J. Webby [Department of Infectious Diseases, St. Jude Children's Research Hospital (SJCRH)] for helpful discussions, access to numerous protocols, and for providing the influenza virus strain used in this work. We also thank D. Smithson (Department of Chemical Biology and Therapeutics (CBT), SJCRH) for his help with instrument training and data analysis, Y. Shao (Protein Production Facility, SJCRH) for the scale-up of PAN construct used in FP assays, T. Jeevan (Department of Infectious Diseases, SJCRH) for the scale-up of virus used in plaque assays, and also C. Jefferies, A. Lemoff and L. Yang (High Throughput Analytical Chemistry Center, CBT, SJCRH) for their help with compound purification and purity determinations. This work was supported by the American Lebanese and Syrian Associated Charities (ALSAC), St. Jude Children's Research Hospital, and a grant from the Children's Infection Defense Center (CIDC) in the Department of Infectious Diseases of St. Jude Children's Research Hospital.
Footnotes
Supporting Information Available: Supporting information for this work includes figures and methods outlining the HTS optimization of the FP assay, photos of plaque assay results, as well as details for the synthesis and characterization of all new compounds presented. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Garibaldi RA. Epidemiology of community-acquired respiratory tract infections in adults. Incidence, etiology, and impact. Am. J. Med. 1985;78:32–37. doi: 10.1016/0002-9343(85)90361-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization . North America circulation of influenza viruses; Influenza update - 21 April 2011. Geneva, Switzerland: 2011. [Google Scholar]
- 3.Patterson KD, Pyle GF. The geography and mortality of the 1918 influenza pandemic. Bull. Hist. Med. 1991;65:4–21. [PubMed] [Google Scholar]
- 4.Demicheli V, Rivetti D, Deeks JJ, Jefferson TO. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst. Rev. 2004:CD001269. doi: 10.1002/14651858.CD001269.pub2. [DOI] [PubMed] [Google Scholar]
- 5.Patriarca PA, Weber JA, Meissner MK, Stricof RL, Dateno B, Braun JE, Arden NH, Kendal AP. Use of influenza vaccine in nursing homes. J. Am. Geriatr. Soc. 1985;33:463–466. doi: 10.1111/j.1532-5415.1985.tb05456.x. [DOI] [PubMed] [Google Scholar]
- 6.Nichol KL, Wuorenma J, von Sternberg T. Benefits of influenza vaccination for low-, intermediate-, and high-risk senior citizens. Arch. Intern. Med. 1998;158:1769–1779. doi: 10.1001/archinte.158.16.1769. [DOI] [PubMed] [Google Scholar]
- 7.Glezen WP, Decker M, Perrotta DM. Survey of underlying conditions of persons hospitalized with acute respiratory disease during influenza epidemics in Houston, 1978-1981. Am. Rev. Respir. Dis. 1987;136:550–555. doi: 10.1164/ajrccm/136.3.550. [DOI] [PubMed] [Google Scholar]
- 8.Stiver G. The treatment of influenza with antiviral drugs. CMAJ. 2003;168:49–56. [PMC free article] [PubMed] [Google Scholar]
- 9.Hayden F. Developing new antiviral agents for influenza treatment: what does the future hold? Clin. Infect. Dis. 2009;48(Suppl 1):S3–S13. doi: 10.1086/591851. [DOI] [PubMed] [Google Scholar]
- 10.De Clercq E, Field HJ. Antiviral Chemistry & Chemotherapy's current antiviral agents FactFile 2008 (2nd edition): RNA viruses. Antiviral Chem. Chemother. 2008;19:63–74. doi: 10.1177/095632020801900204. [DOI] [PubMed] [Google Scholar]
- 11.Lüscher-Mattli M. Influenza chemotherapy: A review of the present state of art and of new drugs in development. Arch. Virol. 2000;145:2233–2248. doi: 10.1007/s007050070017. [DOI] [PubMed] [Google Scholar]
- 12.Moscona A. Medical management of influenza infection. Annu. Rev. Med. 2008;59:397–413. doi: 10.1146/annurev.med.59.061506.213121. [DOI] [PubMed] [Google Scholar]
- 13.Wilson JC, von Itzstein M. Recent strategies in the search for new anti-influenza therapies. Curr. Drug Targets. 2003;4:389–408. doi: 10.2174/1389450033491019. [DOI] [PubMed] [Google Scholar]
- 14.Beigel J, Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 2008;78:91–102. doi: 10.1016/j.antiviral.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leyssen P, De Clercq E, Neyts J. Molecular strategies to inhibit the replication of RNA viruses. Antiviral Res. 2008;78:9–25. doi: 10.1016/j.antiviral.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boltz DA, Aldridge JR, Jr., Webster RG, Govorkova EA. Drugs in development for influenza. Drugs. 2010;70:1349–1362. doi: 10.2165/11537960-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010;17:530–538. doi: 10.1038/nsmb.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hooker L, Strong R, Adams R, Handa B, Merrett JH, Martin JA, Klumpp K. A sensitive, single-tube assay to measure the enzymatic activities of influenza RNA polymerase and other poly(A) polymerases: application to kinetic and inhibitor analysis. Nucleic Acids Res. 2001;29:2691–2698. doi: 10.1093/nar/29.13.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honda A, Ueda K, Nagata K, Ishihama A. Identification of the RNA polymerase-binding site on genome RNA of influenza virus. J. Biochem. 1987;102:1241–1249. doi: 10.1093/oxfordjournals.jbchem.a122163. [DOI] [PubMed] [Google Scholar]
- 20.Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc. Natl. Acad. Sci. U. S. A. 1987;84:8140–8144. doi: 10.1073/pnas.84.22.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seong BL, Brownlee GG. Nucleotides 9 to 11 of the influenza A virion RNA promoter are crucial for activity in vitro. J. Gen. Virol. 1992;73:3115–3124. doi: 10.1099/0022-1317-73-12-3115. [DOI] [PubMed] [Google Scholar]
- 22.Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 23.Crépin T, Dias A, Palencia A, Swale C, Cusack S, Ruigrok RW. Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J. Virol. 2010;84:9096–9104. doi: 10.1128/JVI.00995-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomassini JE, Davies ME, Hastings JC, Lingham R, Mojena M, Raghoobar SL, Singh SB, Tkacz JS, Goetz MA. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996;40:1189–1193. doi: 10.1128/aac.40.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao C, Lou Z, Guo Y, Ma M, Chen Y, Liang S, Zhang L, Chen S, Li X, Liu Y, Bartlam M, Rao Z. Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J. Virol. 2009;83:9024–9030. doi: 10.1128/JVI.00911-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature. 2009;458:909–913. doi: 10.1038/nature07720. [DOI] [PubMed] [Google Scholar]
- 27.Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- 28.Gilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crepin T, Sehr P, Lewis J, Ruigrok RW, Ortin J, Hart DJ, Cusack S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008;15:500–506. doi: 10.1038/nsmb.1421. [DOI] [PubMed] [Google Scholar]
- 29.Tomassini J, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings J, Hazuda D, Lewis J, McClements W, Ponticello G, Radzilowski E, Smith G, Tebben A, Wolfe A. Inhibition of Cap (M(7)GPPPXM)-Dependent Endonucelase of Influenza-Virus by 4-subsituted 2,4-dioxobutanoic Acid Compounds. Antimicrob. Agents Chemother. 1994;38:2827–2837. doi: 10.1128/aac.38.12.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hastings JC, Selnick H, Wolanski B, Tomassini JE. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996;40:1304–1307. doi: 10.1128/aac.40.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reguera J, Weber F, Cusack S. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010 doi: 10.1371/journal.ppat.1001101. Epub Sept 16, 2010. DOI: 10.1371/journal.ppat.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin-Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc. Natl. Acad. Sci. U. S. A. 2010;107:19151–19156. doi: 10.1073/pnas.1013592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouloy M, Plotch SJ, Krug RM. Globin Messenger-Rnas Are Primers for Transcription of Influenza Viral-Rna Invitro. Proc. Natl. Acad. Sci. U. S. A. 1978;75:4886–4890. doi: 10.1073/pnas.75.10.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster RG, Salomon R, Franks J, Govorkova EA, Ilyushina NA, Yen HL, Hulse-Post DJ, Humberd J, Trichet M, Rehg JE, Webby RJ, Mann EH. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J. Exp. Med. 2006;203:689–697. doi: 10.1084/jem.20051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwai Y, Takahashi H, Hatakeyama D, Motoshima K, Ishikawa M, Sugita K, Hashimoto Y, Harada Y, Itamura S, Odagiri T, Tashiro M, Sei Y, Yamaguchi K, Kuzuhara T. Anti-influenza activity of phenethylphenylphthalimide analogs derived from thalidomide. Bioorg. Med. Chem. 2010;18:5379–5390. doi: 10.1016/j.bmc.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 36.Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D, Wang S. Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J. Med. Chem. 2001;44:4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 37.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Organic Synthesis Toward Small-Molecule Probes and Drugs Special Feature: Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6811–6816. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carme Mulero M, Orzaez M, Messeguer J, Messeguer A, Perez-Paya E, Perez-Riba M. A fluorescent polarization-based assay for the identification of disruptors of the RCAN1-calcineurin A protein complex. Anal. Biochem. 2010;398:99–103. doi: 10.1016/j.ab.2009.10.045. [DOI] [PubMed] [Google Scholar]
- 39.Chen T, Kablaoui N, Little J, Timofeevski S, Tschantz WR, Chen P, Feng J, Charlton M, Stanton R, Bauer P. Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem. J. 2009;420:283–294. doi: 10.1042/BJ20081899. [DOI] [PubMed] [Google Scholar]
- 40.Evelyn CR, Ferng T, Rojas RJ, Larsen MJ, Sondek J, Neubig RR. High-throughput screening for small-molecule inhibitors of LARG-stimulated RhoA nucleotide binding via a novel fluorescence polarization assay. J. Biomol. Screen. 2009;14:161–172. doi: 10.1177/1087057108328761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology. 2010;399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gradinaru CC, Marushchak DO, Samim M, Krull UJ. Fluorescence anisotropy: from single molecules to live cells. Analyst. 2010;135:452–459. doi: 10.1039/b920242k. [DOI] [PubMed] [Google Scholar]
- 43.Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, Hobbs D, Ellenberger T, Cusumano CK, Hultgren SJ, Janetka JW. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010;53:4779–4792. doi: 10.1021/jm100438s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harikumar KG, Cawston EE, Miller LJ. Fluorescence Polarization Screening for Allosteric Small Molecule Ligands of the Cholecystokinin Receptor. Assay Drug Dev. Technol. 2011;9:394–402. doi: 10.1089/adt.2010.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang X, Aulabaugh A. Application of fluorescence polarization in HTS assays. Methods Mol. Biol. (N. Y.) 2009;565:127–143. doi: 10.1007/978-1-60327-258-2_6. [DOI] [PubMed] [Google Scholar]
- 46.Kimple AJ, Yasgar A, Hughes M, Jadhav A, Willard FS, Muller RE, Austin CP, Inglese J, Ibeanu GC, Siderovski DP, Simeonov A. A high throughput fluorescence polarization assay for inhibitors of the GoLoco motif/G-alpha interaction. Comb. Chem. High Throughput Screening. 2008;11:396–409. doi: 10.2174/138620708784534770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neher TM, Shuck SC, Liu JY, Zhang JT, Turchi JJ. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem. Biol. 2010;5:953–965. doi: 10.1021/cb1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharlow ER, Leimgruber S, Yellow-Duke A, Barrett R, Wang QJ, Lazo JS. Development, validation and implementation of immobilized metal affinity for phosphochemicals (IMAP)-based high-throughput screening assays for low-molecular-weight compound libraries. Nat. Protoc. 2008;3:1350–1363. doi: 10.1038/nprot.2008.111. [DOI] [PubMed] [Google Scholar]
- 49.Thompson S, Messick T, Schultz DC, Reichman M, Lieberman PM. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J. Biomol. Screen. 2010;15:1107–1115. doi: 10.1177/1087057110379154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burke TJ, Loniello KR, Beebe JA, Ervin KM. Development and application of fluorescence polarization assays in drug discovery. Comb. Chem. High Throughput Screening. 2003;6:183–194. doi: 10.2174/138620703106298365. [DOI] [PubMed] [Google Scholar]
- 51.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 52.Owicki JC. Fluorescence polarization and anisotropy in high throughput screening: perspectives and primer. J. Biomol. Screen. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 53.Parkes KEB, Ermert P, Fassler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2003;46:1153–1164. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 54.Roehrl MH, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 55.Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today. 2005;10:464–492. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 56.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 57.Liu-Young G, Kozal MJ. Hepatitis C protease and polymerase inhibitors in development. AIDS Patient Care ST. 2008;22:449–457. doi: 10.1089/apc.2007.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Paz OG, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 59.Boyd VA, Mason J, Hanumesh P, Price J, Russell CJ, Webb TR. 2-substituted-4,5-dihydroxypyrimidine-6-carboxamide antiviral targeted libraries. J. Comb. Chem. 2009;11:1100–1104. doi: 10.1021/cc900111u. [DOI] [PubMed] [Google Scholar]
- 60.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johns BA. HIV-1 Integrase Strand Transfer Inhibitors. Annual Reports Med. Chem. 2010;45:262–276. [Google Scholar]
- 62.Nakazawa M, Kadowaki SE, Watanabe I, Kadowaki Y, Takei M, Fukuda H. PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antiviral Res. 2008;78:194–201. doi: 10.1016/j.antiviral.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi K, Furuta Y, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Shiraki K. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antiviral Chem. Chemother. 2003;14:235–241. doi: 10.1177/095632020301400502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.